

Chiral Thianthrenes



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
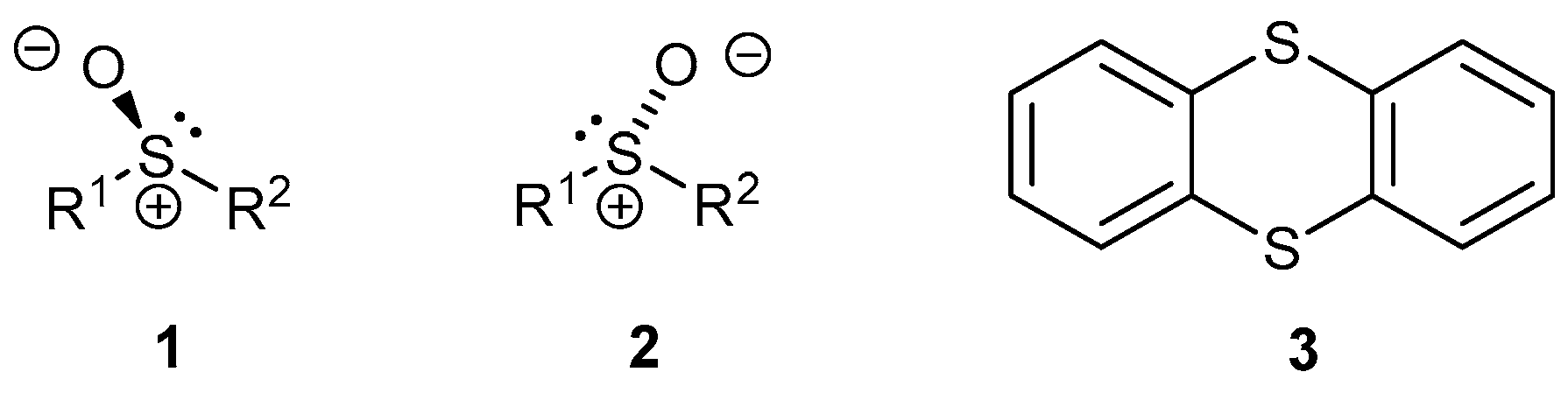
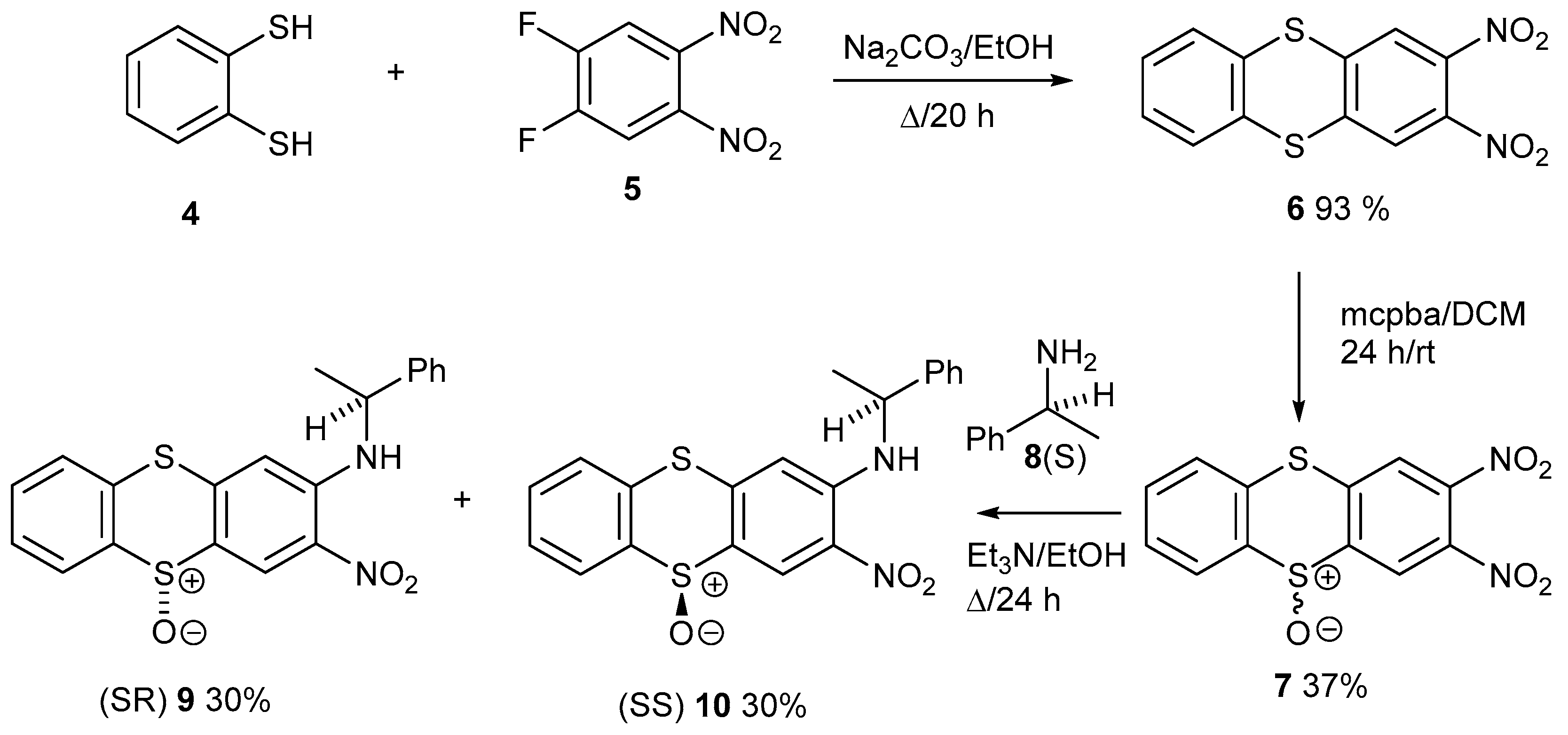
1. Introduction
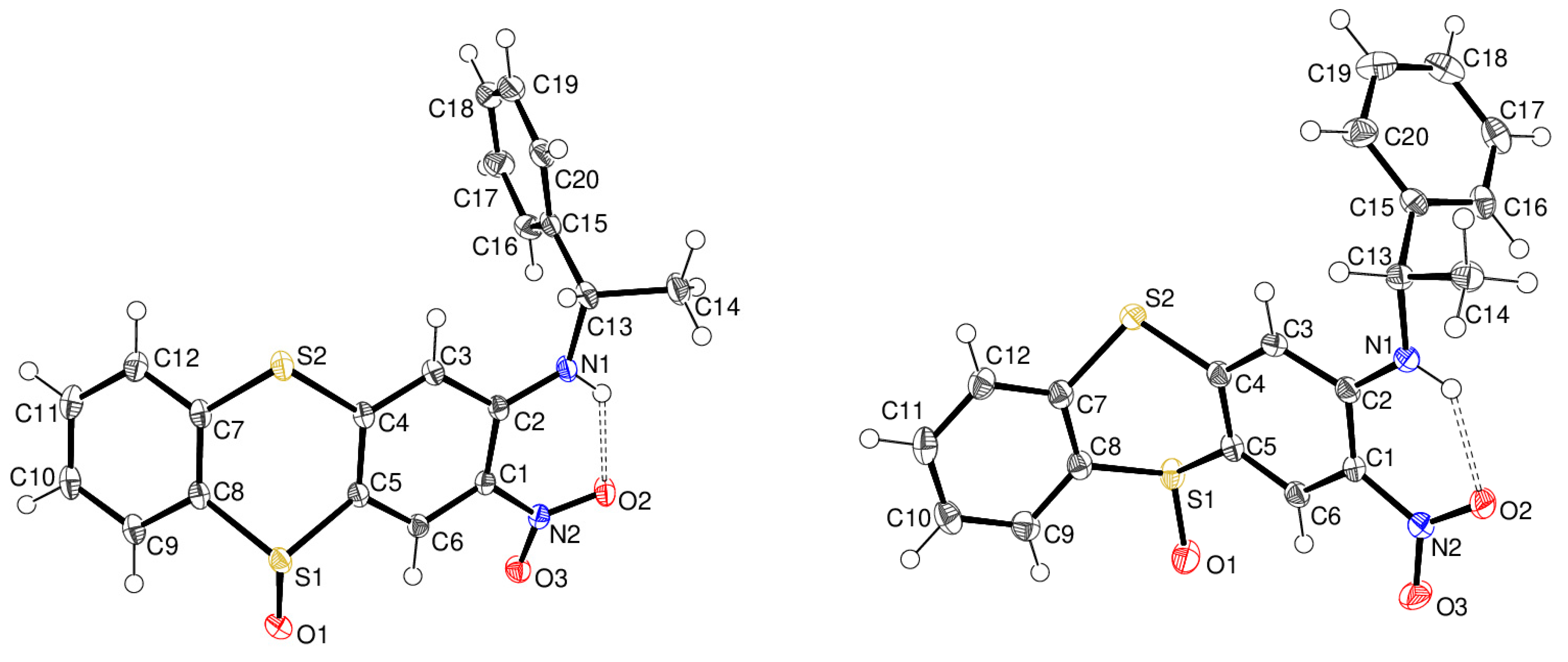
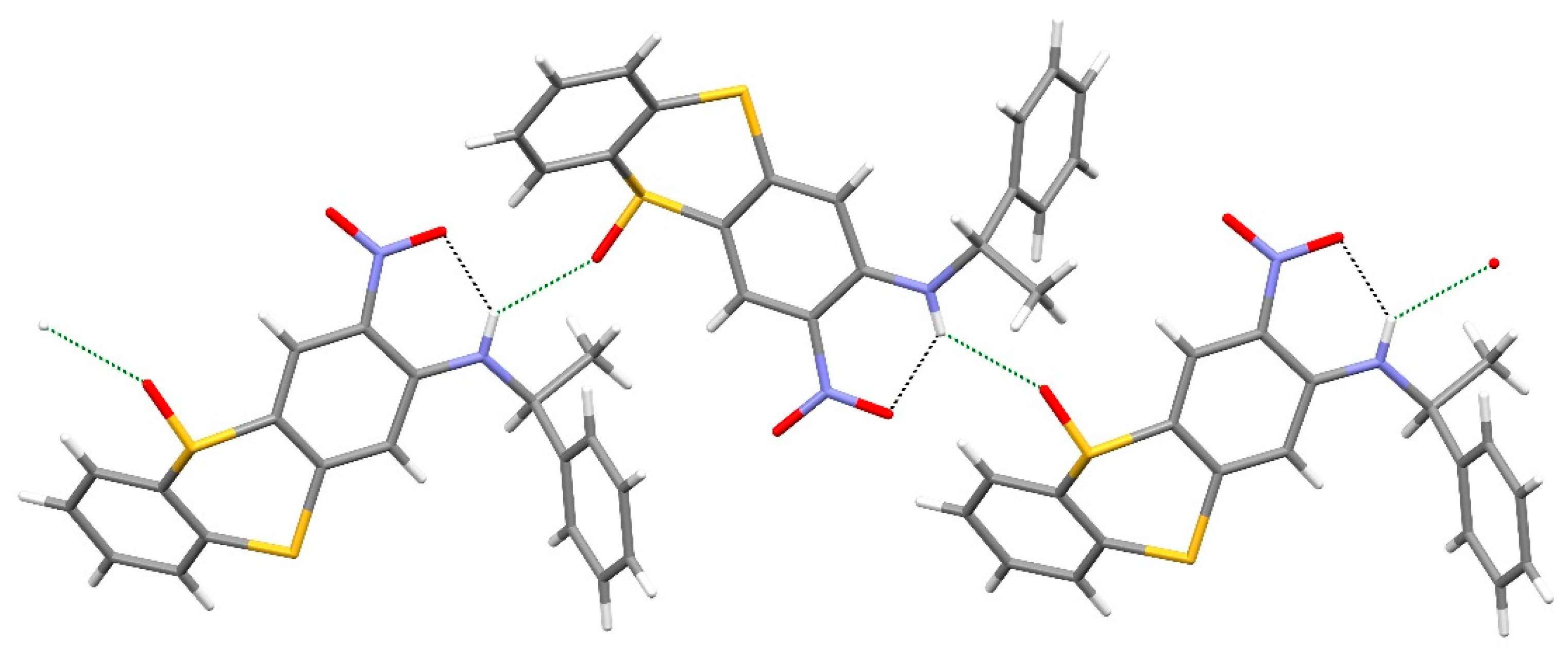
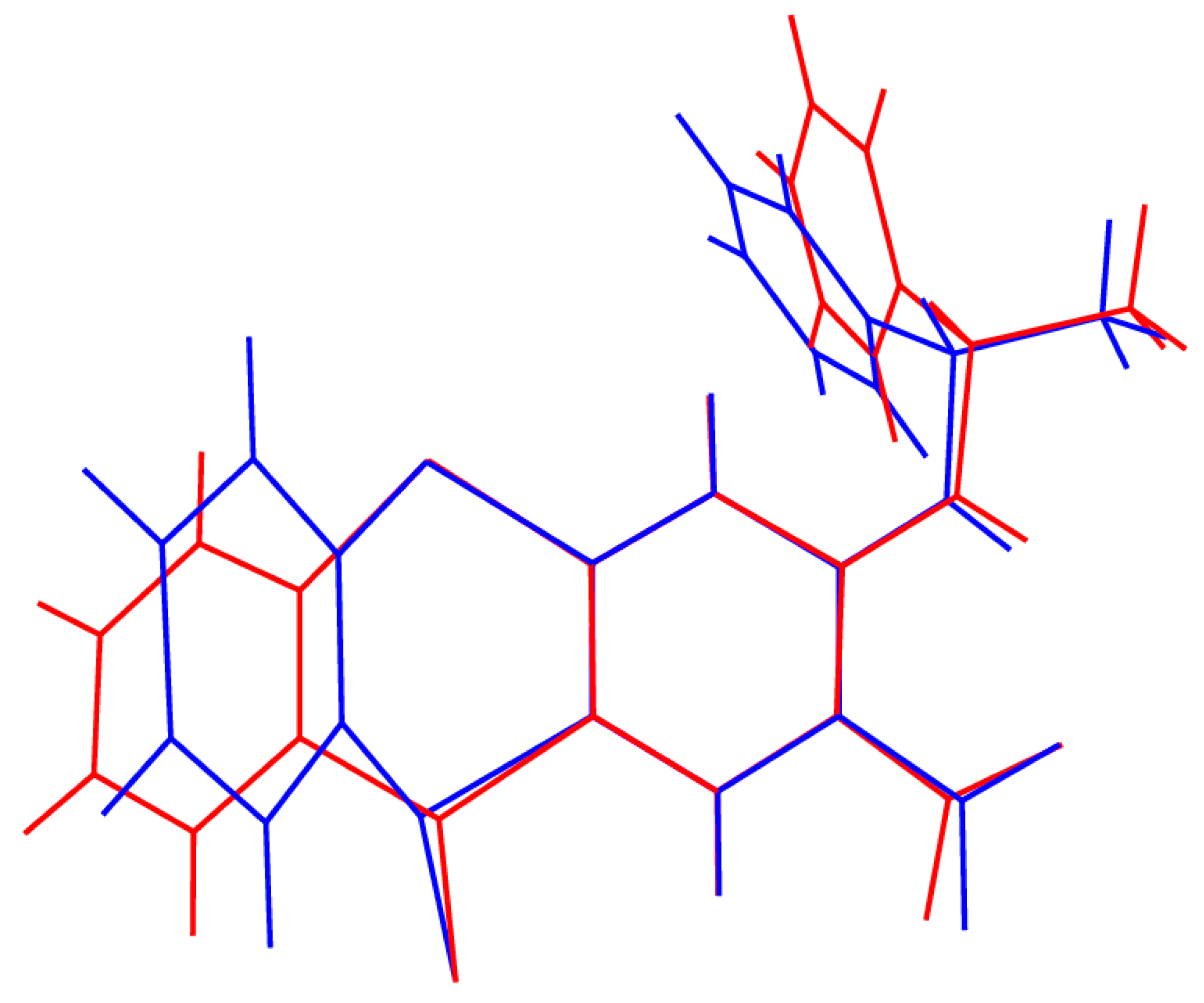
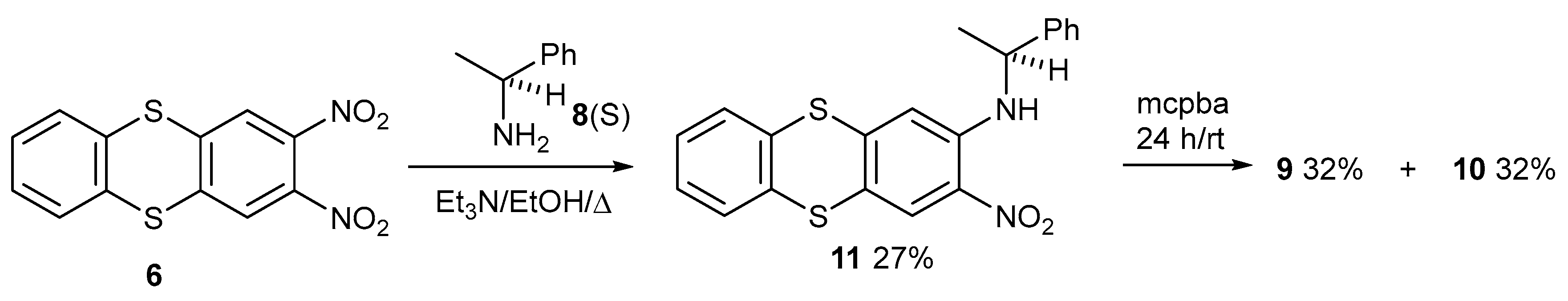
2. Results and Discussion
3. Materials and Methods
Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pellissier, H. Use of chiral sulfoxides in asymmetric synthesis. Tetrahedron 2006, 62, 5559–5601. [Google Scholar] [CrossRef]
- Fernandez, I.; Khiar, N. Recent Developments in the Synthesis and Utilization of Chiral Sulfoxides. Chem. Rev. 2003, 103, 3651–3705. [Google Scholar] [CrossRef] [PubMed]
- Carreno, M.C. Applications of Sulfoxides to Asymmetric Synthesis of Biologically Active Compounds. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar] [CrossRef]
- Harrison, P.W.B.; Kenyon, J.; Phillips, H. CCLXX1X—The Dependence of Rotatory Power on Chemical Constitution. Part XXIX. The Resolution of Sulphoxides into their Optically Active Forms. J. Chem. Soc. 1926, 129, 2079–2090. [Google Scholar] [CrossRef]
- Gilman, H.; Robinson, J.; Beaber, N.J. The reaction between organomagnesium halides and the esters of some sulfur acids. J. Am. Chem. Soc. 1926, 48, 2715–2718. [Google Scholar] [CrossRef]
- Andersen, K.K.; Gaffield, W.; Papanikolaou, N.E.; Foley, J.; Perkins, R.I. Optically Active Sulfoxides. The Synthesis and Rotatory Dispersion of Some Diaryl Sulfoxides. J. Am. Chem. Soc. 1964, 86, 5637–5646. [Google Scholar] [CrossRef]
- Andersen, K.K. Configurational Relationships among Some Sulfoxides. J. Org. Chem. 1964, 29, 1953–1956. [Google Scholar] [CrossRef]
- Andersen, K.K. Synthesis of (+)ethyl-e-tolyl sulfoxide from (-)menthyl(-)-e-toluenesulfinate. Tetrahedron Lett. 1962, 3, 93–95. [Google Scholar] [CrossRef]
- Mellah, M.; Voituriez, A.; Schulz, E. Chiral Sulfur Ligands for Asymmetric Catalysis. Chem. Rev. 2007, 107, 5133–5209. [Google Scholar] [CrossRef]
- Mei, H.; Xie, C.; Han, J.; Soloshonok, V.A. N-tert-Butylsulfinyl-3,3,3-trifluoroacetaldimine: Versatile Reagent for Asymmetric Synthesis of Trifluoromethyl-Containing Amines and Amino Acids of Pharmaceutical Importance. Eur. J. Org. Chem. 2016, 2016, 5917–5932. [Google Scholar] [CrossRef]
- Pellissier, H. Chiral sulfur-containing ligands for asymmetric catalysis. Tetrahedron 2007, 63, 1297–1330. [Google Scholar] [CrossRef]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef] [PubMed]
- Agranat, I.; Caner, H. Intellectual property and chirality of drugs. Drug Discov. Today 1999, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R. Role of sulfur chirality in the chemical processes of biology. Chem. Soc. Rev. 2005, 34, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Kagan, H.B. Asymmetric Synthesis of Chiral Sulfoxides. In Organosulfur Chemistry in Asymmetric Synthesis; Toru, T., Bolm, C., Eds.; Wiley Online Library: New York, NY, USA, 2008; pp. 1–29. [Google Scholar]
- O’Mahony, G.E.; Kelly, P.; Lawrence, S.E.; Maguire, A.R. Synthesis of enantioenriched sulfoxides. ARKIVOC 2011, 1, 1–110. [Google Scholar] [CrossRef]
- Otocka, S.; Kwiatkowska, M.; Madalinska, L.; Kiełbasinski, P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef]
- O’Mahony, G.E.; Ford, A.; Maguire, A.R. Asymmetric oxidation of sulfides. J. Sulfur Chem. 2013, 34, 301–341. [Google Scholar] [CrossRef]
- Wojaczynska, E.; Wojaczynski, J. Enantioselective Synthesis of Sulfoxides: 2000−2009. Chem. Rev. 2010, 110, 4303–4356. [Google Scholar] [CrossRef]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Klika, K.D. Terminology related to the phenomenon ‘self-disproportionation of enantiomers’ (SDE). Helv. Chim. Acta. 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Diter, P.; Taudien, S.; Samuel, O.; Kagan, H.B. Enantiomeric Enrichment of Sulfoxides by Preparative Flash Chromatography on an Achiral Phase. J. Org. Chem. 1994, 59, 370–373. [Google Scholar] [CrossRef]
- Girard, C.; Kagan, H.B. On diastereomeric perturbations. Can. J. Chem. 2000, 78, 816–828. [Google Scholar] [CrossRef]
- Kagan, H.B.; Riant, O. Advances in Asymmetric Synthesis; Elsevier B.V.: Amsterdam, The Netherlands, 1997; Volume 2, pp. 189–235. [Google Scholar]
- Brunel, J.-M.; Kagan, H.B. Catalytic enantioselective oxidation of sulfides with a chiral titanium complex. Bull. Soc. Chim. Fr. 1996, 133, 1109–1115. [Google Scholar]
- Girard, C.; Kagan, H.B. Nonlinear Effects in Asymmetric Synthesis and Stereoselective Reactions: Ten Years of Investigation. Angew. Chem. Int. Ed. 1998, 37, 2923–2959. [Google Scholar] [CrossRef]
- Brunel, J.-M.; Luukas, T.O.; Kagan, H.B. Nonlinear effects as ‘indicators’ in the tuning of asymmetric catalysts. Tetrahedron Asymmetry 1998, 9, 1941–1946. [Google Scholar] [CrossRef]
- Fenwick, D.R.; Kagan, H.B. Top. Stereochem; Denmark, S.E., Ed.; Wiley: Hoboken, NJ, USA, 1999; Volume 22, pp. 257–296. [Google Scholar]
- Fujita, T.; Kamiyama, H.; Osawa, Y.; Kawaguchi, H.; Kim, B.J.; Tatami, A.; Kawashima, W.; Maeda, T.; Nakanishi, A.; Morita, H. Photo SN-bond cleavage and related reactions of thianthrene sulfilimine derivatives. Tetrahedron 2007, 63, 7708–7716. [Google Scholar] [CrossRef]
- Fries, V.; Volk, W. Uber Thianthrene. Chem. Ber. 1909, 42, 1170–1176. [Google Scholar] [CrossRef]
- Naomichi, F.; Takeshi, K.; Yoji, H.; Satoshi, O. A convenient preparation of sterically crowded 1,9-disubstituted dibenzothiophenes and 3,3′-disubstituted diaryl sulfides. Heterocycles 1991, 32, 675–678. [Google Scholar]
- Bosch, E.; Kochi, J.K. Catalytic oxidation of chlorpromazine and related phenothiazines. Cation radicals as the reactive intermediates in sulfoxide formation. J. Chem. Soc. Perkin Trans. I 1995, 8, 1057–1064. [Google Scholar] [CrossRef]
- Mata, E.G. Recent advances in the synthesis of sulfoxides from sulfides. Phosphorus Sulfur Silicon Relat. Elem. 1996, 117, 231–286. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishikawa, H.; Uchida, T.; Katsuki, T. Photopromoted Ru-Catalyzed Asymmetric Aerobic Sulfide Oxidation and Epoxidation Using Water as a Proton Transfer Mediator. J. Am. Chem. Soc. 2010, 132, 12034–12041. [Google Scholar] [CrossRef] [PubMed]
- Sprout, C.M.; Seto, C.T. Using Enzyme Inhibition as a High Throughput Method to Measure the Enantiomeric Excess of a Chiral Sulfoxide. Org. Lett. 2005, 7, 5099–5102. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, C.; Dai, Z.; Yang, M.; Pan, Y.; Hu, H. Efficient Asymmetric Oxidation of Sulfides and Kinetic Resolution of Sulfoxides Catalyzed by a Vanadium−Salan System. J. Org. Chem. 2004, 69, 8500–8503. [Google Scholar] [CrossRef] [PubMed]
- Legros, J.; Bolm, C. Investigations on the Iron-Catalyzed Asymmetric Sulfide Oxidation. Chem.–Eur. J. 2005, 11, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.-Q.; Shine, H.J.; Guzman-Jimenez, I.Y.; Thurston, J.H.; Whitmire, K.H. Mono- and Bis-adducts from the Addition of Thianthrene Cation Radical Salts to Cycloalkenes and Alkenes. J. Org. Chem. 2002, 67, 4030–4039. [Google Scholar] [CrossRef]
- Holst, D.E.; Dorval, C.; Winter, C.K.; Guzei, I.A.; Wickens, Z.K. Regiospecific Alkene Aminofunctionalisation via an Electrogenerated Dielectrophile. J. Am. Chem. Soc. 2023, 145, 8299–8307. [Google Scholar]
- Plater, M.J.; Harrison, W.T.A. A potential iterative approach to 1,4-dihydro-N-heteroacene arrays. ChemistryOpen 2022, 11, e202100150. [Google Scholar] [CrossRef]
- Plater, M.J.; Harrison, W.T.A. Potential building blocks for 1,4-dihydro-N-heteroacenes. ChemistryOpen 2022, 11, e202200092. [Google Scholar] [CrossRef]
- Plater, M.J.; Harrison, W.T.A. New funtionalised phenoxazines and phenothiazines. ACS Omega 2023, 8, 44163–44171. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.K. Specification of Configuration about Quadricovalent Asymmetric Atoms. J. Chem. Soc. 1951, 612–622. [Google Scholar]
- Cahn, R.S.; Ingold, C.K.; Prelog, V. The Specification of Asymmetric Configuration in Organic Chemistry. Experientia 1956, 15, 81–94. [Google Scholar] [CrossRef]
- Clegg, W. X-Ray Crystallography, 2nd ed.; Oxford University Press (Oxford Chemistry Primer): Oxford, UK, 2015. [Google Scholar]
- Eccles, K.S.; Morrison, R.E.; Stokes, S.P.; O’Mahony, G.E.; Hayes, J.A.; Kelly, D.M.; O’Boyle, N.M.; Fabian, L.; Moynihan, H.A.; Maguire, A.R.; et al. Utilizing Sulfoxide Iodine Halogen Bonding for Cocrystallization. Cryst. Growth Des. 2012, 12, 2969–2977. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Gans, J.S.; Shalloway, D. Qmol: A program for molecular visualization on Windows-based PCs. J. Mol. Graph. Model. 2001, 19, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plater, M.J.; Harrison, W.T.A. Chiral Thianthrenes. Int. J. Mol. Sci. 2024, 25, 4311. https://doi.org/10.3390/ijms25084311
Plater MJ, Harrison WTA. Chiral Thianthrenes. International Journal of Molecular Sciences. 2024; 25(8):4311. https://doi.org/10.3390/ijms25084311
Chicago/Turabian StylePlater, M. John, and William T. A. Harrison. 2024. "Chiral Thianthrenes" International Journal of Molecular Sciences 25, no. 8: 4311. https://doi.org/10.3390/ijms25084311
APA StylePlater, M. J., & Harrison, W. T. A. (2024). Chiral Thianthrenes. International Journal of Molecular Sciences, 25(8), 4311. https://doi.org/10.3390/ijms25084311