
Exploring the Role of Gut Microbiota in Patients with Alopecia Areata


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics
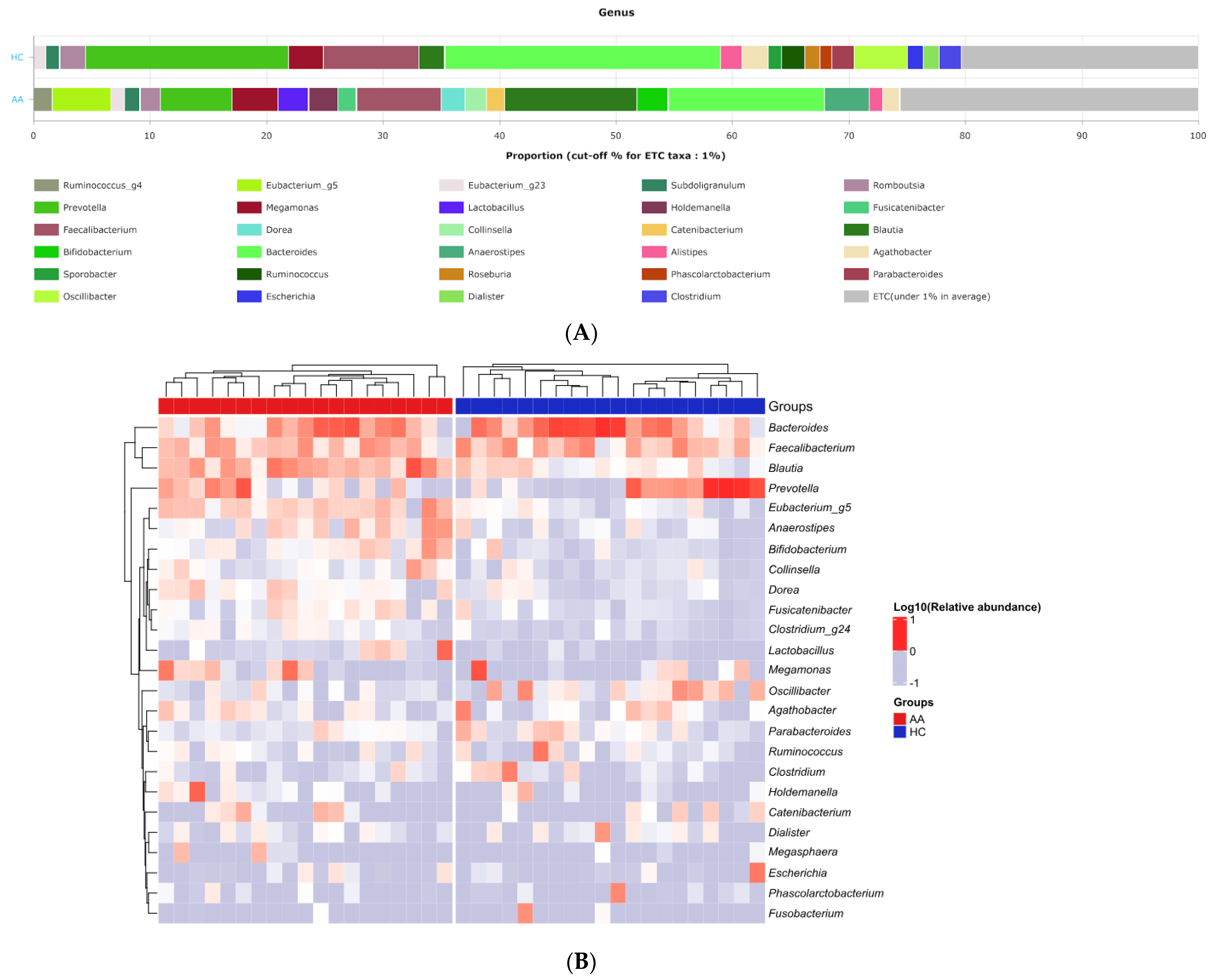
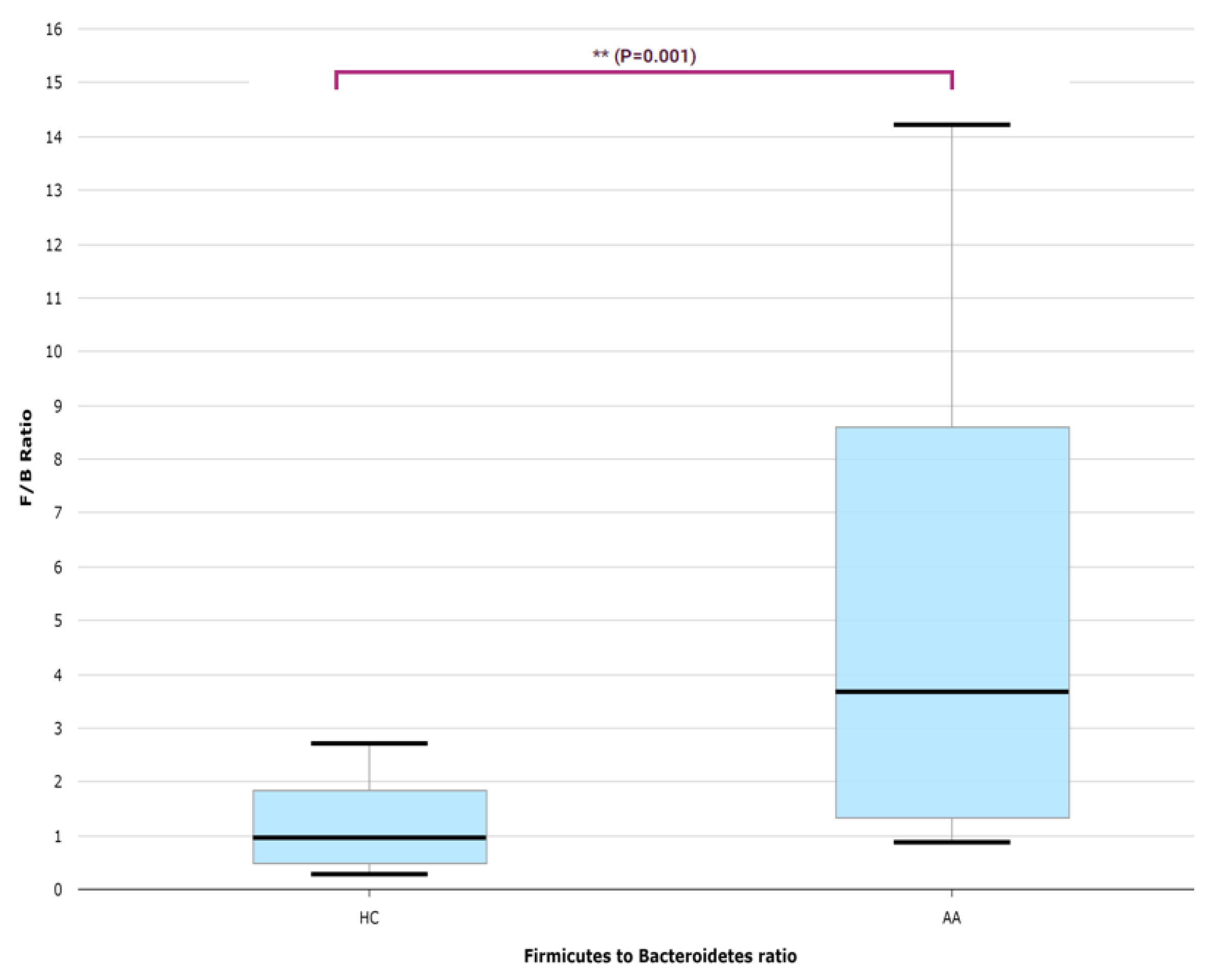
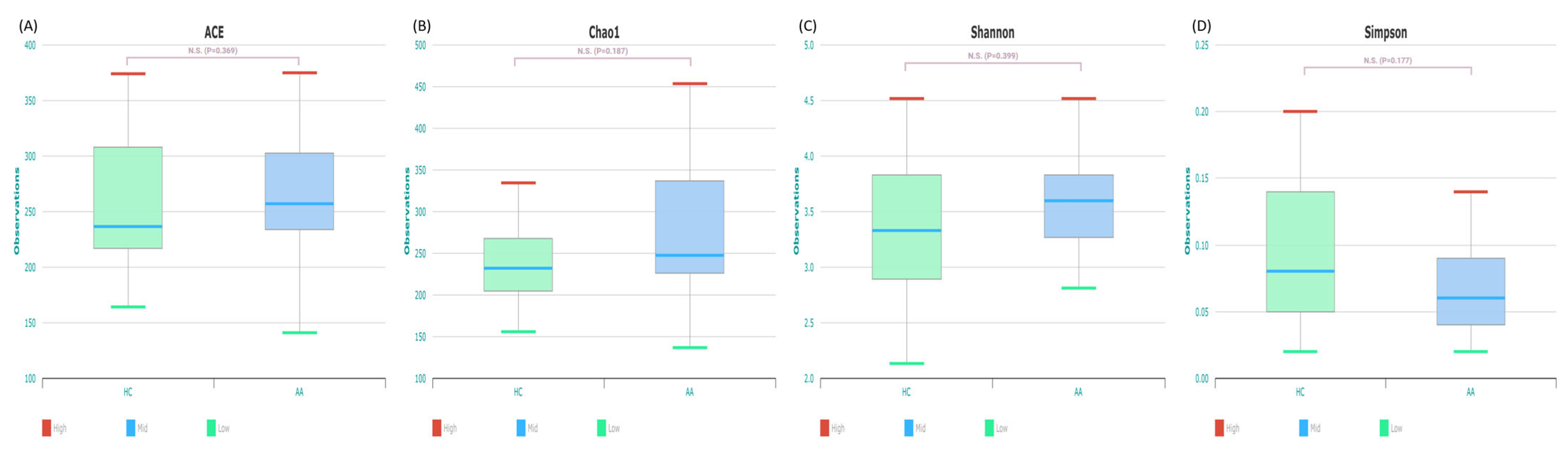
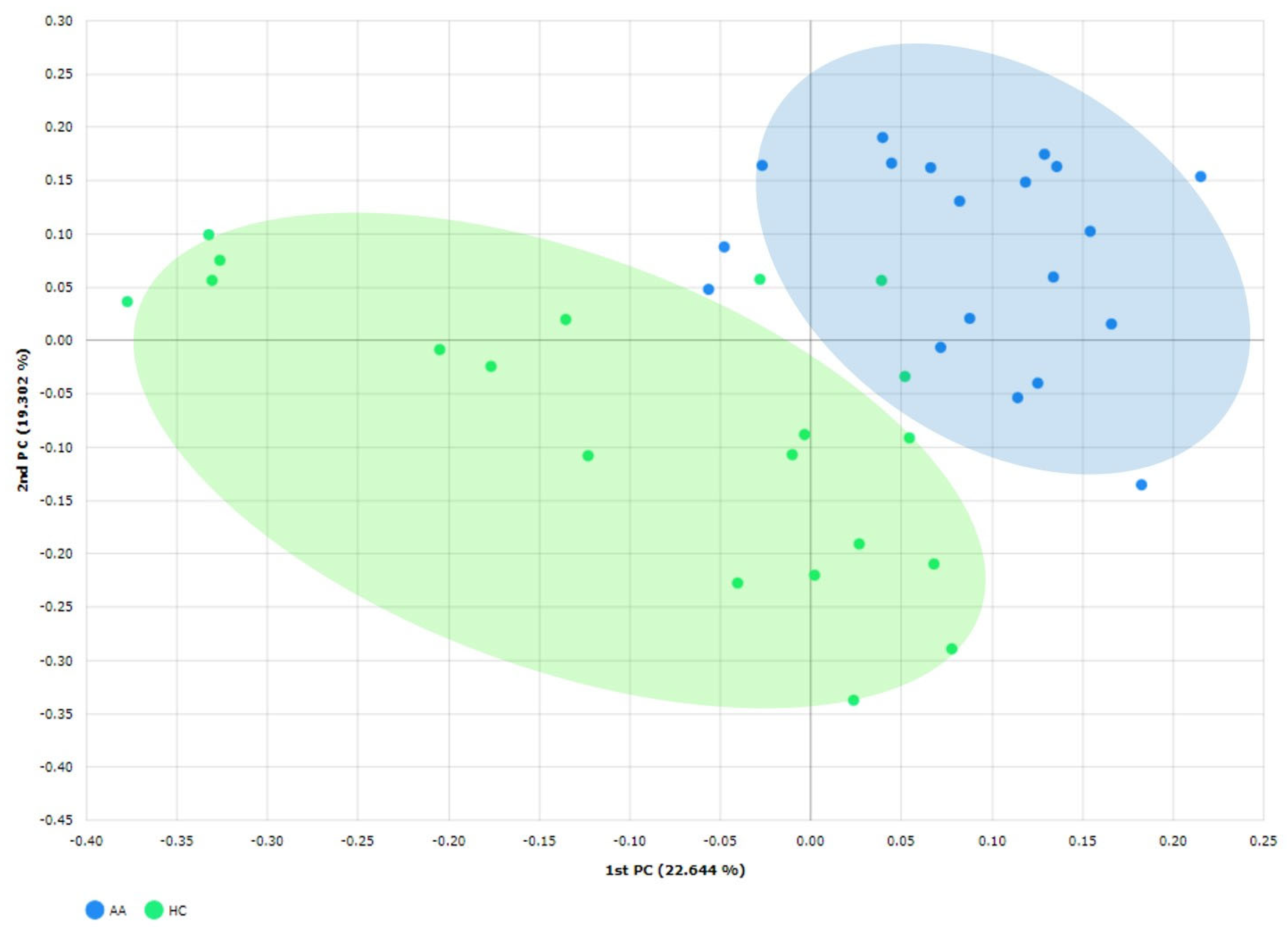
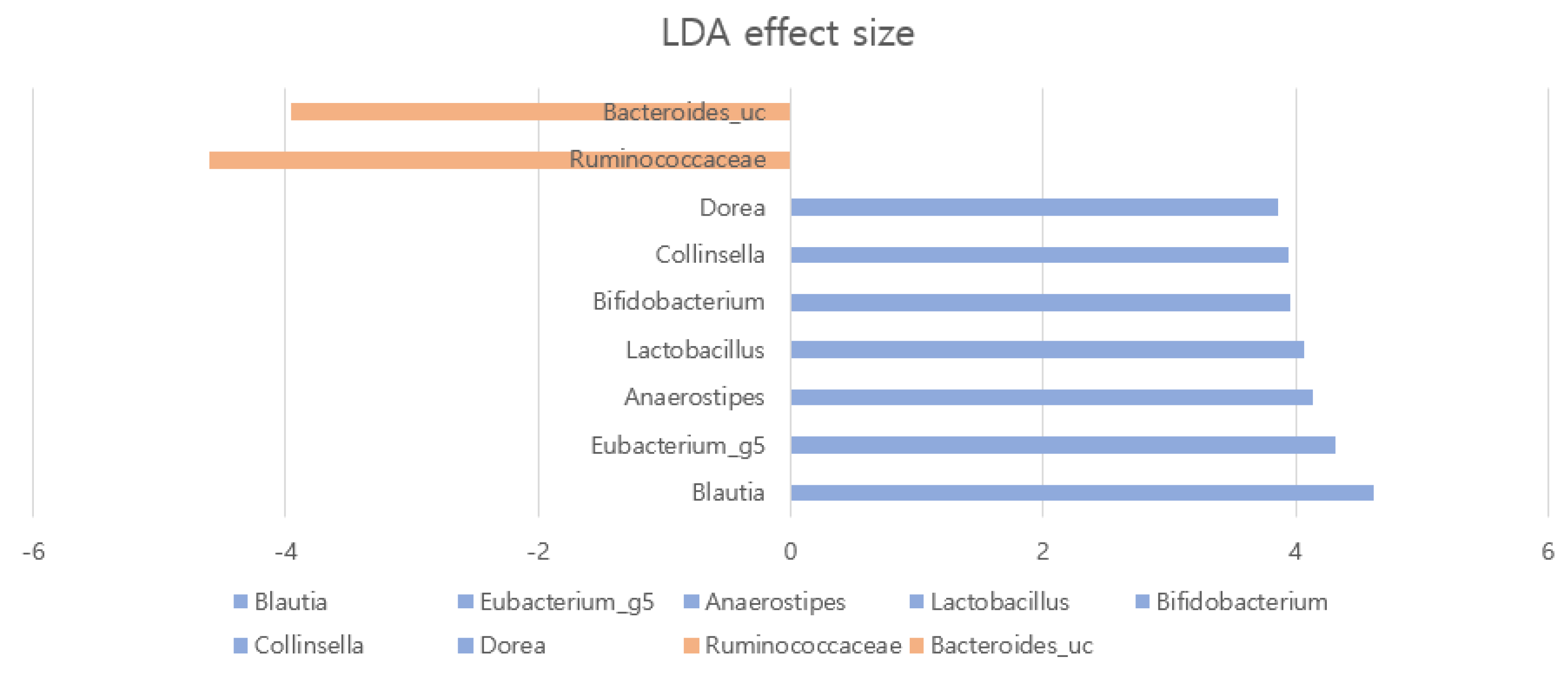
2.2. Gut Microbial Composition and Diversity in AA Patients and Controls
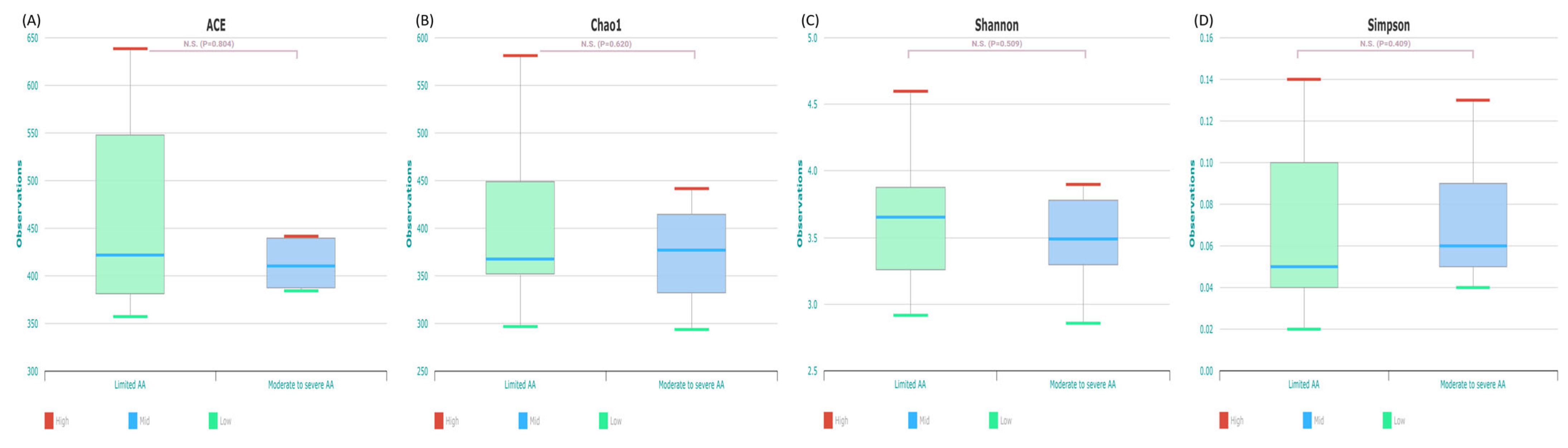
2.3. Gut Microbial Composition and Diversity in Patients with Limited AA and Moderate to Severe AA
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Stool Sample Collection and DNA Extraction
4.3. Statistical and Bioinformatic Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villasante Fricke, A.C.; Miteva, M. Epidemiology and burden of alopecia areata: A systematic review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Gilhar, A.; Etzioni, A.; Paus, R. Alopecia areata. N. Engl. J. Med. 2012, 366, 1515–1525. [Google Scholar] [CrossRef]
- Wang, E.C.E.; Christiano, A.M. The Changing Landscape of Alopecia Areata: The Translational Landscape. Adv. Ther. 2017, 34, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ito, T.; Ohyama, M. Alopecia areata: Current understanding of the pathophysiology and update on therapeutic approaches, featuring the Japanese Dermatological Association guidelines. J. Dermatol. 2022, 49, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.M.; Sheng, Y.Y.; Xu, F.; Qi, S.S.; Liu, X.J.; Hu, R.M.; Miao, Y.; Huang, G.Q.; Yang, Q.P. Imbalance of T-helper 17 and regulatory T cells in patients with alopecia areata. J. Dermatol. 2015, 42, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Tie, Y.; Huang, Y.; Chen, R.; Li, L.; Chen, M.; Zhang, S. Current insights on the roles of gut microbiota in inflammatory bowel disease-associated extra-intestinal manifestations: Pathophysiology and therapeutic targets. Gut Microbes 2023, 15, 2265028. [Google Scholar] [CrossRef]
- Hevia, A.; Milani, C.; Lopez, P.; Cuervo, A.; Arboleya, S.; Duranti, S.; Turroni, F.; Gonzalez, S.; Suarez, A.; Gueimonde, M.; et al. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 2014, 5, e01548-14. [Google Scholar] [CrossRef]
- Zhao, H.; Yuan, L.; Zhu, D.; Sun, B.; Du, J.; Wang, J. Alterations and Mechanism of Gut Microbiota in Graves’ Disease and Hashimoto’s Thyroiditis. Pol. J. Microbiol. 2022, 71, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Rebello, D.; Wang, E.; Yen, E.; Lio, P.A.; Kelly, C.R. Hair Growth in Two Alopecia Patients after Fecal Microbiota Transplant. ACG Case Rep. J. 2017, 4, e107. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.R.; Yang, X.Y.; Xia, H.H.; Wu, L.H.; He, X.X. Hair regrowth following fecal microbiota transplantation in an elderly patient with alopecia areata: A case report and review of the literature. World J. Clin. Cases 2019, 7, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- McElwee, K.J.; Niiyama, S.; Freyschmidt-Paul, P.; Wenzel, E.; Kissling, S.; Sundberg, J.P.; Hoffmann, R. Dietary soy oil content and soy-derived phytoestrogen genistein increase resistance to alopecia areata onset in C3H/HeJ mice. Exp. Dermatol. 2003, 12, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.; Dai, Z.; Christiano, A.M. 649 Gut microbiota plays a role in the development of alopecia areata. J. Investig. Dermatol. 2017, 137, S112. [Google Scholar] [CrossRef]
- Rangu, S.; Lee, J.J.; Hu, W.; Bittinger, K.; Castelo-Soccio, L. Understanding the Gut Microbiota in Pediatric Patients with Alopecia Areata and their Siblings: A Pilot Study. JID Innov. 2021, 1, 100051. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, P.; Hu, R.; Qi, S.; Zhao, Y.; Miao, Y.; Han, Y.; Zhou, L.; Yang, Q. Gut microbiota characterization in Chinese patients with alopecia areata. J. Dermatol. Sci. 2021, 102, 109–115. [Google Scholar] [CrossRef]
- Moreno-Arrones, O.M.; Serrano-Villar, S.; Perez-Brocal, V.; Saceda-Corralo, D.; Morales-Raya, C.; Rodrigues-Barata, R.; Moya, A.; Jaen-Olasolo, P.; Vano-Galvan, S. Analysis of the gut microbiota in alopecia areata: Identification of bacterial biomarkers. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Brzychcy, K.; Dróżdż, I.; Skoczylas, S.; Płoszaj, T.; Sobolewska-Sztychny, D.; Skibińska, M.; Narbutt, J.; Lesiak, A. Gut microbiota in alopecia areata. Postepy Dermatol. Alergol. 2022, 39, 1162–1170. [Google Scholar] [CrossRef]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- González-Bosch, C.; Boorman, E.; Zunszain, P.A.; Mann, G.E. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 2021, 47, 102165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Feng, J.; Li, J.; Zhao, L.; Liu, Y.; Chen, H.; Jin, Y.; Zhu, B.; Wei, Y. Alterations of the Gut Microbiota in Hashimoto’s Thyroiditis Patients. Thyroid 2018, 28, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Cantabrana, C.; Gomez, J.; Delgado, S.; Requena-Lopez, S.; Queiro-Silva, R.; Margolles, A.; Coto, E.; Sanchez, B.; Coto-Segura, P. Gut microbiota dysbiosis in a cohort of patients with psoriasis. Br. J. Dermatol. 2019, 181, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Salem, F.; Kindt, N.; Marchesi, J.R.; Netter, P.; Lopez, A.; Kokten, T.; Danese, S.; Jouzeau, J.Y.; Peyrin-Biroulet, L.; Moulin, D. Gut microbiome in chronic rheumatic and inflammatory bowel diseases: Similarities and differences. United Eur. Gastroenterol. J. 2019, 7, 1008–1032. [Google Scholar] [CrossRef]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhi, F. Lower Level of Bacteroides in the Gut Microbiota Is Associated with Inflammatory Bowel Disease: A Meta-Analysis. Biomed. Res. Int. 2016, 2016, 5828959. [Google Scholar] [CrossRef]
- Sobolewska-Wlodarczyk, A.; Wlodarczyk, M.; Fichna, J.; Wisniewska-Jarosinska, M. Alopecia areata in patients with inflammatory bowel disease: An overview. Folia Med. Cracov 2016, 56, 5–12. [Google Scholar] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Ho, H.J.; Tseng, C.H.; Lai, Z.L.; Shieh, J.J.; Wu, C.Y. Intestinal microbiota profiling and predicted metabolic dysregulation in psoriasis patients. Exp. Dermatol. 2018, 27, 1336–1343. [Google Scholar] [CrossRef]
- Polak, K.; Bergler-Czop, B.; Szczepanek, M.; Wojciechowska, K.; Fratczak, A.; Kiss, N. Psoriasis and Gut Microbiome-Current State of Art. Int. J. Mol. Sci. 2021, 22, 4529. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Amiri, P.; Hosseini, S.A.; Ghaffari, S.; Tutunchi, H.; Ghaffari, S.; Mosharkesh, E.; Asghari, S.; Roshanravan, N. Role of Butyrate, a Gut Microbiota Derived Metabolite, in Cardiovascular Diseases: A comprehensive narrative review. Front. Pharmacol. 2021, 12, 837509. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.; Kim, B.S.; Kim, J.W.; Kim, J.S.; Koh, S.J.; Kim, B.G.; Lee, K.L.; Chun, J. The Effect of Probiotics on Gut Microbiota during the Helicobacter pylori Eradication: Randomized Controlled Trial. Helicobacter 2016, 21, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.E.; Kang, W.; Choi, S.; Park, Y.; Chalita, M.; Kim, H.; Lee, J.H.; Hyun, D.W.; Ryu, K.J.; Sung, H.; et al. The influence of microbial dysbiosis on immunochemotherapy-related efficacy and safety in diffuse large B-cell lymphoma. Blood 2023, 141, 2224–2238. [Google Scholar] [CrossRef] [PubMed]
- Seong, G.; Kim, N.; Joung, J.G.; Kim, E.R.; Chang, D.K.; Chun, J.; Hong, S.N.; Kim, Y.H. Changes in the Intestinal Microbiota of Patients with Inflammatory Bowel Disease with Clinical Remission during an 8-Week Infliximab Infusion Cycle. Microorganisms 2020, 8, 874. [Google Scholar] [CrossRef]
- Lee, S.H.; Yoon, S.H.; Jung, Y.; Kim, N.; Min, U.; Chun, J.; Choi, I. Emotional well-being and gut microbiome profiles by enterotype. Sci. Rep. 2020, 10, 20736. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Shin, J.H.; Kim, J.Y.; Ju, H.J.; Kim, G.M. Exploring the Role of Gut Microbiota in Patients with Alopecia Areata. Int. J. Mol. Sci. 2024, 25, 4256. https://doi.org/10.3390/ijms25084256
Lee JH, Shin JH, Kim JY, Ju HJ, Kim GM. Exploring the Role of Gut Microbiota in Patients with Alopecia Areata. International Journal of Molecular Sciences. 2024; 25(8):4256. https://doi.org/10.3390/ijms25084256
Chicago/Turabian StyleLee, Ji Hae, Ji Hae Shin, Ji Yoon Kim, Hyun Jeong Ju, and Gyong Moon Kim. 2024. "Exploring the Role of Gut Microbiota in Patients with Alopecia Areata" International Journal of Molecular Sciences 25, no. 8: 4256. https://doi.org/10.3390/ijms25084256
APA StyleLee, J. H., Shin, J. H., Kim, J. Y., Ju, H. J., & Kim, G. M. (2024). Exploring the Role of Gut Microbiota in Patients with Alopecia Areata. International Journal of Molecular Sciences, 25(8), 4256. https://doi.org/10.3390/ijms25084256