
Precision Medicine Approaches in Acute Myeloid Leukemia with Adverse Genetics


Abstract
1. Background
2. Genetics of HR AML
3. Immune Landscapes of AML with Adverse Genetics
4. Immunotherapy in AML with Adverse Genetics
5. Rationales for Combining Targeted Therapy with Immunotherapy in AML with Adverse Genetics
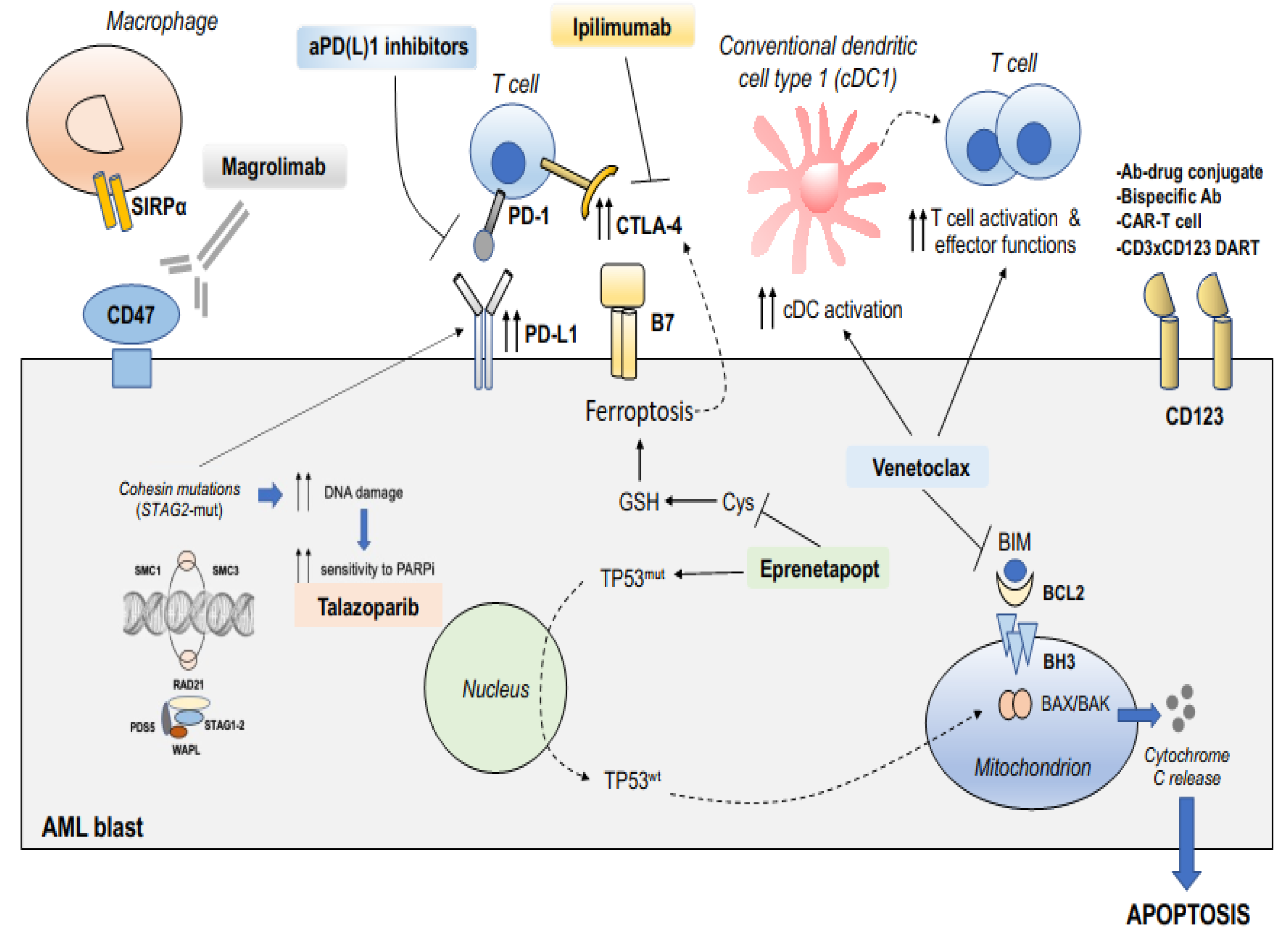
5.1. Exploiting BCL2 Inhibition for Innate and Adaptive Immune Reactivation
5.2. Targeting TP53-Dependent or -Independent Mechanisms of Apoptosis with APR-246/Eprenetapopt
5.3. Targeting CD47 Phagocytic Immune Checkpoint in Adverse-Risk AML
5.4. Targeting Poly(ADP-Ribose) Polymerase in STAG2-Mutated AML
5.5. Splice-Site-Creating Mutations and Sensitivity to Immune Checkpoint Inhibition
6. Current Treatment Strategies for AML with Adverse Genetics
Venetoclax Plus Azacytidine
7. Promising Targeted Approaches for the Treatment of AML with Adverse Genetics
8. Novel Investigational Strategies Combining Immunotherapy and Target Therapy in AML with Adverse Genetics
8.1. APR-246-Based Combinations
8.2. Innate and Adaptive Immune Checkpoint Inhibition in AML with Adverse Genetics
8.3. Poly(ADP-Ribose) Polymerase (PARP)-Inhibitor-Based Combinations
8.4. Regimens Including Menin Inhibitors for KMT2A-Mutated AML
8.5. Combinatorial Strategies Targeting the Interleukin 3 Receptor CD123
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Mendes, A.; Fahrenkrog, B. NUP214 in Leukemia: It’s More than Transport. Cells 2019, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Pérez, C.; de la Torre, E.P.; Sanchez-Garcia, J.; Martín-Calvo, C.; Martínez-Losada, C.; Casaño-Sanchez, J.; Serrano-López, J.; Serrano, J. Evolving Risk Classifications in AML in a Real-Life Scenario: After Changes upon Changes, Is It More and More Adverse? Cancers 2023, 15, 1425. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Pratz, K.W.; DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Recher, C.; Schuh, A.C.; Babu, S.; Dail, M.; et al. ELN Risk Stratification Is Not Predictive of Outcomes for Treatment-Naïve Patients with Acute Myeloid Leukemia Treated with Venetoclax and Azacitidine. Blood 2022, 140, 1441–1444. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Labopin, M.; Socie, G.; Volin, L.; Passweg, J.; Chevallier, P.; Beelen, D.; Milpied, N.; Blaise, D.; Cornelissen, J.J.; et al. Relapse and survival after transplantation for complex karyotype acute myeloid leukemia: A report from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation and the University of Texas MD Anderson Cancer Center. Cancer 2018, 124, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, M.C.; Zhang, M.-J.; Medeiros, B.C.; Armand, P.; Hu, Z.-H.; Nishihori, T.; Aljurf, M.D.; Akpek, G.; Cahn, J.-Y.; Cairo, M.S.; et al. Hematopoietic Cell Transplantation Outcomes in Monosomal Karyotype Myeloid Malignancies. Biol. Blood Marrow Transplant. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Middeke, J.M.; Herold, S.; Rücker-Braun, E.; Berdel, W.E.; Stelljes, M.; Kaufmann, M.; Schäfer-Eckart, K.; Baldus, C.D.; Stuhlmann, R.; Ho, A.D.; et al. TP53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2016, 172, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Heshmati, Y.; Kalfon, J.; Perez, M.W.; Ferrucio, J.X.; Ewers, J.; Engler, B.H.; Kossenkov, A.; Ellegast, J.M.; Yi, J.S.; et al. A distinct core regulatory module enforces oncogene expression in KMT2A-rearranged leukemia. Genes Dev. 2022, 36, 368–389. [Google Scholar] [CrossRef] [PubMed]
- Stasik, C.; Ganguly, S.; Cunningham, M.T.; Hagemeister, S.; Persons, D.L. Infant acute lymphoblastic leukemia with t(11;16)(q23;p13.3) and lineage switch into acute monoblastic leukemia. Cancer Genet. Cytogenet. 2006, 168, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Neuendorff, N.R.; Hemmati, P.; Arnold, R.; Ihlow, J.; Dörken, B.; Müller-Tidow, C.; Westermann, J. BCR-ABL + acute myeloid leukemia: Are we always dealing with a high-risk disease? Blood Adv. 2018, 2, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Soupir, C.P.; Vergilio, J.-A.; Cin, P.D.; Muzikansky, A.; Kantarjian, H.; Jones, D.; Hasserjian, R.P. Philadelphia chromosome–positive acute myeloid leukemia: A rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am. J. Clin. Pathol. 2007, 127, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Konoplev, S.; Yin, C.C.; Kornblau, S.M.; Kantarjian, H.M.; Konopleva, M.; Andreeff, M.; Lu, G.; Zuo, Z.; Luthra, R.; Medeiros, L.J.; et al. Molecular characterization ofde novoPhiladelphia chromosome-positive acute myeloid leukemia. Leuk. Lymphoma 2013, 54, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Nacheva, E.P.; Grace, C.D.; Brazma, D.; Gancheva, K.; Howard-Reeves, J.; Rai, L.; Gale, R.E.; Linch, D.C.; Hills, R.K.; Russell, N.; et al. Does BCR/ABL1 positive Acute Myeloid Leukaemia Exist? Br. J. Haematol. 2013, 161, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Lamble, A.J.; Hagiwara, K.; Gerbing, R.B.; Smith, J.L.; Kolekar, P.; Ries, E.R.; Kolb, A.E.; Alonzo, T.; Ma, X.; Meshinchi, S. CREBBP alterations are associated with a poor prognosis in de novo AML. Blood 2023, 141, 2156–2159. [Google Scholar] [CrossRef] [PubMed]
- Lavallée, V.-P.; Gendron, P.; Lemieux, S.; D’angelo, G.; Hébert, J.; Sauvageau, G. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood 2015, 125, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Birdwell, C.; Fiskus, W.; Kadia, T.M.; DiNardo, C.D.; Mill, C.P.; Bhalla, K.N. EVI1 dysregulation: Impact on biology and therapy of myeloid malignancies. Blood Cancer J. 2021, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Lugthart, S.; Gröschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.; van Zelderen-Bhola, S.L.; Ossenkoppele, G.J.; Vellenga, E.; Ruiter, E.v.D.B.-D.; Schanz, U.; et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef] [PubMed]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Honda, H.; Matsui, H. The enigma of monosomy 7. Blood 2018, 131, 2891–2898. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Mrózek, K.; Eisfeld, A.-K.; Kohlschmidt, J.; Carroll, A.J.; Walker, C.J.; Nicolet, D.; Blachly, J.S.; Bill, M.; Papaioannou, D.; Wang, E.S.; et al. Complex karyotype in de novo acute myeloid leukemia: Typical and atypical subtypes differ molecularly and clinically. Leukemia 2019, 33, 1620–1634. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.T.; Park, H.J.; Kim, B.K.; An, H.Y.; Choi, J.Y.; Kang, H.J. Post-Transplantation Cyclophosphamide-Based Haploidentical versus Matched Unrelated Donor Peripheral Blood Hematopoietic Stem Cell Transplantation Using Myeloablative Targeted Busulfan-Based Conditioning for Pediatric Acute Leukemia. Transplant. Cell. Ther. 2022, 28, 195.e1–195.e7. [Google Scholar] [CrossRef] [PubMed]
- Breems, D.A.; Van Putten, W.L.; De Greef, G.E.; Van Zelderen-Bhola, S.L.; Gerssen-Schoorl, K.B.; Mellink, C.H.; Nieuwint, A.; Jotterand, M.; Hagemeijer, A.; Beverloo, H.B.; et al. Monosomal karyotype in acute myeloid leukemia: A better indicator of poor prognosis than a complex karyotype. J. Clin. Oncol. 2008, 26, 4791–4797. [Google Scholar] [CrossRef] [PubMed]
- Anelli, L.; Pasciolla, C.; Zagaria, A.; Specchia, G.; Albano, F. Monosomal karyotype in myeloid neoplasias: A literature review. OncoTargets Ther. 2017, 10, 2163–2171. [Google Scholar] [CrossRef]
- Tang, J.-L.; Hou, H.-A.; Chen, C.-Y.; Liu, C.-Y.; Chou, W.-C.; Tseng, M.-H.; Huang, C.-F.; Lee, F.-Y.; Liu, M.-C.; Yao, M.; et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: Prognostic implication and interaction with other gene alterations. Blood 2009, 114, 5352–5361. [Google Scholar] [CrossRef] [PubMed]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microrna expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef]
- Hanaki, S.; Shimada, M. Targeting EZH2 as cancer therapy. J. Biochem. 2021, 170, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in Myeloid Malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dai, H.; Wang, Q.; Wang, Q.; Xu, Y.; Wang, Y.; Sun, A.; Ruan, J.; Chen, S.; Wu, D. EZH2 Mutations Are Related to Low Blast Percentage in Bone Marrow and -7/del(7q) in De Novo Acute Myeloid Leukemia. PLoS ONE 2013, 8, e61341. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 mutations and impact on clinical outcome: An analysis in 1,604 patients with newly diagnosed acute myeloid leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, A.C.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; LaFave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 leads to myelodysplastic syndrome–like disease in mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Kitaura, J.; Togami, K.; Nishimura, K.; Enomoto, Y.; Uchida, T.; Kagiyama, Y.; Kawabata, K.C.; Nakahara, F.; Izawa, K.; et al. Myelodysplastic syndromes are induced by histone methylation–Altering ASXL1 mutations. J. Clin. Investig. 2013, 123, 4627–4640. [Google Scholar] [CrossRef] [PubMed]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 2019, 76, 2511–2523. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.-A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Sorcini, D.; Falini, B. BCORgene alterations in hematologic diseases. Blood 2021, 138, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Class, C.A.; Sasaki, K.; Ravandi, F.; Cortes, J.E.; Daver, N.; Takahashi, K.; Short, N.J.; DiNardo, C.D.; et al. Outcomes of acute myeloid leukemia with myelodysplasia related changes depend on diagnostic criteria and therapy. Am. J. Hematol. 2020, 95, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Tiacci, E.; Holmes, A.B.; Kohlmann, A.; Martelli, M.P.; Kern, W.; Spanhol-Rosseto, A.; Klein, H.-U.; Dugas, M.; Schindela, S.; et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 2011, 118, 6153–6163. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Sorcini, D.; Guzman, A.G.; Reyes, J.M.; Stella, A.; Marra, A.; Sartori, S.; Brunetti, L.; Rossi, R.; Del Papa, B.; et al. Bcor deficiency perturbs erythro-megakaryopoiesis and cooperates with Dnmt3a loss in acute erythroid leukemia onset in mice. Leukemia 2021, 35, 1949–1963. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.J.; So, J.; Rogers, A.J.; Gregory, G.; Li, J.; Zethoven, M.; Gearhart, M.D.; Bardwell, V.J.; Johnstone, R.W.; Vervoort, S.J.; et al. Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat. Commun. 2019, 10, 1347. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, C.; Lussana, F.; Salmoiraghi, S.; Cavagna, R.; Buklijas, K.; Elidi, L.; Zanghi’, P.; Michelato, A.; Delaini, F.; Oldani, E.; et al. Clinical significance of chromatin-spliceosome acute myeloid leukemia: A report from the Northern Italy Leukemia Group (NILG) randomized trial 02/06. Haematologica 2021, 106, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin–RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Craddock, C.F. Full-intensity and reduced-intensity allogeneic stem cell transplantation in AML. Bone Marrow Transplant. 2008, 41, 415–423. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1 K700E Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Saez, B.; Walter, M.J.; Graubert, T.A. Splicing factor gene mutations in hematologic malignancies. Blood 2017, 129, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Ochi, Y.; Ogawa, S. Chromatin-Spliceosome Mutations in Acute Myeloid Leukemia. Cancers 2021, 13, 1232. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. Cancer 2020, 20, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Hou, H.-A.; Tang, J.-L.; Kuo, Y.-Y.; Chiu, Y.-C.; Liu, C.-Y.; Tseng, M.-H.; Lin, T.-Y.; Liu, M.-C.; Liu, C.-W.; et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Eckardt, J.-N.; Stasik, S.; Röllig, C.; Sauer, T.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; Steffen, B.; et al. Alterations of cohesin complex genes in acute myeloid leukemia: Differential co-mutations, clinical presentation and impact on outcome. Blood Cancer J. 2023, 13, 1–9. [Google Scholar] [CrossRef]
- Black, H.E.; Jhujh, S.; Stewart, G.S.; Savage, I.K.; Mills, I.K. STAG2 Loss Gives Rise to Therapeutically Targetable DNA Damage Repair Defects and Altered Replication Fork Dynamics in Acute Myeloid Leukaemia. Blood 2019, 134, 1255. [Google Scholar] [CrossRef]
- Jacqueline, S.G. A Pilot Proof-of-Concept Study of Talazoparib for Cohesin-Mutated AML and MDS with Excess Blasts. 2021. Available online: https://clinicaltrials.gov/show/NCT03974217 (accessed on 7 April 2024).
- Perri, F.; Pisconti, S.; Scarpati, G.D.V. P53 mutations and cancer: A tight linkage. Ann. Transl. Med. 2016, 4, 522. [Google Scholar] [CrossRef]
- George, B.; Kantarjian, H.; Baran, N.; Krocker, J.D.; Rios, A. TP53 in Acute Myeloid Leukemia: Molecular Aspects and Patterns of Mutation. Int. J. Mol. Sci. 2021, 22, 10782. [Google Scholar] [CrossRef] [PubMed]
- Sallman, A.D.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia 2015, 30, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Nadorp, B.; Fornerod, M.; Nicolet, D.; Wu, H.; Walker, C.J.; Sun, Z.; Witkowski, M.T.; Tikhonova, A.N.; Guillamot-Ruano, M.; et al. An inflammatory state remodels the immune microenvironment and improves risk stratification in acute myeloid leukemia. Nat. Cancer 2023, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Vadakekolathu, J.; Mazziotta, F.; Reeder, S.; Yau, T.-O.; Mukhopadhyay, R.; Dickins, B.; Altmann, H.; Kramer, M.; Knaus, H.A.; et al. Immune dysfunction signatures predict outcomes and define checkpoint blockade–unresponsive microenvironments in acute myeloid leukemia. J. Clin. Investig. 2022, 132, e159579. [Google Scholar] [CrossRef] [PubMed]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020, 12, 546. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Dufva, O.; Pölönen, P.; Brück, O.; Keränen, M.A.; Klievink, J.; Mehtonen, J.; Huuhtanen, J.; Kumar, A.; Malani, D.; Siitonen, S.; et al. Immunogenomic Landscape of Hematological Malignancies. Cancer Cell 2020, 38, 380–399. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Limongello, R.; Marra, A.; Mancusi, A.; Bonato, S.; Hoxha, E.; Ruggeri, L.; Hui, S.; Velardi, A.; Pierini, A. Novel Immune Cell-Based Therapies to Eradicate High-Risk Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 695051. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.d.l.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol. 2021, 11, 656218. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2015, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Barbullushi, K.; Rampi, N.; Serpenti, F.; Sciumè, M.; Fabris, S.; De Roberto, P.; Fracchiolla, N.S. Vaccination Therapy for Acute Myeloid Leukemia: Where Do We Stand? Cancers 2022, 14, 2994. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Schmitt, A.; Rojewski, M.T.; Chen, J.; Giannopoulos, K.; Fei, F.; Yu, Y.; Götz, M.; Heyduk, M.; Ritter, G.; et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 2008, 111, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Rothe, M.; Deiser, K.; Altmann, T.; Bücklein, V.L.; Köhnke, T.; Augsberger, C.; Konstandin, N.P.; Spiekermann, K.; et al. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: Results of a phase I trial. Clin. Transl. Immunol. 2020, 9, e1117. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Pratz, K.W.; Wei, A.H.; Pullarkat, V.; Jonas, B.A.; Recher, C.; Babu, S.; Schuh, A.C.; Dail, M.; Sun, Y.; et al. Outcomes in Patients with Poor-Risk Cytogenetics with or without TP53 Mutations Treated with Venetoclax and Azacitidine. Clin. Cancer Res. 2022, 28, 5272–5279. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Yang, D.; Pillai, R.; Sanchez, J.F.; Mei, M.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Salhotra, A.; et al. Association of leukemia genetics with response to venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Am. J. Hematol. 2019, 94, E253–E255. [Google Scholar] [CrossRef] [PubMed]
- Cherry, E.M.; Abbott, D.; Amaya, M.; McMahon, C.; Schwartz, M.; Rosser, J.; Sato, A.; Schowinsky, J.T.; Inguva, A.; Minhajuddin, M.; et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021, 5, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, N.E.; Ramachandra, N.; Sahu, S.; Gitego, N.; Lopez, A.; Pradhan, K.; Bhagat, T.D.; Gordon-Mitchell, S.; Pena, B.R.; Kazemi, M.; et al. ASXL1 mutations are associated with distinct epigenomic alterations that lead to sensitivity to venetoclax and azacytidine. Blood Cancer J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, P.; Mao, M.; Zhang, S.; Bigenwald, C.; Dutertre, C.-A.; Lehmann, C.H.; Pan, H.; Paulhan, N.; Amon, L.; et al. BCL2 Inhibition Reveals a Dendritic Cell–Specific Immune Checkpoint That Controls Tumor Immunosurveillance. Cancer Discov. 2023, 13, 2448–2469. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.V.; Jitkova, Y.; et al. Venetoclax enhances T cell-mediated anti-leukemic activity by increasing ROS production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Haribhai, D.; Mathew, R.; Duggan, R.; Ellis, P.A.; Wang, R.; Lasater, E.A.; Shi, Y.; Dave, N.; Riehm, J.J.; et al. Venetoclax increases intratumoral effector t cells and antitumor efficacy in combination with immune checkpoint blockade. Cancer Discov. 2021, 11, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2021, 107, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free. Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.; Sun, Z.; Wang, Y.; Wang, L.; Liu, Z.; Han, X. Ferroptosis: A double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef] [PubMed]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Z.; Zhang, W.; Meng, L.; Wang, J.; Lv, Z.; Xia, H.; Wu, M.; Zhang, Y.; Wang, J. Ferroptosis Mediation Patterns Reveal Novel Tool to Implicate Immunotherapy and Multi-Omics Characteristics in Bladder Cancer. Front. Cell Dev. Biol. 2022, 10, 791630. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, A.T.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Akarca, A.U.; Martino, G.; Ramsay, A.; Ascani, S.; Perriello, V.M.; O’nions, J.; Wilson, A.J.; Gupta, R.; Childerhouse, A.; et al. CD47 expression in acute myeloid leukemia varies according to genotype. Haematologica 2023, 108, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef] [PubMed]
- Oreskovic, E.; Wheeler, E.C.; Mengwasser, K.E.; Fujimura, E.; Martin, T.D.; Tothova, Z.; Elledge, S.J. Genetic analysis of cancer drivers reveals cohesin and CTCF as suppressors of PD-L1. Proc. Natl. Acad. Sci. USA 2022, 119, e2120540119. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Colombo, N.; Arrowsmith, E.; Narayan, V.; Yonemori, K.; Scambia, G.; Zelnak, A.; Bauer, T.M.; Jin, N.; Ulahannan, S.V.; et al. Avelumab Plus Talazoparib in Patients With BRCA1/2- or ATM-Altered Advanced Solid Tumors. JAMA Oncol. 2023, 9, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-González, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.-M.; Yao, F.-Y.; Liu, J.; Li, M.-Y.; Jiang, J.-Y.; Cheng, Y.; Xu, S.; Li, S.-Q.; Zhang, N.; Huang, B.; et al. Splicing factor-mediated regulation patterns reveals biological characteristics and aid in predicting prognosis in acute myeloid leukemia. J. Transl. Med. 2023, 21, 1–17. [Google Scholar] [CrossRef]
- Chiche, E.; Rahmé, R.; Bertoli, S.; Dumas, P.-Y.; Micol, J.-B.; Hicheri, Y.; Pasquier, F.; Peterlin, P.; Chevallier, P.; Thomas, X.; et al. Real-life experience with CPX-351 and impact on the outcome of high-risk AML patients: A multicentric French cohort. Blood Adv. 2021, 5, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hui, G.; Ladha, A.; Cheung, E.; Berube, C.; Coutre, S.; Gotlib, J.; Liedtke, M.; Zhang, T.Y.; Muffly, L.S.; Mannis, G.N. Routine Use of Gemtuzumab Ozogamicin in 7+3-Based Inductions for All "Non-Adverse" Risk AML. Blood 2020, 136, 36–37. [Google Scholar] [CrossRef]
- Loke, J.; Buka, R.; Craddock, C. Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia: Who, When, and How? Front. Immunol. 2021, 12, 659595. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Atallah, E.L.; Shallis, R.M.; Saliba, A.N.; Stahl, M.F.; Bewersdorf, J.P.; Grenet, J.; Patel, A.A.; Abaza, Y.; Murthy, G.S.G.; et al. Predictors of Long-Term Outcome in TP53-Mutated Acute Myeloid Leukemia Patients Receiving Allogeneic Stem Cell Transplant after First- or Second-Line Therapy: Results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Blood 2022, 140, 1435–1437. [Google Scholar] [CrossRef]
- Britt, A.; Mohyuddin, G.R.; McClune, B.; Singh, A.; Lin, T.; Ganguly, S.; Abhyankar, S.; Shune, L.; McGuirk, J.; Skikne, B.; et al. Acute myeloid leukemia or myelodysplastic syndrome with chromosome 17 abnormalities and long-term outcomes with or without hematopoietic stem cell transplantation. Leuk. Res. 2020, 95, 106402. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; McCullough, K.; Johnson, I.; Al-Kali, A.; Begna, K.H.; Patnaik, M.M.; Litzow, M.R.; Hogan, W.; Shah, M.; Alkhateeb, H.; et al. Real-world experience with venetoclax and hypomethylating agents in myelodysplastic syndromes with excess blasts. Am. J. Hematol. 2022, 97, E214–E216. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Altman, J.K.; Assi, R.; Bixby, D.; Fathi, A.T.; Foran, J.M.; Gojo, I.; Hall, A.C.; Jonas, B.A.; Kishtagari, A.; et al. Acute Myeloid Leukemia, Version 3.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Madamsetty, V.S.; Mukherjee, A.; Mukherjee, S. Recent Trends of the Bio-Inspired Nanoparticles in Cancer Theranostics. Front. Pharmacol. 2019, 10, 1264. [Google Scholar] [CrossRef]
- Wang, X.; Huang, R.; Fang, K.; Zhang, X. PB1795: Bio-Inspired Nanomedicine for Targeted Acute Myeloid Leukemia Immunotherapy. HemaSphere 2022, 6, 1675–1676. [Google Scholar] [CrossRef]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Neldeborg, S.; Soerensen, J.F.; Møller, C.T.; Bill, M.; Gao, Z.; Bak, R.O.; Holm, K.; Sorensen, B.; Nyegaard, M.; Luo, Y.; et al. Dual intron-targeted CRISPR-Cas9-mediated disruption of the AML RUNX1-RUNX1T1 fusion gene effectively inhibits proliferation and decreases tumor volume in vitro and in vivo. Leukemia 2023, 37, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-C.; Kim, H.S.; Chen, Y.; Li, Y.; LaMere, M.W.; Chen, C.; Wang, H.; Gong, J.; Palumbo, C.D.; Ashton, J.M.; et al. Scaffold-mediated CRISPR-Cas9 delivery system for acute myeloid leukemia therapy. Sci. Adv. 2021, 7, eabg3217. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Komrokji, R.S.; DeZern, A.E.; Sebert, M.; Garcia-Manero, G.; Rahmé, R.; Steensma, D.P.; Che, J.L.; Roboz, G.J.; Madelaine, I.; et al. Long Term Follow-up and Combined Phase 2 Results of Eprenetapopt (APR-246) and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML). Blood 2021, 138, 246. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Sweet, K.; Miller, C.; Wennborg, A.; et al. Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: A phase 1, dose-finding and expansion study. Lancet Haematol. 2023, 10, e272–e283. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.A.; Asch, A.S.; Mannis, G.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and Efficacy of the Anticluster of Differentiation 47 Antibody Magrolimab Combined with Azacitidine in Patients With Previously Untreated AML: Phase Ib Results. J. Clin. Oncol. 2023, 41, 4893–4904. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int. J. Clin. Exp. Pathol. 2014, 7, 6880–6888. [Google Scholar] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients (Pts) with Very High/High-Risk Myelodysplastic Syndrome (vHR/HR-MDS) and Acute Myeloid Leukemia (AML): Final Analysis from a Phase Ib Study. Blood 2021, 138 (Suppl. 1), 244. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Westermann, J.; Kovacsovics, T.; Assouline, S.; Schuh, A.C.; Kim, H.-J.; Macias, G.R.; Sanford, D.; Luskin, M.R.; Stein, E.M.; et al. P582: First Results of a Phase Ii Study (Stimulus-Aml1) Investigating Sabatolimab + Azacitidine + Venetoclax in Patients with Newly Diagnosed Acute Myeloid Leukemia. HemaSphere 2022, 6, 481–482. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, A.P.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A single-arm, phase 2 study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef] [PubMed]
- Reville, P.K.; Kantarjian, H.M.; Ravandi, F.; Jabbour, E.; DiNardo, C.D.; Daver, N.; Pemmaraju, N.; Ohanian, M.; Alvarado, Y.; Xiao, L.; et al. Nivolumab maintenance in high-risk acute myeloid leukemia patients: A single-arm, open-label, phase II study. Blood Cancer J. 2021, 11, 1–5. [Google Scholar] [CrossRef]
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.; Morrow, J.; DeZern, A.E.; Foster, M.C.; Levis, M.J.; Coombs, C.C.; et al. Multi-Center Phase 2 Study of Pembroluzimab (Pembro) and Azacitidine (AZA) in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Newly Diagnosed (≥65 Years) AML Patients. Blood 2019, 134, 832. [Google Scholar] [CrossRef]
- Goswami, M.; Gui, G.; Dillon, L.W.; Lindblad, E.K.; Thompson, J.; Valdez, J.; Kim, D.-Y.; Ghannam, J.Y.; Oetjen, A.K.; Destefano, C.B.; et al. Pembrolizumab and decitabine for refractory or relapsed acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e003392. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Vincent, B.G.; Ivanova, A.; Moore, D.; McKinnon, K.P.; Wilkinson, A.D.; Mukhopadhyay, R.; Mazziotta, F.; Knaus, H.A.; Foster, M.C.; et al. Phase II Trial of Pembrolizumab after High-Dose Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Tschernia, N.P.; Kumar, V.; Moore, D.T.; Vincent, B.G.; Coombs, C.C.; Van Deventer, H.; Foster, M.C.; DeZern, A.E.; Luznik, L.; Riches, M.L.; et al. Safety and Efficacy of Pembrolizumab Prior to Allogeneic Stem Cell Transplantation for Acute Myelogenous Leukemia. Transplant. Cell. Ther. 2021, 27, 1021.e1–1021.e5. [Google Scholar] [CrossRef] [PubMed]
- Chien, K.S.; Kim, K.; Nogueras-Gonzalez, G.M.; Borthakur, G.; Naqvi, K.; Daver, N.G.; Montalban-Bravo, G.; Cortes, J.E.; DiNardo, C.D.; Jabbour, E.; et al. Phase II study of azacitidine with pembrolizumab in patients with intermediate-1 or higher-risk myelodysplastic syndrome. Br. J. Haematol. 2021, 195, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Popat, R.; Mattison, R.J.; Menne, T.; Bloor, A.; Gaymes, T.; Khwaja, A.; Juckett, M.; Chen, Y.; Cotter, M.J.; et al. A Phase I trial of talazoparib in patients with advanced hematologic malignancies. Int. J. Hematol. Oncol. 2021, 10, IJH35. [Google Scholar] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O’Connell, C.L.; Youngblood, B.A.; et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Portwood, S.M.; Cantella, M.C.; Cronin, T.L.; Wang, E.S. Addition of the PARP Inhibitor, Talazoparib, to Gemtuzumab Ozogamicin Significantly Enhances Anti-Leukemic Activity in Human CD33+ Acute Myeloid Leukemia. Blood 2019, 134, 1371. [Google Scholar] [CrossRef]
- Aldoss, I.; Issa, G.C.; Thirman, M.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. LBA-5 Revumenib Monotherapy in Patients with Relapsed/Refractory KMT2Ar Acute Leukemia: Topline Efficacy and Safety Results from the Pivotal Augment-101 Phase 2 Study; American Society of Hematology: Washington, DC, USA, 2023; Available online: https://ash.confex.com/ash/2023/webprogram/Paper192042.html (accessed on 7 April 2024).
- Carter, B.Z.; Tao, W.; Mak, P.Y.; Ostermann, L.B.; Mak, D.H.; McGeehan, G.M.; Ordentlich, P.; Andreeff, M. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood 2021, 138, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Mughal, T.I.; Brooks, C.; Lindsay, R.; Pemmaraju, N. Targeting CD123 in hematologic malignancies: Identifying suitable patients for targeted therapy. Leuk. Lymphoma 2021, 62, 2568–2586. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Garcia, J.S.; Raulston, E.G.; Garzon, J.L.; Galinsky, I.; Baxter, E.W.; Leonard, R.; DeAngelo, D.J.; Luskin, M.R.; Reilly, C.R.; et al. Tagraxofusp in Combination with Azacitidine and Venetoclax in Newly Diagnosed CD123+ Acute Myeloid Leukemia, Expansion Cohort of a Phase 1b Multicenter Trial. Blood 2023, 142 (Suppl. 1), 4277. [Google Scholar] [CrossRef]
- Daver, N.G.; Montesinos, P.; Aribi, A.M.; Martinelli, G.; Wang, E.S.; Altman, J.K.; Roboz, G.J.; Burke, P.W.; Walter, R.B.; Begna, K.; et al. A phase 1b/2 study of pivekimab sunirine (PVEK, IMGN632) in combination with venetoclax/azacitidine or magrolimab for patients with CD123-positive acute myeloid leukemia (AML). J. Clin. Oncol. 2023, 41, TPS7073. [Google Scholar] [CrossRef]


{kind=link}
| High-Risk Genetic Features |
|---|
| t(6;9)(p23.3;q34.1)/DEK::NUP214 t(v;11q23.3)KMT2A-rearranged t(9;22)(q34.1;q11.2)/BCR::ABL1 (BCR-ABL+) t(8;16)(p11.2;p13.3)/KAT6A::CREBBP inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2) GATA2, MECOM(EVI1) t(3q26.2;v)/MECOM(EVI1)-rearranged −5 or del(5q); −7; −17/abn(17p) Complex karyotype (CK) Monosomal karyotype (MK) Mutated RUNX1 Mutated EZH2 Mutated ASXL1 Mutated BCOR Spliceosome mutations (SRSF2, SF3B1, U2AF1, ZRSR2) Mutated STAG2 Mutated TP53 |
| Drug | Combinations | Mutation | Clinical Trials | Disease | Outcomes | |
|---|---|---|---|---|---|---|
APR-246 small molecule that targets TP53-mutated cancers | APR-246 + AZA APR-246 + AZA APR-246 + VEN + AZA | TP53 TP53 TP53 | NCT03072043 (PHASE IB-II) NCT03588078 (PHASE II) NCT04214860 (PHASE I) | 40 MDS 11 AML 34 MDS 18 AML 49 AML | 1 1 1 | ORR 71%, CR 44 Median OS 10.8 mo ORR 52% CR 37% Median OS 12.1 mo ORR 64% CR 38% |
MAGROLIMAB monoclonal antibody against CD47 and macrophage checkpoint inhibitor | MAGROLIMAB + AZA MAGROLIMAB + AZA vs. VEN-AZA or chemo MAGROLIMAB + AZA-VEN vs. placebo + AZA + VEN | TP53 TP53 TP53 | NCT03248479 (PHASE I) NCT04778397 (PHASE III) NCT05079230 (PHASE III) | 87 AML (82.8% TP53) Ongoing Ongoing | 1 1 1 | ORR 47.2% CR 31.9% median OS 9.8 mo Ongoing Ongoing |
SABATOLIMAB Checkpoint inhibitor anti TIM3 monoclonal antibody | SABATOLIMAB + HMA SABATOLIMAB + AZA + VEN | All;HR AML All | NCT03066648 (PHASE Ib) NCT04150029 (PHASE II) | 53 MDS 48 AML Ongoing | 1 1 | ORR AML 40% CR30%; HR AML ORR 53% median duration of response 12 months Ongoing |
NIVOLUMAB Checkpoint inhibitor antiPD-1 monoclonal antibody approved for different types of cancers | NIVOLUMAB + AZA NIVOLUMAB + AZA + IPILIMUMAB NIVOLUMAB + CHEMO | All All All (50% HR) | NCT02397720 (PHASE II) NCT02397720 (PHASE II) NCT02464657 (PHASE II) | 70 AML 31 AML 42 AML | >1 >1 1 | ORR 33%, CR22% median OS 6.2 mo (ASLX1 better response) ORR 46%, CR36% median OS 10.5 mo ORR 80%, CR64% median OS 18.5 mo |
PEMBROLIZUMAB Checkpoint inhibitor antiPD-1 monoclonal antibody approved for different types of cancers | PEMBRO + AZA PEMBRO + ARA C PEMBRO + DEC +/− VEN PEMBRO + AZA + VEN PEMBRO + CHEMO | All All All All All | NCT02845297 (PHASE II) NCT02768792 (PHASE II) NCT03969446 (PHASE II) NCT04284787 (PHASE II) NCT04214249 (PHASE II) | 37 AML(17 first line) 37 AML Ongoing Ongoing Ongoing | ≥1 >1 ≥1 ≥1 1 | ORR 55%, CR14% median OS 10.8 mo newly diagnosed ORR94%, CR47% median OS 13 mo ORR46%, CR38% median OS 11 mo (ASLX1 better response) Ongoing Ongoing |
TALAZOPARIB PARP inhibitor approved for breast cancer | TALAZOPARIB + DEC TALAZOPARIB BASED TALAZOPARIB + GO | All Cohesin mutated Cd33+ | NCT02878785 (PHASE I) NCT03974217 (PHASE I) NCT04207190 (PHASE I) | 24 AML Ongoing Ongoing | >1 ≥1 >1 | CR 8% Ongoing Ongoing |
REVUNEMIB Menin inhibitor FDA approved in adult and pediatric relapsed or refractory (R/R) KMT2A-rearranged acute leukemia | REVUNEMIB + VEN + ASX727 REVUNEMIB + VEN + AZA | All All | NCT05360160 (PHASE II) NCT06177067 (PHASE II) | Ongoing Ongoing | 1 >1 | Ongoing Ongoing |
TAGRAXOFUSP CD123-directed cytotoxin approved as monotherapy for the treatment of blastic plasmacytoid dendritic cell neoplasm PIVEKIMAB Antibody drug conjugate targeting CD-123 | TAGRAXOFUSP + AZA + VEN PIVEKIMAB + AZA + VEN | HR AML Cd123+ | NCT03113643 (PHASE IB) NCT04086264 (PHASE IB-II) | Ongoing (preliminary results 26 AML HR) Ongoing | 1 ≥1 | Ongoing preliminary results CR 39% median OS 14 mo; median OS TP53 9.5 mo Ongoing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoro, N.; Salutari, P.; Di Ianni, M.; Marra, A. Precision Medicine Approaches in Acute Myeloid Leukemia with Adverse Genetics. Int. J. Mol. Sci. 2024, 25, 4259. https://doi.org/10.3390/ijms25084259
Santoro N, Salutari P, Di Ianni M, Marra A. Precision Medicine Approaches in Acute Myeloid Leukemia with Adverse Genetics. International Journal of Molecular Sciences. 2024; 25(8):4259. https://doi.org/10.3390/ijms25084259
Chicago/Turabian StyleSantoro, Nicole, Prassede Salutari, Mauro Di Ianni, and Andrea Marra. 2024. "Precision Medicine Approaches in Acute Myeloid Leukemia with Adverse Genetics" International Journal of Molecular Sciences 25, no. 8: 4259. https://doi.org/10.3390/ijms25084259
APA StyleSantoro, N., Salutari, P., Di Ianni, M., & Marra, A. (2024). Precision Medicine Approaches in Acute Myeloid Leukemia with Adverse Genetics. International Journal of Molecular Sciences, 25(8), 4259. https://doi.org/10.3390/ijms25084259