
Effects of Mild Closed-Head Injury and Subanesthetic Ketamine Infusion on Microglia, Axonal Injury, and Synaptic Density in Sprague–Dawley Rats

 , ,
, , 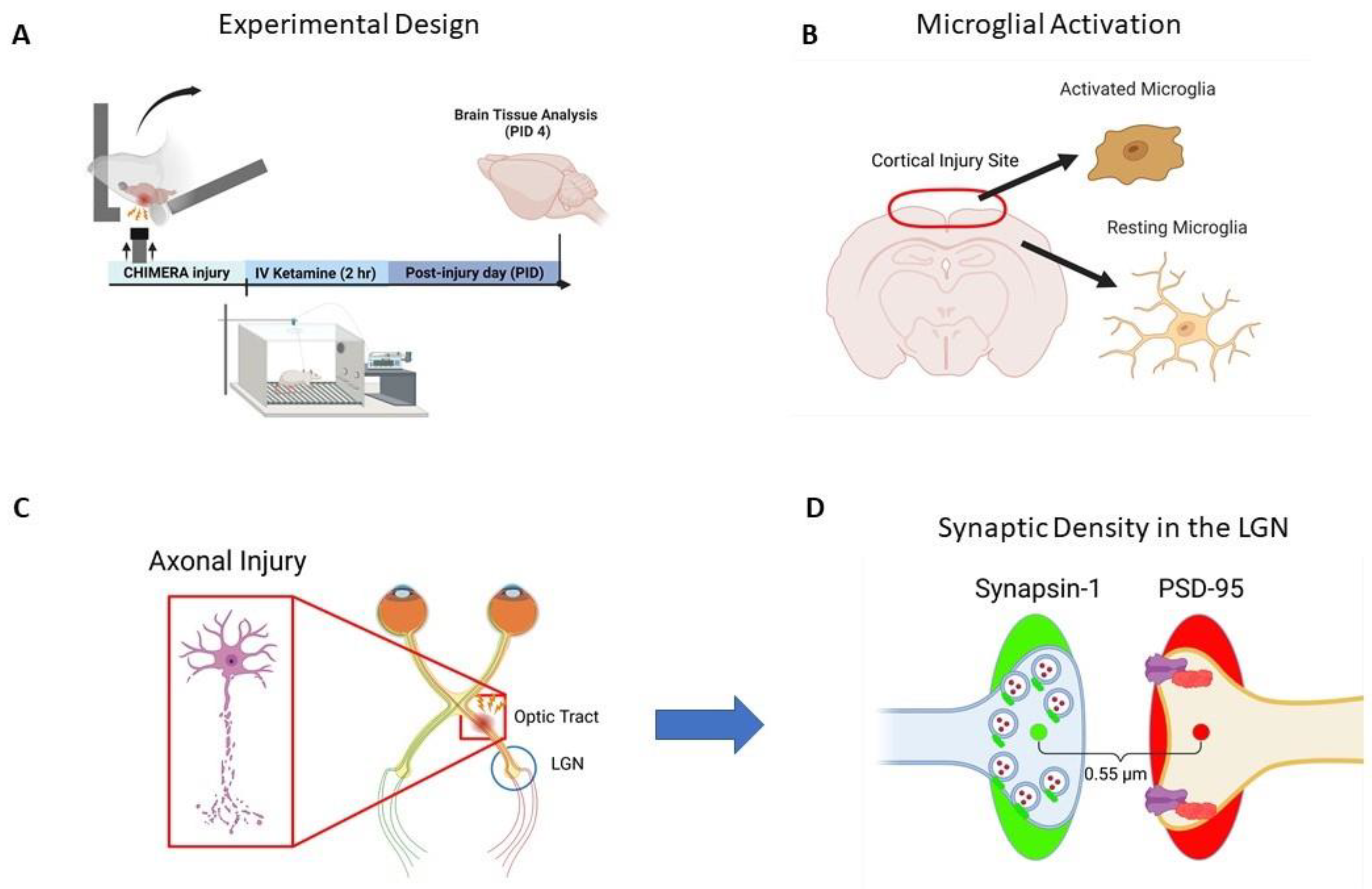{kind=link}
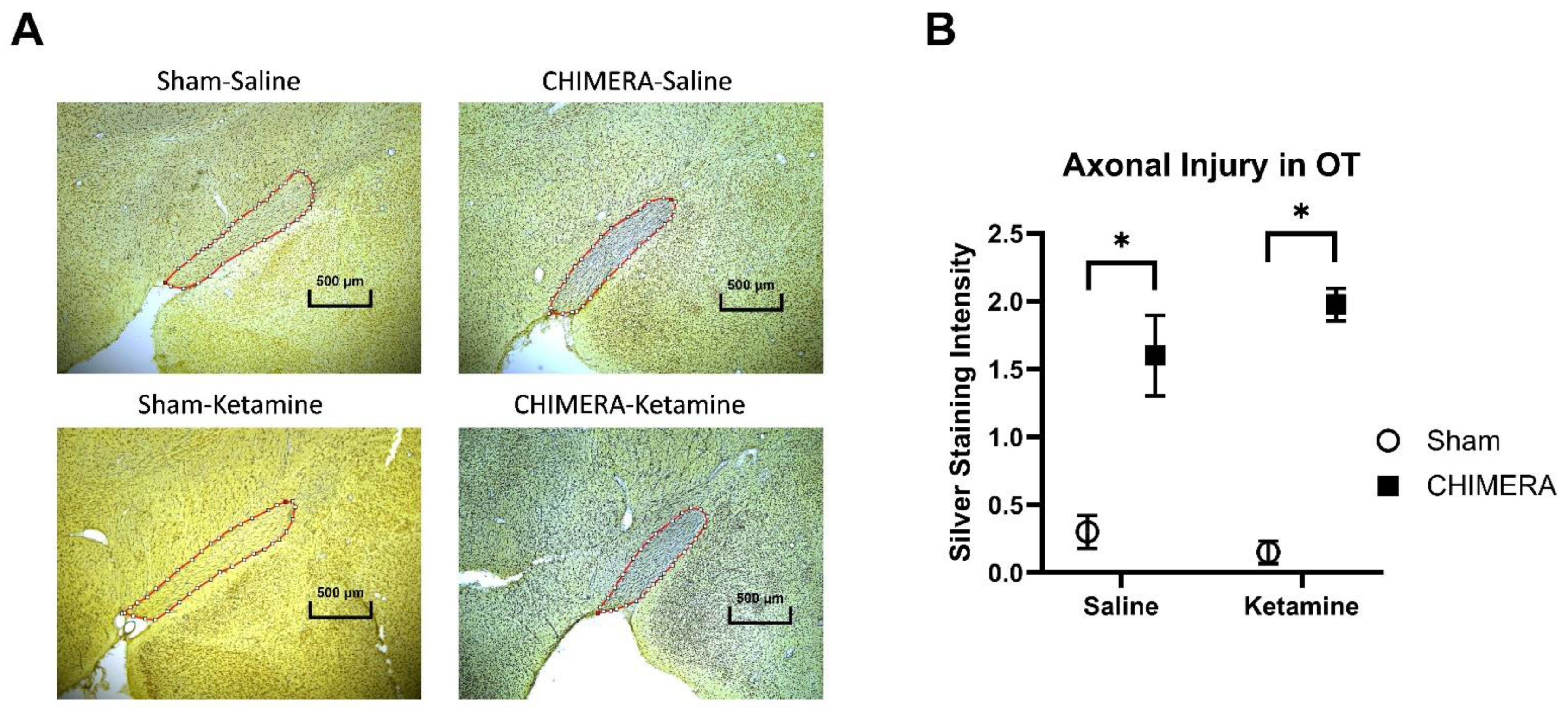{kind=link}
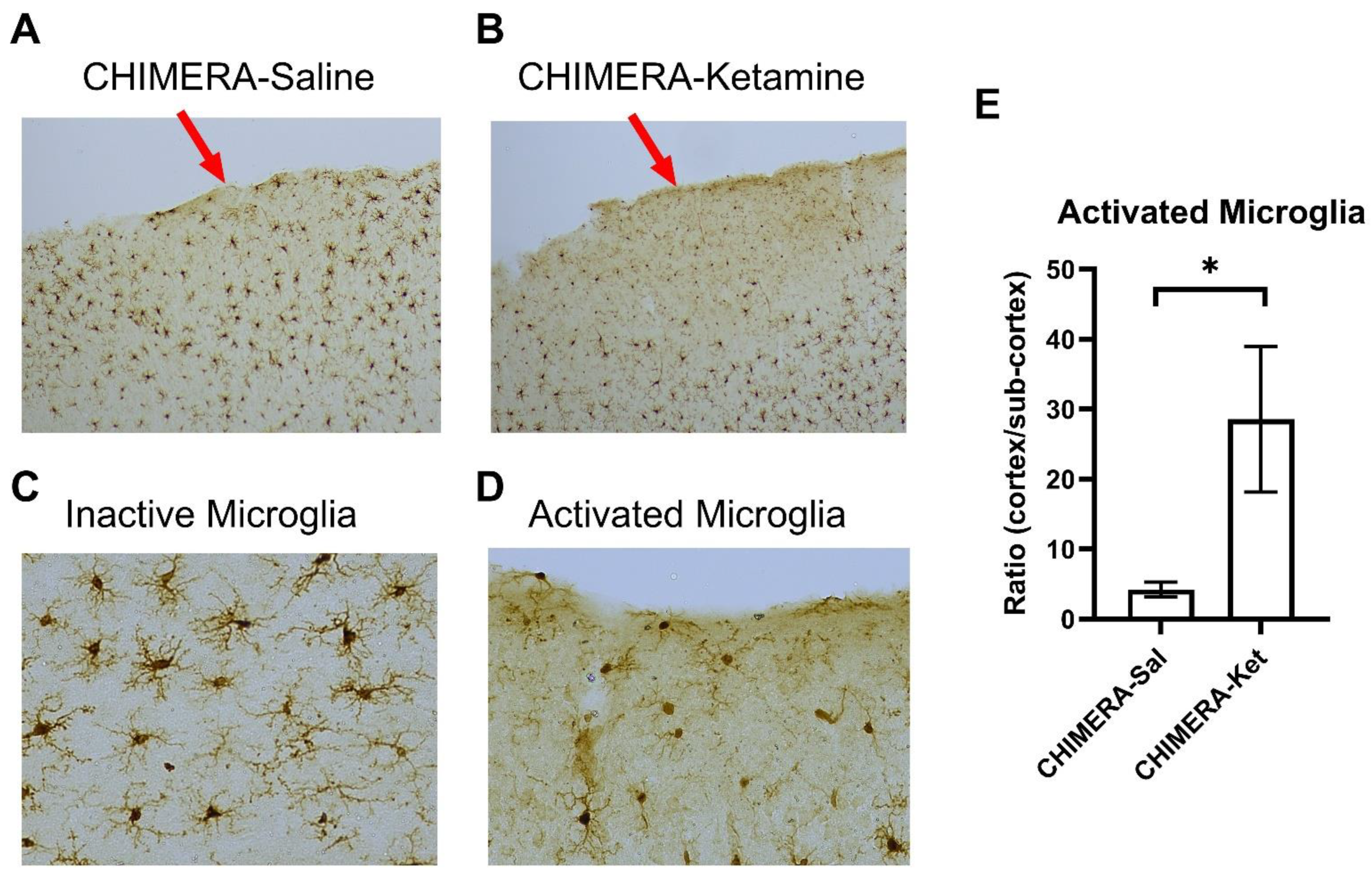{kind=link}
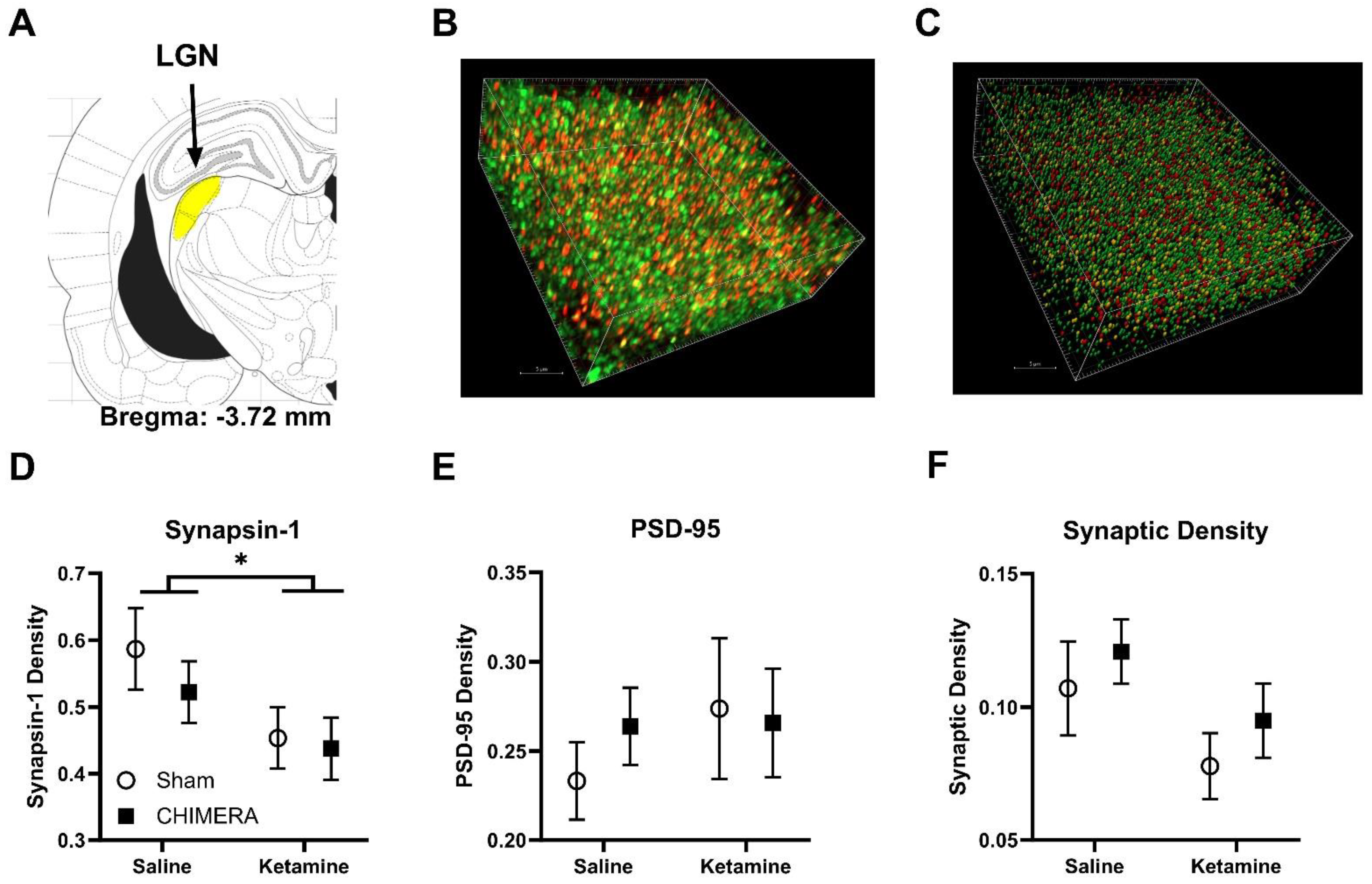{kind=link}
Abstract
1. Introduction
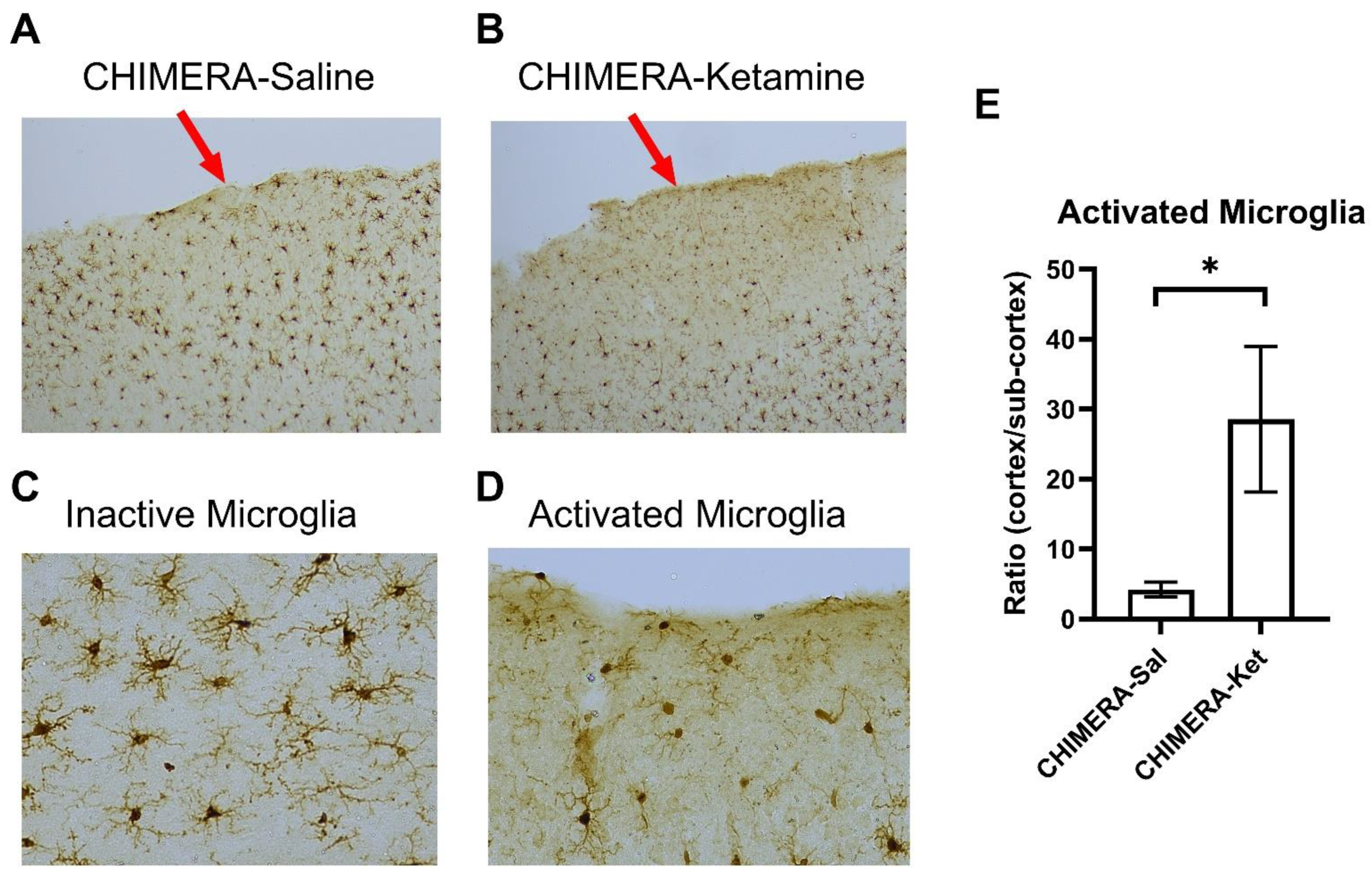
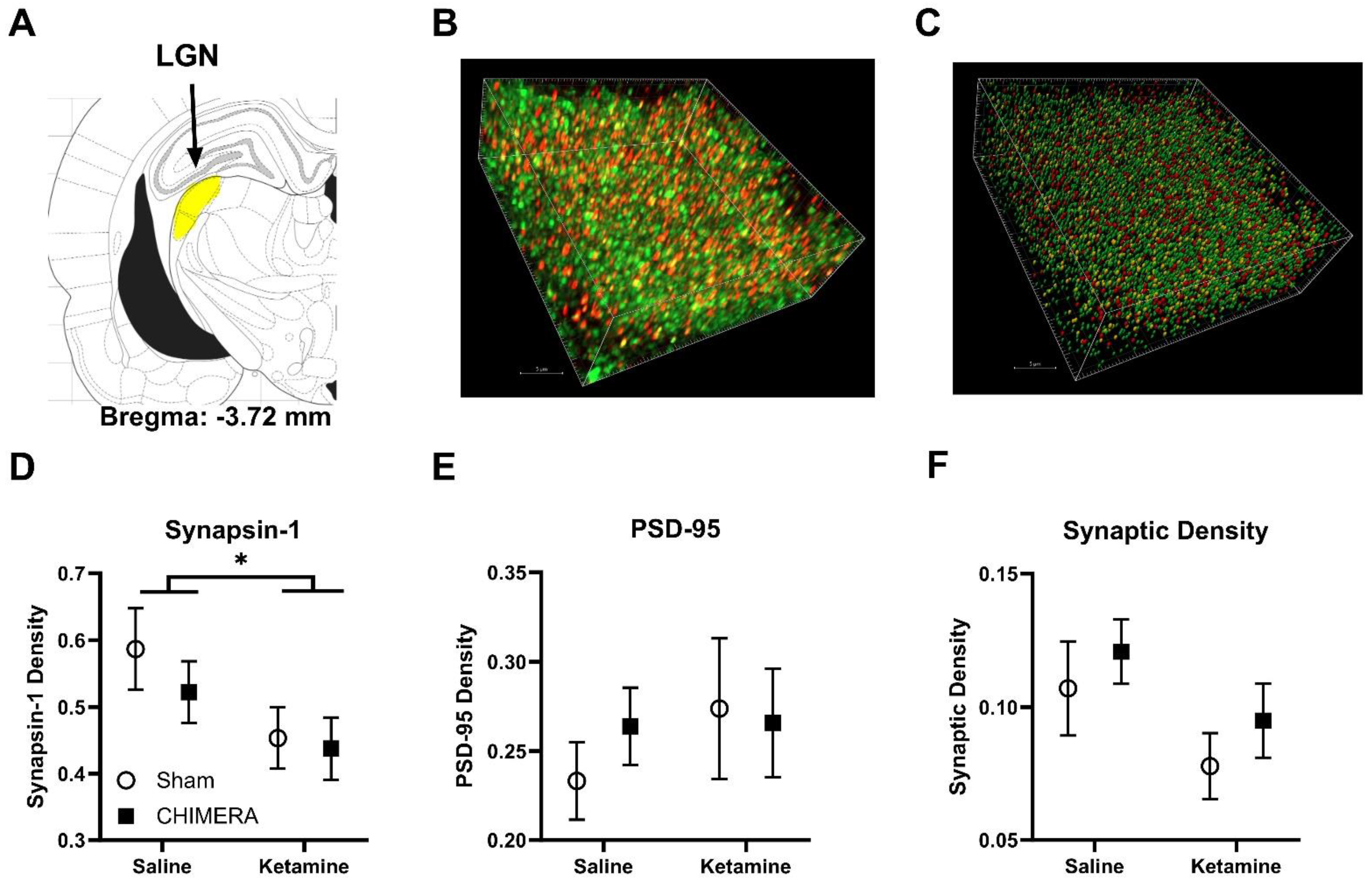
2. Results
3. Discussion
4. Methods
4.1. Animals
4.2. CHIMERA and Ketamine Infusion
4.3. Brain Tissue Collection
4.4. Silver Staining
4.5. Iba-1 Immunohistochemistry
4.6. Synapsin and PSD-95 Immunohistochemistry
4.7. SEQUIN
4.8. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC. Report to Congress on Traumatic Brain Injury Epidemiology and Rehabilitation; CDC: Atlanta, GA, USA, 2015. [Google Scholar]
- McInnes, K.; Friesen, C.L.; MacKenzie, D.E.; Westwood, D.A.; Boe, S.G. Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review. PLoS ONE 2017, 12, e0174847. [Google Scholar] [CrossRef] [PubMed]
- Cancelliere, C.; Verville, L.; Stubbs, J.L.; Yu, H.; Hincapie, C.A.; Cassidy, J.D.; Wong, J.J.; Shearer, H.M.; Connell, G.; Southerst, D.; et al. Post-Concussion Symptoms and Disability in Adults with Mild Traumatic Brain Injury: A Systematic Review and Meta-Analysis. J. Neurotrauma 2023, 40, 1045–1059. [Google Scholar] [CrossRef]
- Va/DoD. VA/DoD Clinical Practice Guideline for the Management and Rehabilitation of Post-Acute Mild Traumatic Brain Injury; Va/DoD: Washington, DC, USA, 2021. [Google Scholar]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.B.; Leite-Morris, K.A.; Wang, L.; Rumbika, K.K.; Heinrichs, S.C.; Zeng, X.; Wu, L.; Arena, D.T.; Teng, Y.D. Pathophysiological Bases of Comorbidity: Traumatic Brain Injury and Post-Traumatic Stress Disorder. J. Neurotrauma 2018, 35, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Spencer, H.F.; Boese, M.; Berman, R.Y.; Radford, K.D.; Choi, K.H. Effects of a Subanesthetic Ketamine Infusion on Inflammatory and Behavioral Outcomes after Closed Head Injury in Rats. Bioengineering 2023, 10, 941. [Google Scholar] [CrossRef]
- Aleksandrova, L.R.; Phillips, A.G. Neuroplasticity as a convergent mechanism of ketamine and classical psychedelics. Trends Pharmacol. Sci. 2021, 42, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Ye, Y.; Chen, F.; Han, W.C.; Sun, J.M.; Lu, X.; Guo, R.; Cao, K.; Zheng, M.J.; Liao, L.C. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience 2017, 343, 30–38. [Google Scholar] [CrossRef]
- CDC. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2014; CDC: Atlanta, GA, USA, 2019. [Google Scholar]
- Namjoshi, D.R.; Cheng, W.H.; McInnes, K.A.; Martens, K.M.; Carr, M.; Wilkinson, A.; Fan, J.; Robert, J.; Hayat, A.; Cripton, P.A.; et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): A novel, surgery-free model of traumatic brain injury. Mol. Neurodegener. 2014, 9, 55. [Google Scholar] [CrossRef]
- Kleiven, S. Why Most Traumatic Brain Injuries are Not Caused by Linear Acceleration but Skull Fractures are. Front. Bioeng. Biotechnol. 2013, 1, 15. [Google Scholar] [CrossRef]
- Malik, S.; Alnaji, O.; Malik, M.; Gambale, T.; Farrokhyar, F.; Rathbone, M.P. Inflammatory cytokines associated with mild traumatic brain injury and clinical outcomes: A systematic review and meta-analysis. Front. Neurol. 2023, 14, 1123407. [Google Scholar] [CrossRef]
- Narayana, P.A. White matter changes in patients with mild traumatic brain injury: MRI perspective. Concussion 2017, 2, CNC35. [Google Scholar] [CrossRef]
- Smith, C.; Gentleman, S.M.; Leclercq, P.D.; Murray, L.S.; Griffin, W.S.; Graham, D.I.; Nicoll, J.A. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol. Appl. Neurobiol. 2013, 39, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Namjoshi, D.R.; Cheng, W.H.; Bashir, A.; Wilkinson, A.; Stukas, S.; Martens, K.M.; Whyte, T.; Abebe, Z.A.; McInnes, K.A.; Cripton, P.A.; et al. Defining the biomechanical and biological threshold of murine mild traumatic brain injury using CHIMERA (Closed Head Impact Model of Engineered Rotational Acceleration). Exp. Neurol. 2017, 292, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Tucker, L.B.; Fu, A.H.; McCabe, J.T. Hippocampal-Dependent Cognitive Dysfunction following Repeated Diffuse Rotational Brain Injury in Male and Female Mice. J. Neurotrauma 2021, 38, 1585–1606. [Google Scholar] [CrossRef] [PubMed]
- Zietlow, J.; Berns, K.; Jenkins, D.; Zietlow, S. Prehospital Use of Ketamine: Effectiveness in Critically Ill and Injured Patients. Mil. Med. 2019, 184, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Reede, K.; Bartholomew, R.; Nielsen, D.; Ahmeti, M.; Zreik, K. Ketamine in Trauma: A Literature Review and Administration Guidelines. Cureus 2023, 15, e48099. [Google Scholar] [CrossRef] [PubMed]
- Eikermann, M.; Grosse-Sundrup, M.; Zaremba, S.; Henry, M.E.; Bittner, E.A.; Hoffmann, U.; Chamberlin, N.L. Ketamine activates breathing and abolishes the coupling between loss of consciousness and upper airway dilator muscle dysfunction. Anesthesiology 2012, 116, 35–46. [Google Scholar] [CrossRef]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Alway, E.J.; et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 2019, 364, eaat8078. [Google Scholar] [CrossRef] [PubMed]
- Sala, N.; Paoli, C.; Bonifacino, T.; Mingardi, J.; Schiavon, E.; La Via, L.; Milanese, M.; Tornese, P.; Datusalia, A.K.; Rosa, J.; et al. Acute Ketamine Facilitates Fear Memory Extinction in a Rat Model of PTSD Along With Restoring Glutamatergic Alterations and Dendritic Atrophy in the Prefrontal Cortex. Front. Pharmacol. 2022, 13, 759626. [Google Scholar] [CrossRef]
- Ardalan, M.; Wegener, G.; Polsinelli, B.; Madsen, T.M.; Nyengaard, J.R. Neurovascular plasticity of the hippocampus one week after a single dose of ketamine in genetic rat model of depression. Hippocampus 2016, 26, 1414–1423. [Google Scholar] [CrossRef]
- Liu, W.X.; Wang, J.; Xie, Z.M.; Xu, N.; Zhang, G.F.; Jia, M.; Zhou, Z.Q.; Hashimoto, K.; Yang, J.J. Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology 2016, 233, 405–415. [Google Scholar] [CrossRef]
- Radford, K.D.; Spencer, H.F.; Zhang, M.; Berman, R.Y.; Girasek, Q.L.; Choi, K.H. Association between intravenous ketamine-induced stress hormone levels and long-term fear memory renewal in Sprague-Dawley rats. Behav. Brain Res. 2020, 378, 112259. [Google Scholar] [CrossRef]
- Radford, K.D.; Park, T.Y.; Jaiswal, S.; Pan, H.; Knutsen, A.; Zhang, M.; Driscoll, M.; Osborne-Smith, L.A.; Dardzinski, B.J.; Choi, K.H. Enhanced fear memories and brain glucose metabolism ((18)F-FDG-PET) following sub-anesthetic intravenous ketamine infusion in Sprague-Dawley rats. Transl. Psychiatry 2018, 8, 263. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Wang, X.; Wu, H.; Wu, Q.; Zhang, J. Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration. Front. Aging Neurosci. 2022, 14, 825086. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef]
- Broussard, J.I.; Acion, L.; De Jesus-Cortes, H.; Yin, T.; Britt, J.K.; Salas, R.; Costa-Mattioli, M.; Robertson, C.; Pieper, A.A.; Arciniegas, D.B.; et al. Repeated mild traumatic brain injury produces neuroinflammation, anxiety-like behaviour and impaired spatial memory in mice. Brain Inj. 2018, 32, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Shukla, D.K.; Armstrong, R.C.; Marion, C.M.; Radomski, K.L.; Selwyn, R.G.; Dardzinski, B.J. Repetitive Model of Mild Traumatic Brain Injury Produces Cortical Abnormalities Detectable by Magnetic Resonance Diffusion Imaging, Histopathology, and Behavior. J. Neurotrauma 2017, 34, 1364–1381. [Google Scholar] [CrossRef]
- Patel, R.K.; Prasad, N.; Kuwar, R.; Haldar, D.; Abdul-Muneer, P.M. Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain Behav. Immun. 2017, 64, 244–258. [Google Scholar] [CrossRef]
- McAteer, K.M.; Corrigan, F.; Thornton, E.; Turner, R.J.; Vink, R. Short and Long Term Behavioral and Pathological Changes in a Novel Rodent Model of Repetitive Mild Traumatic Brain Injury. PLoS ONE 2016, 11, e0160220. [Google Scholar] [CrossRef]
- Zhang, N.; Yao, L.; Wang, P.; Liu, Z. Immunoregulation and antidepressant effect of ketamine. Transl. Neurosci. 2021, 12, 218–236. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wilde, E.A.; Li, X.; Hunter, J.V.; Narayana, P.A.; Hasan, K.; Biekman, B.; Swank, P.; Robertson, C.; Miller, E.; McCauley, S.R.; et al. Loss of Consciousness Is Related to White Matter Injury in Mild Traumatic Brain Injury. J. Neurotrauma 2016, 33, 2000–2010. [Google Scholar] [CrossRef] [PubMed]
- Sorg, S.F.; Delano-Wood, L.; Luc, N.; Schiehser, D.M.; Hanson, K.L.; Nation, D.A.; Lanni, E.; Jak, A.J.; Lu, K.; Meloy, M.J.; et al. White matter integrity in veterans with mild traumatic brain injury: Associations with executive function and loss of consciousness. J. Head. Trauma. Rehabil. 2014, 29, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Hayes, J.P.; Lafleche, G.; Salat, D.H.; Verfaellie, M. White matter abnormalities are associated with overall cognitive status in blast-related mTBI. Brain Imaging Behav. 2017, 11, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Hayes, J.P.; Lafleche, G.; Salat, D.H.; Verfaellie, M. White matter abnormalities are associated with chronic postconcussion symptoms in blast-related mild traumatic brain injury. Hum. Brain Mapp. 2016, 37, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Mohammadian, M.; Roine, T.; Hirvonen, J.; Kurki, T.; Posti, J.P.; Katila, A.J.; Takala, R.S.K.; Tallus, J.; Maanpaa, H.R.; Frantzen, J.; et al. Alterations in Microstructure and Local Fiber Orientation of White Matter Are Associated with Outcome after Mild Traumatic Brain Injury. J. Neurotrauma 2020, 37, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Gazdzinski, L.M.; Mellerup, M.; Wang, T.; Adel, S.A.A.; Lerch, J.P.; Sled, J.G.; Nieman, B.J.; Wheeler, A.L. White Matter Changes Caused by Mild Traumatic Brain Injury in Mice Evaluated Using Neurite Orientation Dispersion and Density Imaging. J. Neurotrauma 2020, 37, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Chen, H.; Kim, H.Y. Multiple Mild Traumatic Brain Injuries Lead to Visual Dysfunction in a Mouse Model. J. Neurotrauma 2020, 37, 286–294. [Google Scholar] [CrossRef]
- Chen, H.; Kevala, K.; Aflaki, E.; Marugan, J.; Kim, H.Y. GPR110 ligands reduce chronic optic tract gliosis and visual deficit following repetitive mild traumatic brain injury in mice. J. Neuroinflamm. 2021, 18, 157. [Google Scholar] [CrossRef]
- De Moraes, C.G. Anatomy of the visual pathways. J. Glaucoma 2013, 22 (Suppl. 5), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Evanson, N.K.; Guilhaume-Correa, F.; Herman, J.P.; Goodman, M.D. Optic tract injury after closed head traumatic brain injury in mice: A model of indirect traumatic optic neuropathy. PLoS ONE 2018, 13, e0197346. [Google Scholar] [CrossRef] [PubMed]
- Hetzer, S.M.; Shalosky, E.M.; Torrens, J.N.; Evanson, N.K. Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses. Cells 2021, 10, 3343. [Google Scholar] [CrossRef]
- Sharma, S.; Chitranshi, N.; Wall, R.V.; Basavarajappa, D.; Gupta, V.; Mirzaei, M.; Graham, S.L.; Klistorner, A.; You, Y. Trans-synaptic degeneration in the visual pathway: Neural connectivity, pathophysiology, and clinical implications in neurodegenerative disorders. Surv. Ophthalmol. 2022, 67, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Eyolfson, E.; Carr, T.; Fraunberger, E.; Khan, A.; Clark, I.; Mychasiuk, R.; Lohman, A.W. Repeated mild traumatic brain injuries in mice cause age- and sex-specific alterations in dendritic spine density. Exp. Neurol. 2022, 357, 114172. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, J. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J. Neuropathol. Exp. Neurol. 2011, 70, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wu, H.; Zeng, J.; Pluimer, B.; Dong, S.; Xie, X.; Guo, X.; Ge, T.; Liang, X.; Feng, S.; et al. Mild traumatic brain injury induces microvascular injury and accelerates Alzheimer-like pathogenesis in mice. Acta Neuropathol. Commun. 2021, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Pilipovic, K.; Rajic Bumber, J.; Dolenec, P.; Grzeta, N.; Jankovic, T.; Kriz, J.; Zupan, G. Long-Term Effects of Repetitive Mild Traumatic Injury on the Visual System in Wild-Type and TDP-43 Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 6584. [Google Scholar] [CrossRef] [PubMed]
- Reitz, S.J.; Sauerbeck, A.D.; Kummer, T.T. SEQUIN: An imaging and analysis platform for quantification and characterization of synaptic structures in mouse. STAR Protoc. 2021, 2, 100268. [Google Scholar] [CrossRef]
- Sauerbeck, A.D.; Gangolli, M.; Reitz, S.J.; Salyards, M.H.; Kim, S.H.; Hemingway, C.; Gratuze, M.; Makkapati, T.; Kerschensteiner, M.; Holtzman, D.M.; et al. SEQUIN Multiscale Imaging of Mammalian Central Synapses Reveals Loss of Synaptic Connectivity Resulting from Diffuse Traumatic Brain Injury. Neuron 2020, 107, 257–273.e5. [Google Scholar] [CrossRef]
- Sullivan, G.M.; Mierzwa, A.J.; Kijpaisalratana, N.; Tang, H.; Wang, Y.; Song, S.K.; Selwyn, R.; Armstrong, R.C. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J. Neuropathol. Exp. Neurol. 2013, 72, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- DeWalt, G.J.; Mahajan, B.; Foster, A.R.; Thompson, L.D.E.; Marttini, A.A.; Schmidt, E.V.; Mansuri, S.; D’Souza, D.; Patel, S.B.; Tenenbaum, M.; et al. Region-specific alterations in astrocyte and microglia morphology following exposure to blasts in the mouse hippocampus. Neurosci. Lett. 2018, 664, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglial response to brain injury: A brief synopsis. Toxicol. Pathol. 2000, 28, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhao, L.; Wang, Y.; Guo, Q.; An, Q.; Geng, J.; Zhang, C.; Guo, Z. Ketamine Regulates the Autophagy Flux and Polarization of Microglia through the HMGB1-RAGE Axis and Exerts Antidepressant Effects in Mice. J. Neuropathol. Exp. Neurol. 2022, 81, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Shimazawa, M.; Inokuchi, Y.; Fukumitsu, H.; Furukawa, S.; Araie, M.; Hara, H. Degenerative alterations in the visual pathway after NMDA-induced retinal damage in mice. Brain Res. 2008, 1212, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Liang, H.F.; Barsamian, B.; Sun, S.W. Amyloid-beta induced retrograde axonal degeneration in a mouse tauopathy model. Neuroimage 2019, 189, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Weyand, T.G. The multifunctional lateral geniculate nucleus. Rev. Neurosci. 2016, 27, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Giarratana, A.O.; Zheng, C.; Reddi, S.; Teng, S.L.; Berger, D.; Adler, D.; Sullivan, P.; Thakker-Varia, S.; Alder, J. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with Bryostatin-1 improves outcomes. Sci. Rep. 2020, 10, 19919. [Google Scholar] [CrossRef] [PubMed]
- McLendon, L.A.; Kralik, S.F.; Grayson, P.A.; Golomb, M.R. The Controversial Second Impact Syndrome: A Review of the Literature. Pediatr. Neurol. 2016, 62, 9–17. [Google Scholar] [CrossRef]
- Marietta, M.P.; White, P.F.; Pudwill, C.R.; Way, W.L.; Trevor, A.J. Biodisposition of ketamine in the rat: Self-induction of metabolism. J. Pharmacol. Exp. Ther. 1976, 196, 536–544. [Google Scholar] [CrossRef]
- Radford, K.D.; Park, T.Y.; Lee, B.H.; Moran, S.; Osborne, L.A.; Choi, K.H. Dose-response characteristics of intravenous ketamine on dissociative stereotypy, locomotion, sensorimotor gating, and nociception in male Sprague-Dawley rats. Pharmacol. Biochem. Behav. 2017, 153, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Phoumthipphavong, V.; Barthas, F.; Hassett, S.; Kwan, A.C. Longitudinal Effects of Ketamine on Dendritic Architecture In Vivo in the Mouse Medial Frontal Cortex. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.W.; Holtmaat, A.; Wilbrecht, L.; Welker, E.; Svoboda, K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat. Neurosci. 2006, 9, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Thelen, C.; Flaherty, E.; Saurine, J.; Sens, J.; Mohamed, S.; Pitychoutis, P.M. Sex Differences in the Temporal Neuromolecular and Synaptogenic Effects of the Rapid-acting Antidepressant Drug Ketamine in the Mouse Brain. Neuroscience 2019, 398, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Radford, K.D.; Berman, R.Y.; Zhang, M.; Wu, T.J.; Choi, K.H. Sex-related differences in intravenous ketamine effects on dissociative stereotypy and antinociception in male and female rats. Pharmacol. Biochem. Behav. 2020, 199, 173042. [Google Scholar] [CrossRef]
- Carrier, N.; Kabbaj, M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology 2013, 70, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Kabbaj, M. Sex Differences in Effects of Ketamine on Behavior, Spine Density, and Synaptic Proteins in Socially Isolated Rats. Biol. Psychiatry 2016, 80, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Tebianian, M.; Tofigh, N.; Taheri, R.S.; Mousavi, S.A.; Naseri, A.; Ahmadi, A.; Munawar, N.; Shahpasand, K. Active immunotherapy against pathogenic Cis pT231-tau suppresses neurodegeneration in traumatic brain injury mouse models. Neuropeptides 2022, 96, 102285. [Google Scholar] [CrossRef] [PubMed]
- Korotzer, A.R.; Pike, C.J.; Cotman, C.W. beta-Amyloid peptides induce degeneration of cultured rat microglia. Brain Res. 1993, 624, 121–125. [Google Scholar] [CrossRef]
- Toshimitsu, M.; Kamei, Y.; Ichinose, M.; Seyama, T.; Imada, S.; Iriyama, T.; Fujii, T. Atomoxetine, a selective norepinephrine reuptake inhibitor, improves short-term histological outcomes after hypoxic-ischemic brain injury in the neonatal male rat. Int. J. Dev. Neurosci. 2018, 70, 34–45. [Google Scholar] [CrossRef]
- Lin, C.T.; Lecca, D.; Yang, L.Y.; Luo, W.; Scerba, M.T.; Tweedie, D.; Huang, P.S.; Jung, Y.J.; Kim, D.S.; Yang, C.H.; et al. 3,6′-dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. eLife 2020, 9, e54726. [Google Scholar] [CrossRef] [PubMed]
- Doorn, K.J.; Goudriaan, A.; Blits-Huizinga, C.; Bol, J.G.; Rozemuller, A.J.; Hoogland, P.V.; Lucassen, P.J.; Drukarch, B.; van de Berg, W.D.; van Dam, A.M. Increased amoeboid microglial density in the olfactory bulb of Parkinson’s and Alzheimer’s patients. Brain Pathol. 2014, 24, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, A.E.; Salm, A.K. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J. Neurosci. 2003, 23, 7759–7766. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C.R. The Rat Brain in Stereotaxic Coordinates, 5th ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2005. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boese, M.; Berman, R.Y.; Qiu, J.; Spencer, H.F.; Radford, K.D.; Choi, K.H. Effects of Mild Closed-Head Injury and Subanesthetic Ketamine Infusion on Microglia, Axonal Injury, and Synaptic Density in Sprague–Dawley Rats. Int. J. Mol. Sci. 2024, 25, 4287. https://doi.org/10.3390/ijms25084287
Boese M, Berman RY, Qiu J, Spencer HF, Radford KD, Choi KH. Effects of Mild Closed-Head Injury and Subanesthetic Ketamine Infusion on Microglia, Axonal Injury, and Synaptic Density in Sprague–Dawley Rats. International Journal of Molecular Sciences. 2024; 25(8):4287. https://doi.org/10.3390/ijms25084287
Chicago/Turabian StyleBoese, Martin, Rina Y. Berman, Jennifer Qiu, Haley F. Spencer, Kennett D. Radford, and Kwang H. Choi. 2024. "Effects of Mild Closed-Head Injury and Subanesthetic Ketamine Infusion on Microglia, Axonal Injury, and Synaptic Density in Sprague–Dawley Rats" International Journal of Molecular Sciences 25, no. 8: 4287. https://doi.org/10.3390/ijms25084287
APA StyleBoese, M., Berman, R. Y., Qiu, J., Spencer, H. F., Radford, K. D., & Choi, K. H. (2024). Effects of Mild Closed-Head Injury and Subanesthetic Ketamine Infusion on Microglia, Axonal Injury, and Synaptic Density in Sprague–Dawley Rats. International Journal of Molecular Sciences, 25(8), 4287. https://doi.org/10.3390/ijms25084287