Development of a Novel, Potent, and Selective Sialyltransferase Inhibitor for Suppressing Cancer Metastasis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
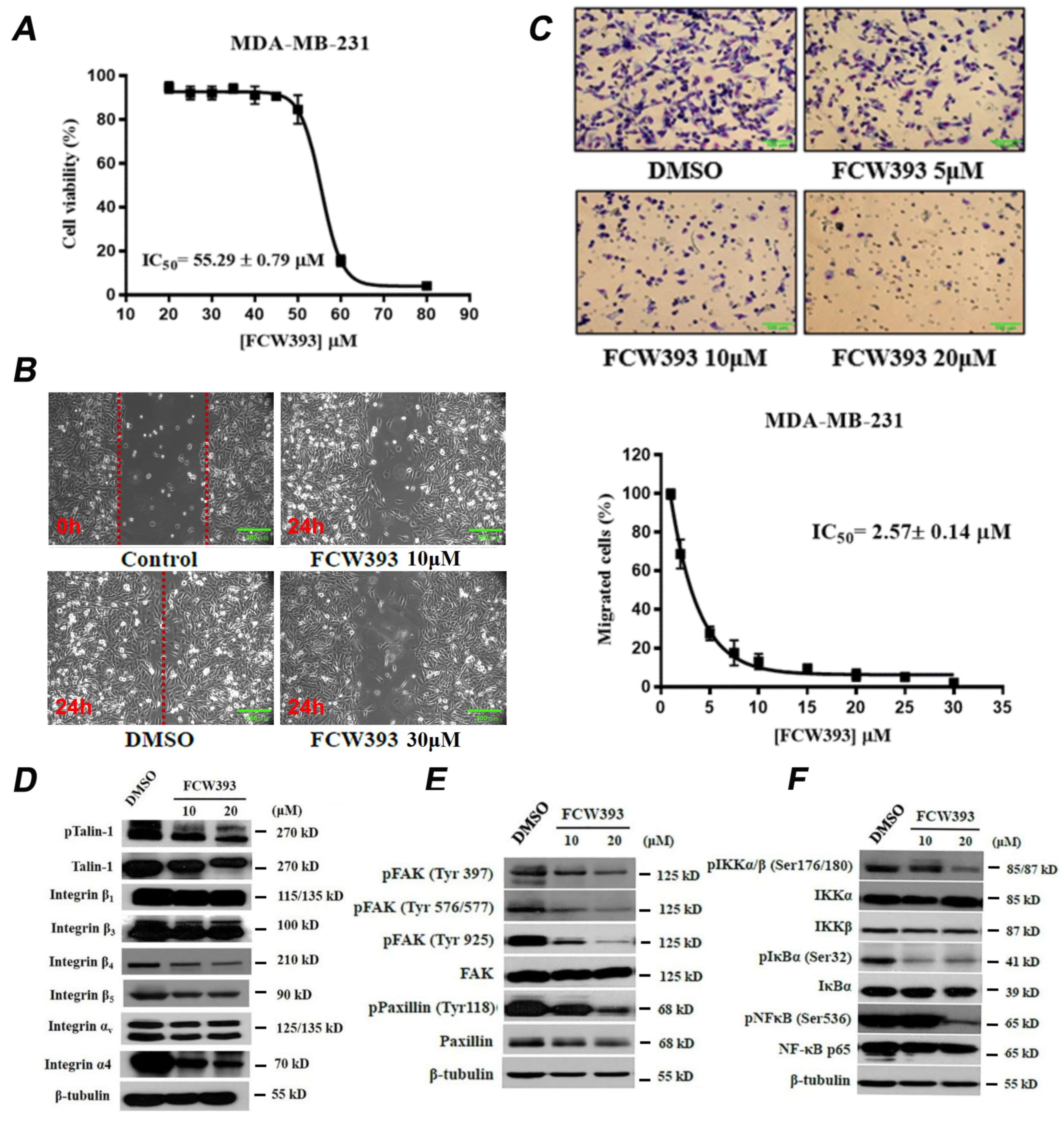
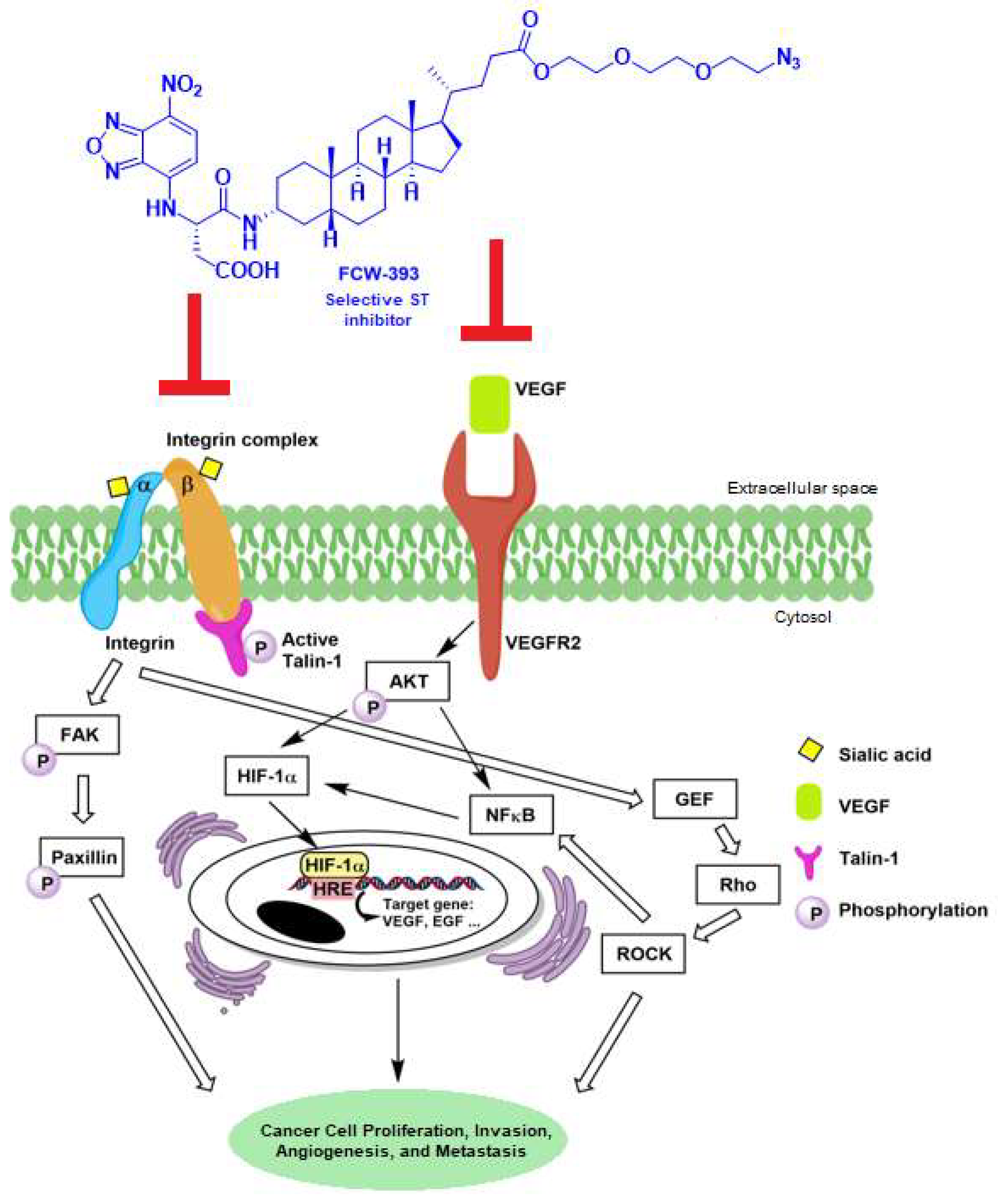
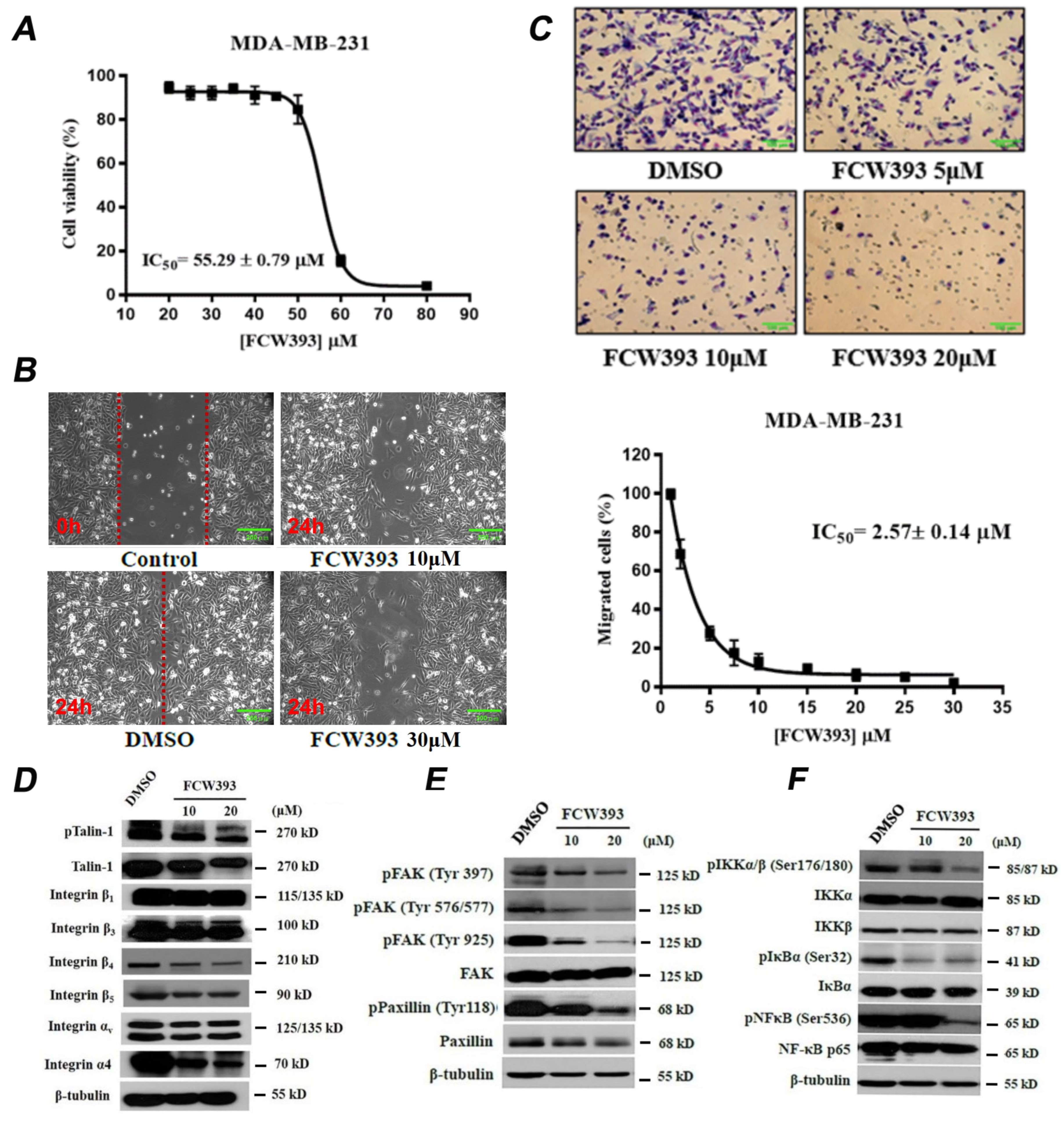
2.1. FCW393 Inhibits Integrin Sialylation and MDA-MB-231 Cell Migration
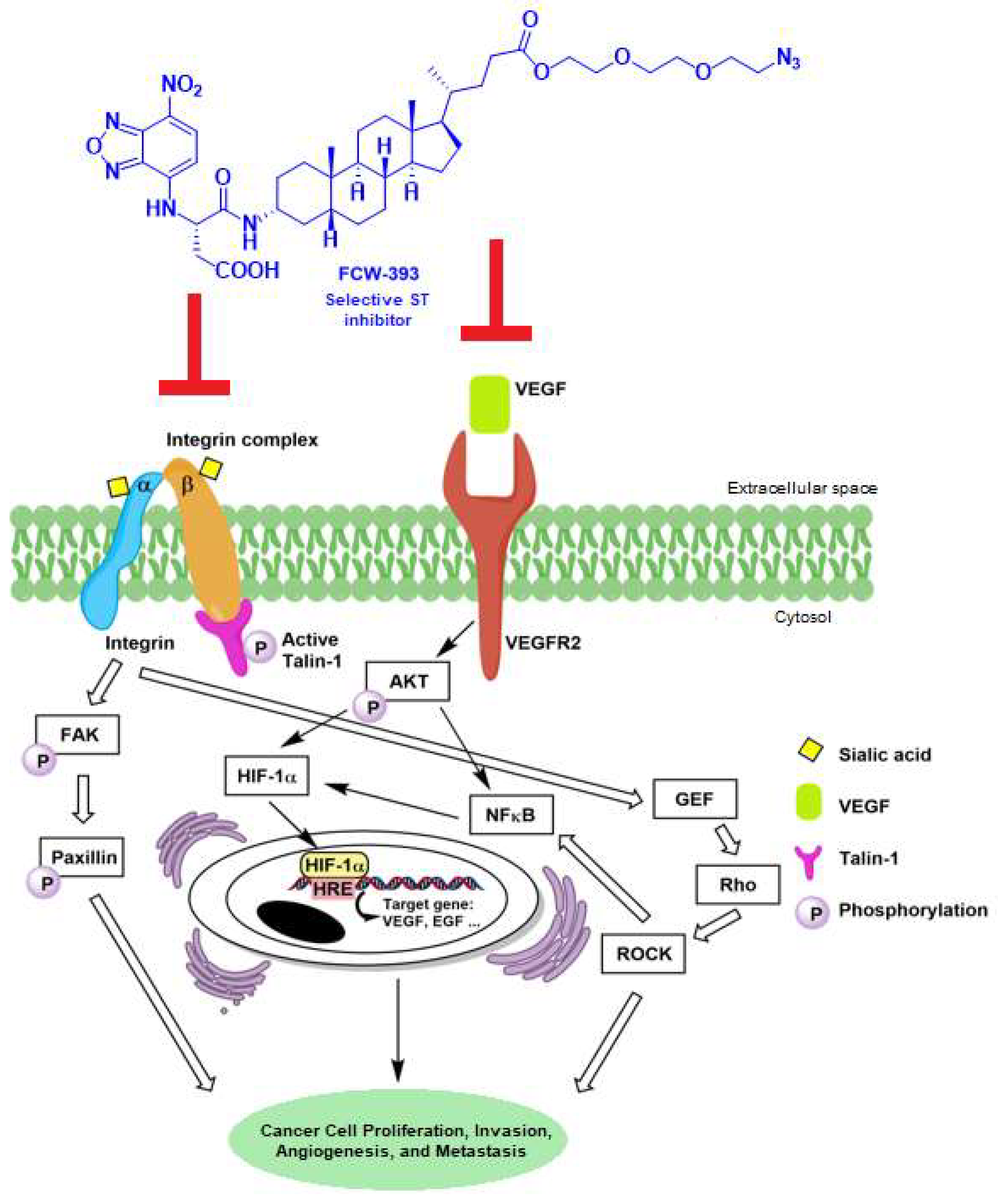
2.2. FCW393 Suppresses Integrin-FAK-Paxillin and NFκB Signal Transduction Pathways in MDA-MB-231 Cells
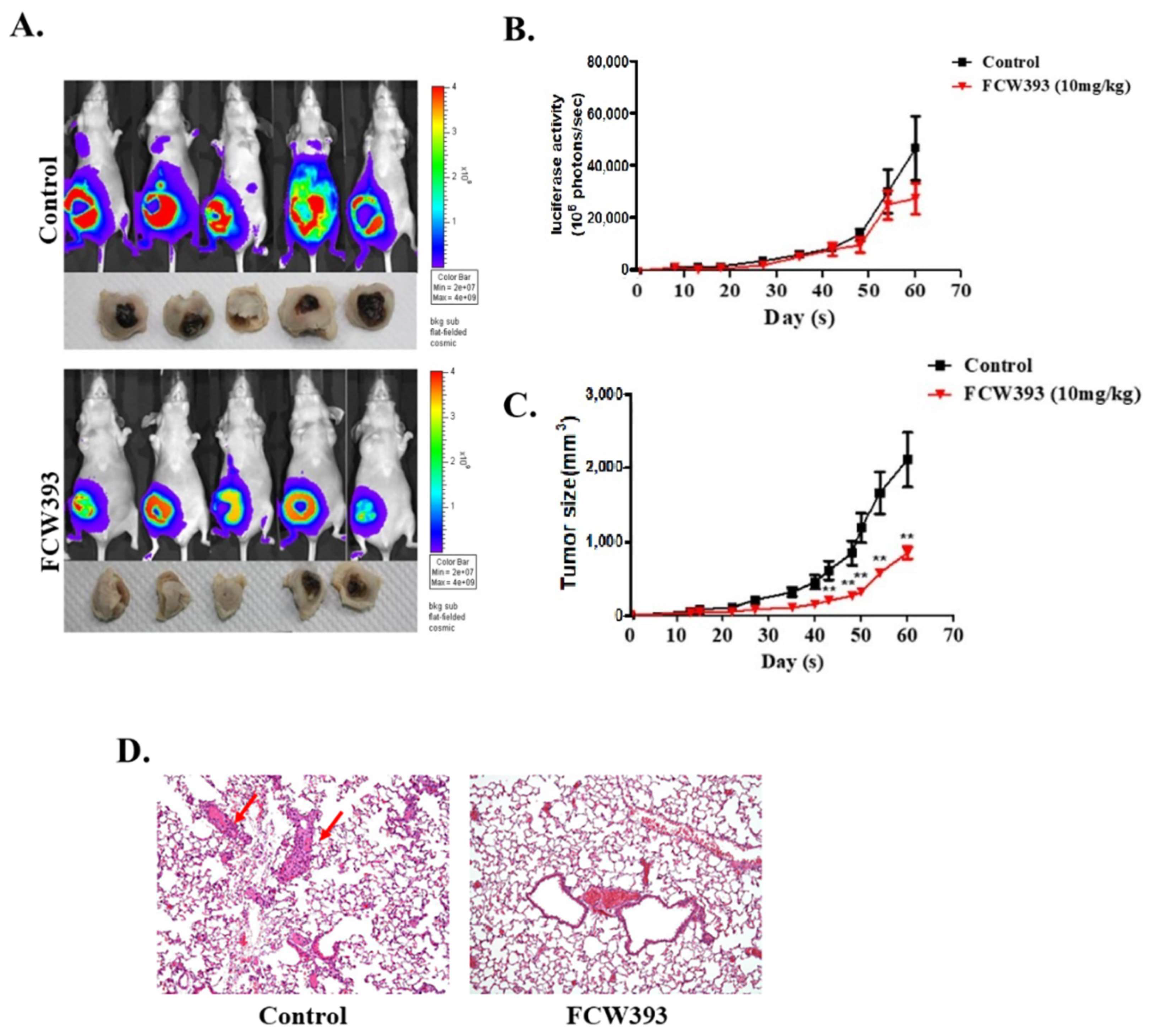
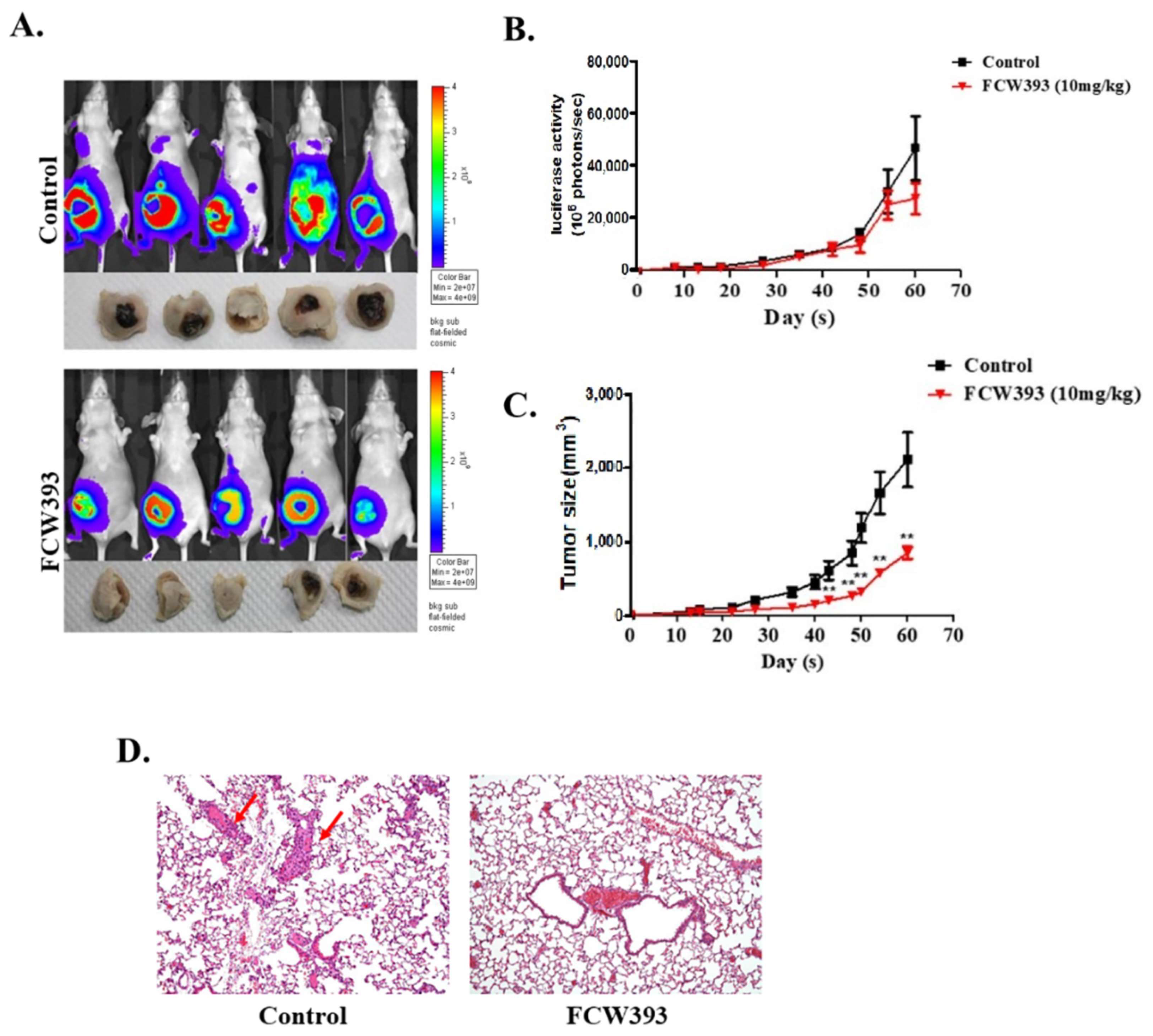
2.3. Antineoplastic Efficacy and Antimetastatic Activity of FCW393 in a Xenograft Model of Human Breast Cancer in MDA-MB-231 Cell-Bearing Mice
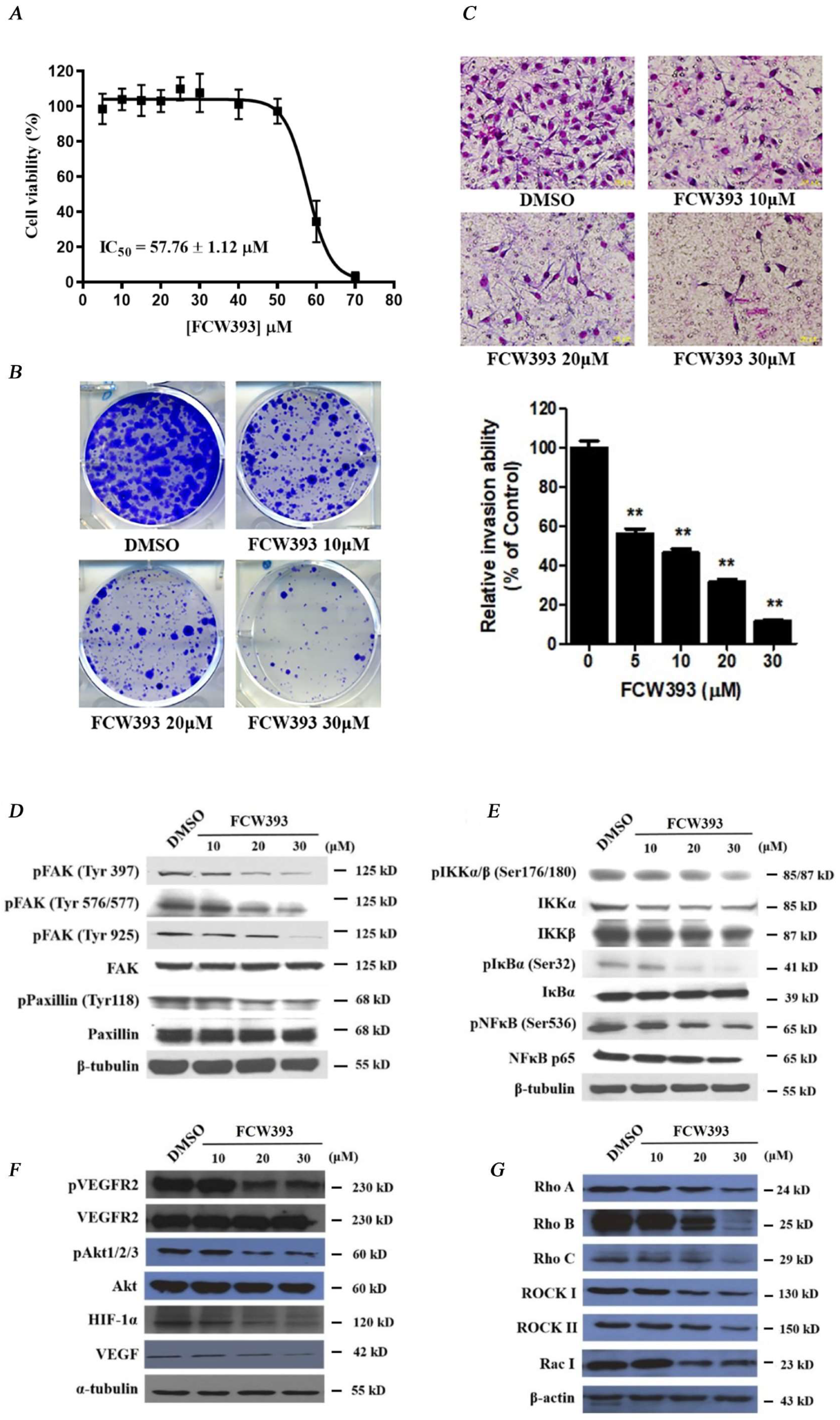
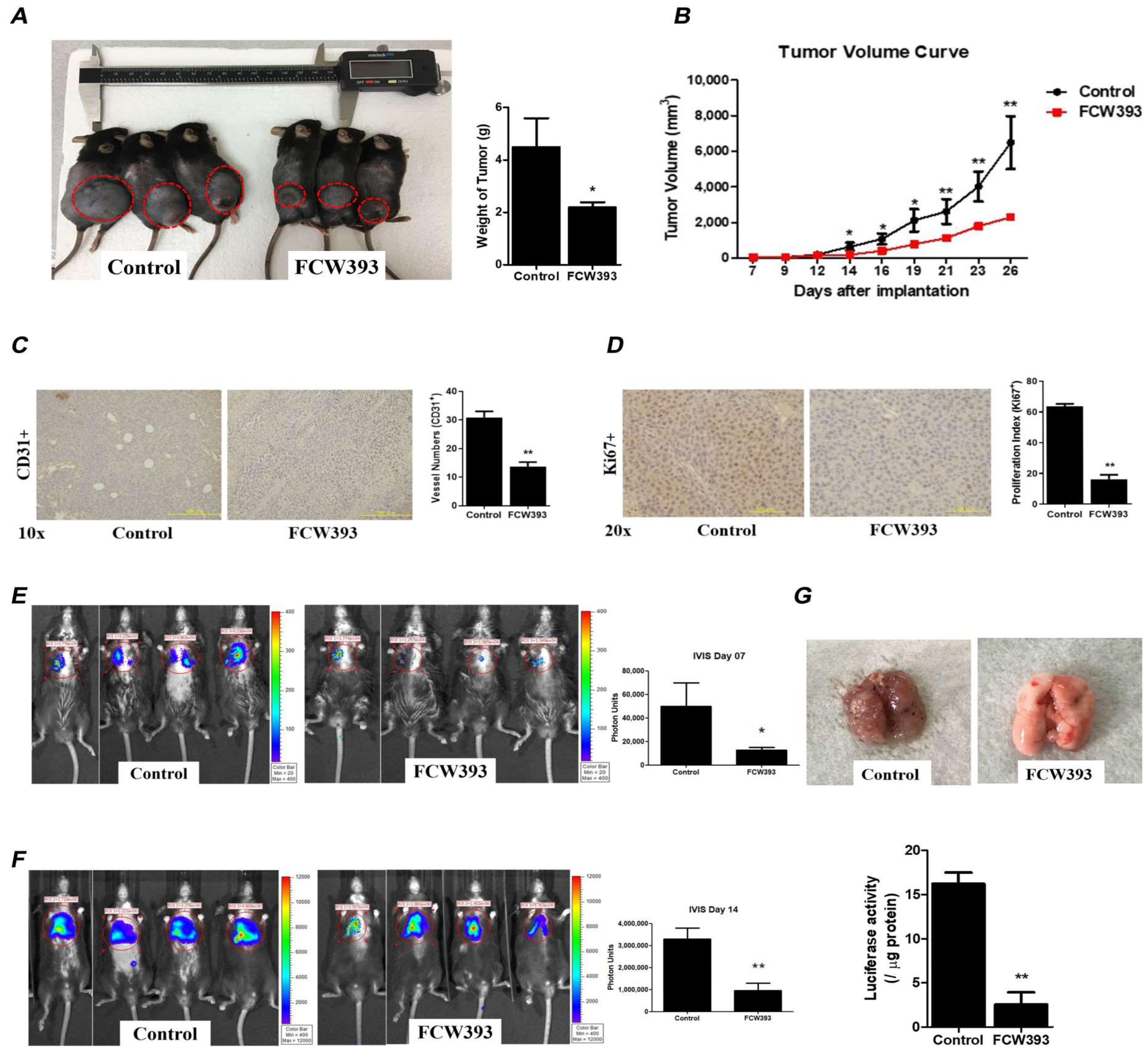
2.4. FCW393 Inhibits B16F10 Cell Colonization and Invasiveness
2.5. FCW393-Inhibition of Signal Transduction Pathways in B16F10 Cells
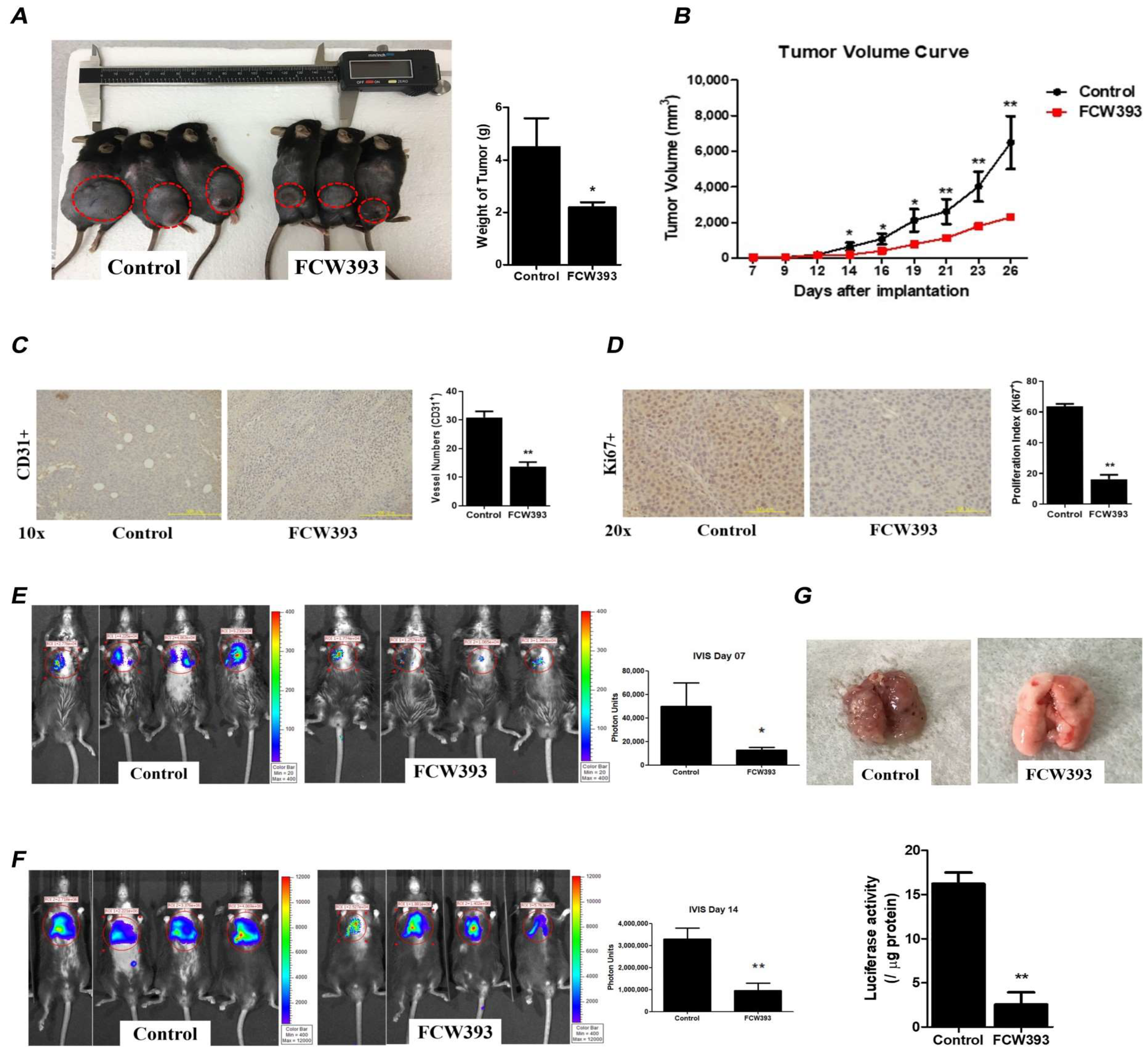
2.6. FCW393 Attenuates Tumor Growth and Delays Lung Metastasis in Mice Bearing Established B16F10 Melanoma
3. Discussion
4. Materials and Methods
4.1. Determination of FCW393 IC50 for In Vitro ST Inhibition
4.2. Cell Culture
4.3. Evaluation of FCW393 Cytotoxicity
4.4. Wound Healing Assay
4.5. Transwell Migration Assay
4.6. Cell Invasion Assay
4.7. Western Blot Analysis
4.8. Lectin Affinity Assay Followed by IP-Western Blotting
4.9. Breast Cancer Mouse Model
4.10. Metastatic Melanoma Mouse Model
4.11. Histological Analysis
4.12. Immunohistochemistry
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bull, C.; den Brok, M.H.; Adema, G.J. Sweet escape: Sialic acids in tumor immune evasion. Biochim. Biophys. Acta 2014, 1846, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Vajaria, B.N.; Patel, P.S. Glycosylation: A hallmark of cancer? Glycoconj. J. 2017, 34, 147–156. [Google Scholar] [CrossRef]
- Garnham, R.; Scott, E.; Livermore, K.E.; Munkley, J. ST6GAL1: A key player in cancer. Oncol. Lett. 2019, 18, 983–989. [Google Scholar] [PubMed]
- Vajaria, B.N.; Patel, K.R.; Begum, R.; Patel, P.S. Sialylation: An avenue to target cancer cells. Pathol. Oncol. Res. 2016, 22, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Laubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Adriaenssens, E.; Ottenberg, K.; Furlan, A.; Courtand, G.; Vercoutter-Edouart, A.-S.; Hanisch, F.-G.; Delannoy, P.; Le Bourhis, X. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology 2006, 16, 54–64. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Glavey, S.V.; Manier, S.; Natoni, A.; Sacco, A.; Moschetta, M.; Reagan, M.R.; Murillo, L.S.; Sahin, I.; Wu, P.; Mishima, Y.; et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood 2014, 124, 1765–1776. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, X.; Liang, L.; Pan, X.; Lv, H.; Zhao, Y. Sialyltransferase ST3GAL6 mediates the effect of microRNA-26a on cell growth, migration, and invasion in hepatocellular carcinoma through the protein kinase B/mammalian target of rapamycin pathway. Cancer Sci. 2017, 108, 267–276. [Google Scholar] [CrossRef]
- Britain, C.M.; Holdbrooks, A.T.; Anderson, J.C.; Willey, C.D.; Bellis, S.L. Sialylation of EGFR by the ST6Gal-I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death. J. Ovarian Res. 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Wichert, B.; Milde-Langosch, K.; Galatenko, V.; Schmalfeldt, B.; Oliveira-Ferrer, L. Prognostic role of the sialyltransferase ST6GAL1 in ovarian cancer. Glycobiology 2018, 28, 898–903. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Wu, L.; Sun, X.L. Sialyltransferase inhibition and recent advances. Biochim. Biophys. Acta 2016, 1864, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Szabo, R.; Skropeta, D. Advancement of Sialyltransferase Inhibitors: Therapeutic Challenges and Opportunities. Med. Res. Rev. 2017, 37, 219–270. [Google Scholar] [CrossRef]
- Pietrobono, S.; Stecca, B. Aberrant Sialylation in CancerL Biomarker and Potential Target for Therapeutic Intervention? Cancers 2021, 13, 2014. [Google Scholar] [CrossRef]
- Perez, S.J.L.P.; Fu, C.W.; Li, W.S. Sialyltransferase Inhibitors for the Treatment of Cancer Metastasis: Current Challenges and Future Perspectives. Molecules 2021, 26, 5673. [Google Scholar] [CrossRef] [PubMed]
- Rillahan, C.D.; Antonopoulos, A.; Lefort, C.T.; Sonon, R.; Azadi, P.; Ley, K.; Dell, A.; Haslam, S.M.; Paulson, J.C. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 2012, 8, 661–668. [Google Scholar] [CrossRef]
- Macauley, M.S.; Arlian, B.M.; Rillahan, C.D.; Pang, P.-C.; Bortell, N.; Marcondes, M.C.G.; Haslam, S.M.; Dell, A.; Paulson, J.C. Systemic blockade of sialylation in mice with a global inhibitor of sialyltransferases. J. Biol. Chem. 2014, 289, 35149–35158. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; Wassink, M.; de Graaf, A.M.A.; van Delft, F.L.; den Brok, M.H.; Adema, G.J. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol. Cancer Ther. 2013, 12, 1935–1946. [Google Scholar] [CrossRef]
- Heise, T.; Pijnenborg, J.F.A.; Büll, C.; van Hilten, N.; Kers-Rebel, E.D.; Balneger, N.; Elferink, H.; Adema, G.J.; Boltje, T.J. Potent metabolic sialylation inhibitors based on C-5-modified fluorinated sialic acids. J. Med. Chem. 2019, 62, 1014–1021. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; van Dinther, E.A.W.; Peters, T.; de Graaf, A.M.A.; Leusen, J.H.W.; Kreutz, M.; Figdor, C.G.; den Brok, M.H.; Adema, G.J. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano. 2015, 9, 733–745. [Google Scholar] [CrossRef]
- Hosoguchi, K.; Maeda, T.; Furukawa, J.; Shinohara, Y.; Hinou, H.; Sekiguchi, M.; Togame, H.; Takemoto, H.; Kondo, H.; Nishimura, S.-I. An efficient approach to the discovery of potent inhibitors against glycosyltransferases. J. Med. Chem. 2010, 53, 5607–5619. [Google Scholar] [CrossRef]
- Danielle, S.; Ralf, S.; Tobias, H.; Schmidt, R.R. Asymmetric synthesis and affinity of potent sialyltransferase inhibitors based on transition-state analogs. Glycoconj. J. 2004, 21, 205–219. [Google Scholar]
- Lee, L.; Chang, K.H.; Valiyev, F.; Liu, H.J.; Li, W.S. Synthesis and biological evaluation of 5’-triazole nucleosides. J. Chin. Chem. Soc. 2006, 53, 1547–1555. [Google Scholar] [CrossRef]
- Miyazaki, T.; Angata, K.; Seeberger, P.H.; Hindsgaul, O.; Fukuda, M. CMP substitutions preferentially inhibit polysialic acid synthesis. Glycobiology 2008, 18, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Wlasichuk, K.B.; Kashem, M.; Nikrad, P.V.; Bird, P.; Jiang, C.; Venot, A.P. Determination of the specificities of rat liver Gal(αl-4)GlcNAc α2,6-sialyltransferase and Gal(α1-3/4)GlcNAc α2,3-sialyltransferase using synthetic modified acceptors. J. Biol. Chem. 1993, 268, 13971–13977. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Xue, J.; Locke, R.D.; Chandrasekaran, E.V.; Srikrishnan, T.; Matta, K.L. Synthesis of fluorinated mucin core branched oligosaccharides with the potential of novel substrates and enzyme inhibitors for glycosyltransferases and sulfotransferases. J. Org. Chem. 2006, 71, 3696–3706. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Brown, S.J.; Register, A.C.; Rosen, H.; Paulson, J.C. High-throughput screening for inhibitors of sialyl- and fucosyltransferases. Angew. Chem. Int. Ed. 2011, 50, 12534–12537. [Google Scholar] [CrossRef]
- Hidari, K.I.P.J.; Oyama, K.; Ito, G.; Nakayama, M.; Inai, M.; Goto, S.; Kanai, Y.; Watanabe, K.; Yoshida, K.; Furuta, T.; et al. Identification and characterization of flavonoids as sialyltransferase inhibitors. Biochem. Biophys. Res. Commun. 2009, 382, 609–613. [Google Scholar] [CrossRef]
- Chang, K.H.; Lee, L.; Chen, J.; Li, W.S. Lithocholic acid analogues, new and potent α-2,3-sialyltransferase inhibitors. Chem. Commun. 2006, 6, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Tang, Y.A.; Huang, S.M.; Juan, H.F.; Wu, L.W.; Sun, Y.C.; Wang, S.C.; Wu, K.W.; Balraj, G.; Chang, T.T.; et al. A novel sialyltransferase inhibitor suppresses FAK/paxillin signaling and cancer angiogenesis and metastasis pathways. Cancer Res. 2011, 71, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Wang, C.H.; Chang, H.C.; More, S.V.; Li, W.S.; Hung, W. A novel sialyltransferase inhibitor AL10 suppresses invasion and metastasis of lung cancer cells by inhibiting integrin-mediated signaling. J. Cell Physiol. 2010, 223, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Shyr, Y.M.; Liao, W.Y.; Chen, T.H.; Wang, S.E.; Lu, P.C.; Lin, P.Y.; Chen, Y.B.; Mao, W.Y.; Han, H.Y.; et al. Elevation of β-galactoside α2,6-sialyltransferase 1 in a fructoseresponsive manner promotes pancreatic cancer metastasis. Oncotarget 2017, 8, 7691–7709. [Google Scholar] [CrossRef] [PubMed]
- Abdu-Allah, H.H.; Chang, T.T.; Li, W.S. Synthesis of B- and C-ring-modified lithocholic acid analogues as potential sialyltransferase inhibitors. Steroids 2016, 112, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Su, M.L.; Chang, T.M.; Chiang, C.H.; Chang, H.C.; Hou, M.F.; Li, W.S.; Hung, W.C. Inhibition of chemokine (C-C motif) receptor 7 sialylation suppresses CCL19-stimulated proliferation, invasion and anti-anoikis. PLoS ONE 2014, 9, e98823. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Chang, W.W.; Chen, C.C.; Tsai, Y.C. Stachybotrydial, a potent inhibitor of fucosyltransferase and sialyltransferase. Biochem. Biophys. Res. Commun. 2005, 331, 953–957. [Google Scholar] [CrossRef]
- Seko, A.; Yamase, T.; Yamashita, K. Polyoxometalates as effective inhibitors for sialyl- and sulfotransferases. J. Inorg. Biochem. 2009, 103, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; McRoberts, K.; Coyner, M.; Andarawewa, K.L.; Frierson, H.F.J.; Sanders, J.M.; Swenson, S.; Markland, F.; Conaway, M.R.; Theodorescu, D. Integrin agonists as adjuvants in chemotherapy for melanoma. Clin. Cancer Res. 2008, 14, 6193–6197. [Google Scholar] [CrossRef]
- Toricelli, M.; Melo, F.H.; Peres, G.B.; Silva, D.C.; Jasiulionis, M.G. Timp1 interacts with beta-1 integrin and CD63 along melanoma genesis and confers anoikis resistance by activating PI3-K signaling pathway independently of Akt phosphorylation. Mol. Cancer. 2013, 12, 22. [Google Scholar] [CrossRef]
- Parvani, J.G.; Gujrati, M.D.; Mack, M.A.; Schiemann, W.P.; Lu, Z.R. Silencing β3 integrin by targeted ECO/siRNA nanoparticles inhibits EMT and metastasis of triple-negative breast cancer. Cancer Res. 2015, 75, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Kahraman, N.; Ashour, A.; Ozpolat, B. FOXM1 transcriptionally regulates expression of integrin β1 in triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 163, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Bergonzini, C.; Kroese, K.; Zweemer, A.J.M.; Danen, E.H.J. Targeting Integrins for Cancer Therapy—Disappointments and Opportunities. Front. Cell Dev. Biol. 2022, 10, 863850. [Google Scholar] [CrossRef] [PubMed]
- Parvani, J.G.; Galliher-Beckley, A.J.; Schiemann, B.J.; Schiemann, W.P. Targeted inactivation of β1 integrin induces β3 integrin switching, which drives breast cancer metastasis by TGF-β. Mol. Biol. Cell. 2013, 24, 3449–3459. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, A.; Zijlstra, A.; Zutter, M.M. Flipping the switch: Integrin switching provides metastatic competence. Sci. Signal. 2014, 7, pe9. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.W.; Tsai, H.E.; Chen, W.S.; Chang, T.T.; Chen, C.L.; Hsiao, P.W.; Li, W.S. Sialyltransferase inhibitors suppress breast cancer metastasis. J. Med. Chem. 2021, 64, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Harduin-Lepers, A.; Vallejo-Ruiz, V.; Krzewinski-Recchi, M.A.; Samyn-Petit, B.; Julien, S.; Delannoy, P. The human sialyltransferase family. Biochimie 2001, 83, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Costell, M.; Fässler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Nader, G.P.; Ezratty, E.J.; Gundersen, G.G. Fak, talin and pipkigamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat. Cell Biol. 2016, 18, 491–503. [Google Scholar] [CrossRef]
- Gu, J.; Taniguchi, N. Regulation of integrin functions by N-glycans. Glycoconj. J. 2004, 21, 9–15. [Google Scholar] [CrossRef]
- Janik, M.E.; Lityńska, A.; Vereecken, P. Cell migration—The role of integrin glycosylation. Biochim. Biophys. Acta 2010, 1800, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Christie, D.R.; Shaikh, F.M.; Lucas, J.A., IV; Lucas, J.A., III; Bellis, S.L. ST6Gal-I expression in ovarian cancer cells promotes an invasive phenotype by altering integrin glycosylation and function. J. Ovarian Res. 2008, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Ng, E.; Varki, A.; Varki, N.M. α2-6-Linked sialic acids on N-glycans modulate carcinoma differentiation in vivo. Cancer Res. 2008, 68, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, F.M.; Seales, E.C.; Clem, W.C.; Hennessy, K.M.; Zhuo, Y.; Bellis, S.L. Tumor cell migration and invasion are regulated by expression of variant integrin glycoforms. Exp. Cell Res. 2008, 314, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Desiniotis, A.; Kyprianou, N. Significance of talin in cancer progression and metastasis. Int. Rev. Cell Mol. Biol. 2011, 289, 117–147. [Google Scholar] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Chuang, H.H.; Zhen, Y.Y.; Tsai, Y.C.; Chuang, C.H.; Hsiao, M.; Huang, M.S.; Yang, C.J. FAK in Cancer: From Mechanisms to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 1726. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer. 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Britain, C.M.; Dorsett, K.A.; Bellis, S.L. The glycosyltransferase ST6Gal-I protects tumor cells against serum growth factor withdrawal by enhancing survival signaling and proliferative potential. J. Biol. Chem. 2017, 292, 4663–4673. [Google Scholar] [CrossRef]
- Kang, N.Y.; Kim, C.H.; Kim, K.S.; Ko, J.H.; Lee, J.H.; Jeong, Y.K.; Lee, Y.C. Expression of the human CMP-NeuAc:GM3 alpha2,8-sialyltransferase (GD3 synthase) gene through the NF-kappaB activation in human melanoma SK-MEL-2 cells. Biochim. Biophys. Acta 2007, 1769, 622–630. [Google Scholar] [CrossRef]
- Rusiniak, M.E.; Punch, P.R.; Hait, N.C.; Maiti, A.; Burns, R.T.; Chapla, D.; Moremen, K.W.; Zhao, P.; Wells, L.; Hoffmeister, K.; et al. Extracellular ST6GAL1 regulates monocyte-macrophage development and survival. Glycobiology 2022, 32, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Morishige, K.; Takahashi, T.; Hashimoto, K.; Ogata, S.; Tsutsumi, S.; Takata, K.; Ohta, T.; Kawagoe, J.; Takahashi, K.; et al. Fasudil inhibits vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Cannon, R.D.; Coates, D.E.; Mei, L. Effect of the Rho-Kinase/ROCK Signaling Pathway on Cytoskeleton Components. Genes 2023, 14, 272. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Tsai, H.E.; Weng, W.T.; Liu, L.F.; Weng, C.H.; Chuang, M.R.; Lam, H.C.; Wu, C.S.; Tee, R.; Wen, Z.H.; et al. Systemic pro-opiomelanocortin expression induces melanogenic differentiation and inhibits tumor angiogenesis in established mouse melanoma. Hum. Gene Ther. 2011, 22, 325–335. [Google Scholar] [CrossRef]
- Tsai, H.E.; Liu, G.S.; Kung, M.L.; Liu, L.F.; Wu, J.C.; Tang, C.H.; Huang, C.H.; Chen, S.C.; Lam, H.C.; Wu, C.S.; et al. Downregulation of hepatoma-derived growth factor contributes to retarded lung metastasis via inhibition of epithelial-mesenchymal transition by systemic POMC gene delivery in melanoma. Mol. Cancer Ther. 2013, 12, 1016–1025. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, H.-E.; Chen, C.-L.; Chang, T.-T.; Fu, C.-W.; Chen, W.-C.; Perez, S.J.L.P.; Hsiao, P.-W.; Tai, M.-H.; Li, W.-S. Development of a Novel, Potent, and Selective Sialyltransferase Inhibitor for Suppressing Cancer Metastasis. Int. J. Mol. Sci. 2024, 25, 4283. https://doi.org/10.3390/ijms25084283
Tsai H-E, Chen C-L, Chang T-T, Fu C-W, Chen W-C, Perez SJLP, Hsiao P-W, Tai M-H, Li W-S. Development of a Novel, Potent, and Selective Sialyltransferase Inhibitor for Suppressing Cancer Metastasis. International Journal of Molecular Sciences. 2024; 25(8):4283. https://doi.org/10.3390/ijms25084283
Chicago/Turabian StyleTsai, Han-En, Chia-Ling Chen, Tzu-Ting Chang, Chih-Wei Fu, Wei-Chia Chen, Ser John Lynon P. Perez, Pei-Wen Hsiao, Ming-Hong Tai, and Wen-Shan Li. 2024. "Development of a Novel, Potent, and Selective Sialyltransferase Inhibitor for Suppressing Cancer Metastasis" International Journal of Molecular Sciences 25, no. 8: 4283. https://doi.org/10.3390/ijms25084283