
Discovery of Strong 3-Nitro-2-Phenyl-2H-Chromene Analogues as Antitrypanosomal Agents and Inhibitors of Trypanosoma cruzi Glucokinase

 , and
, and 
Abstract
1. Introduction
2. Results and Discussion
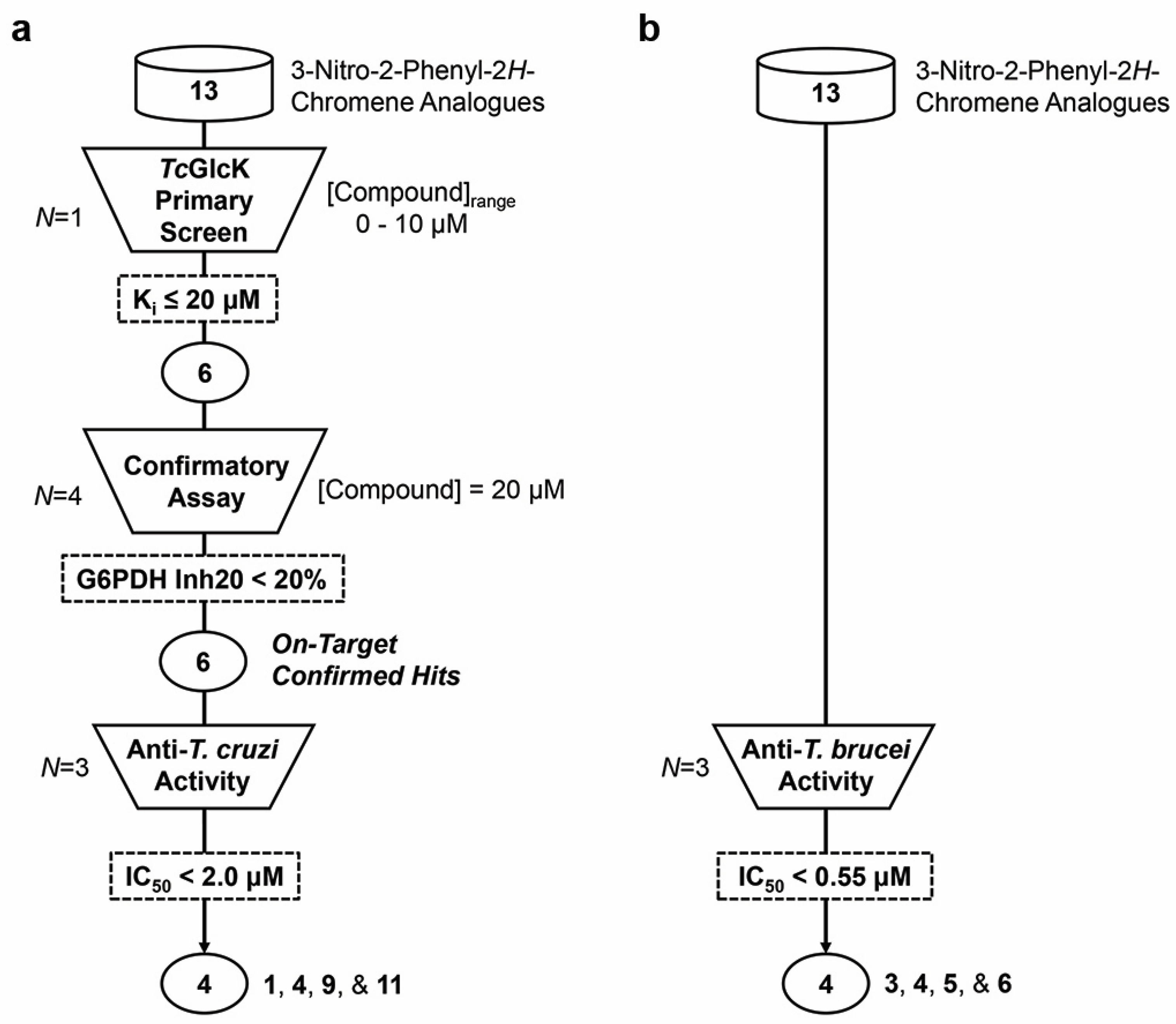
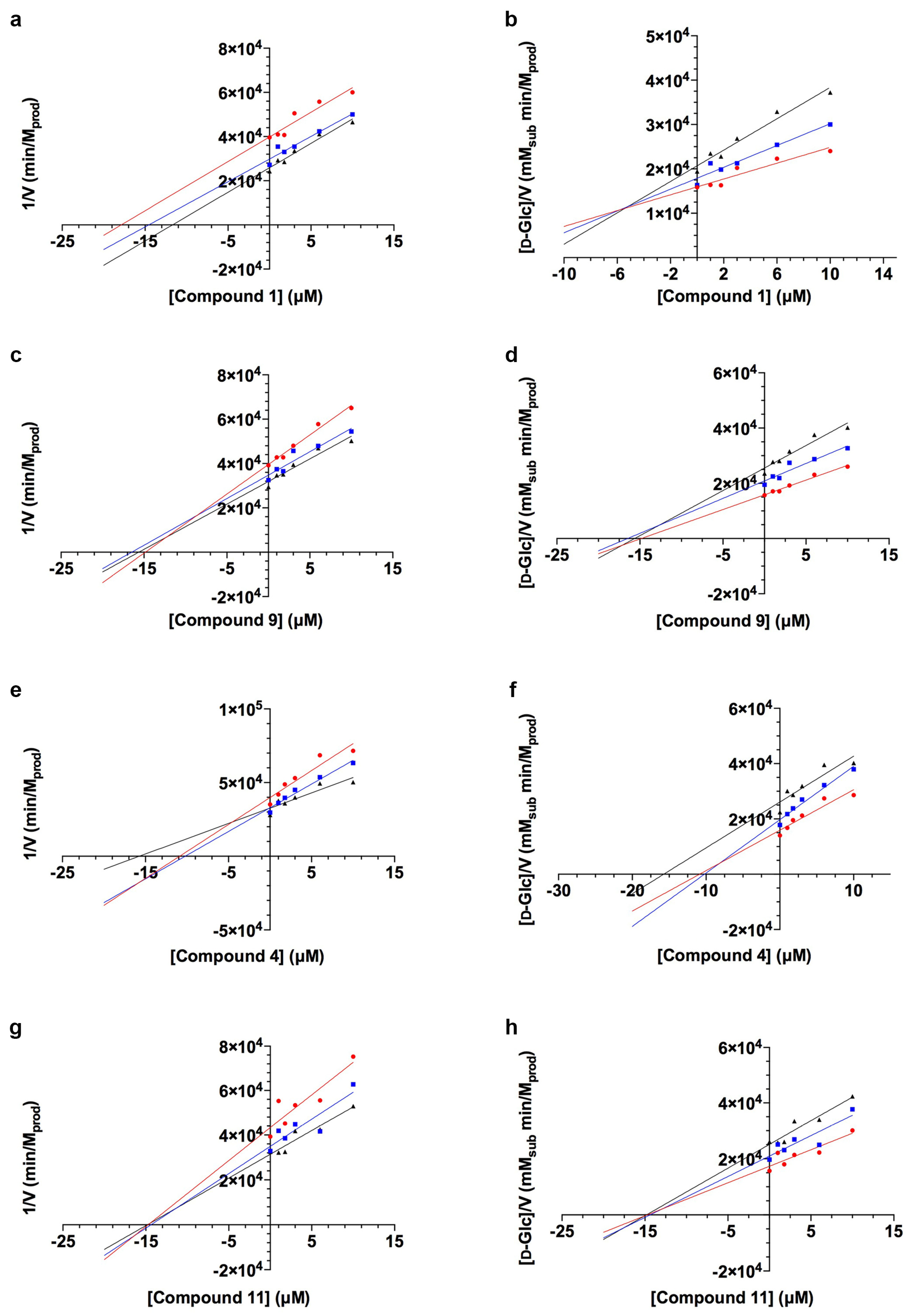
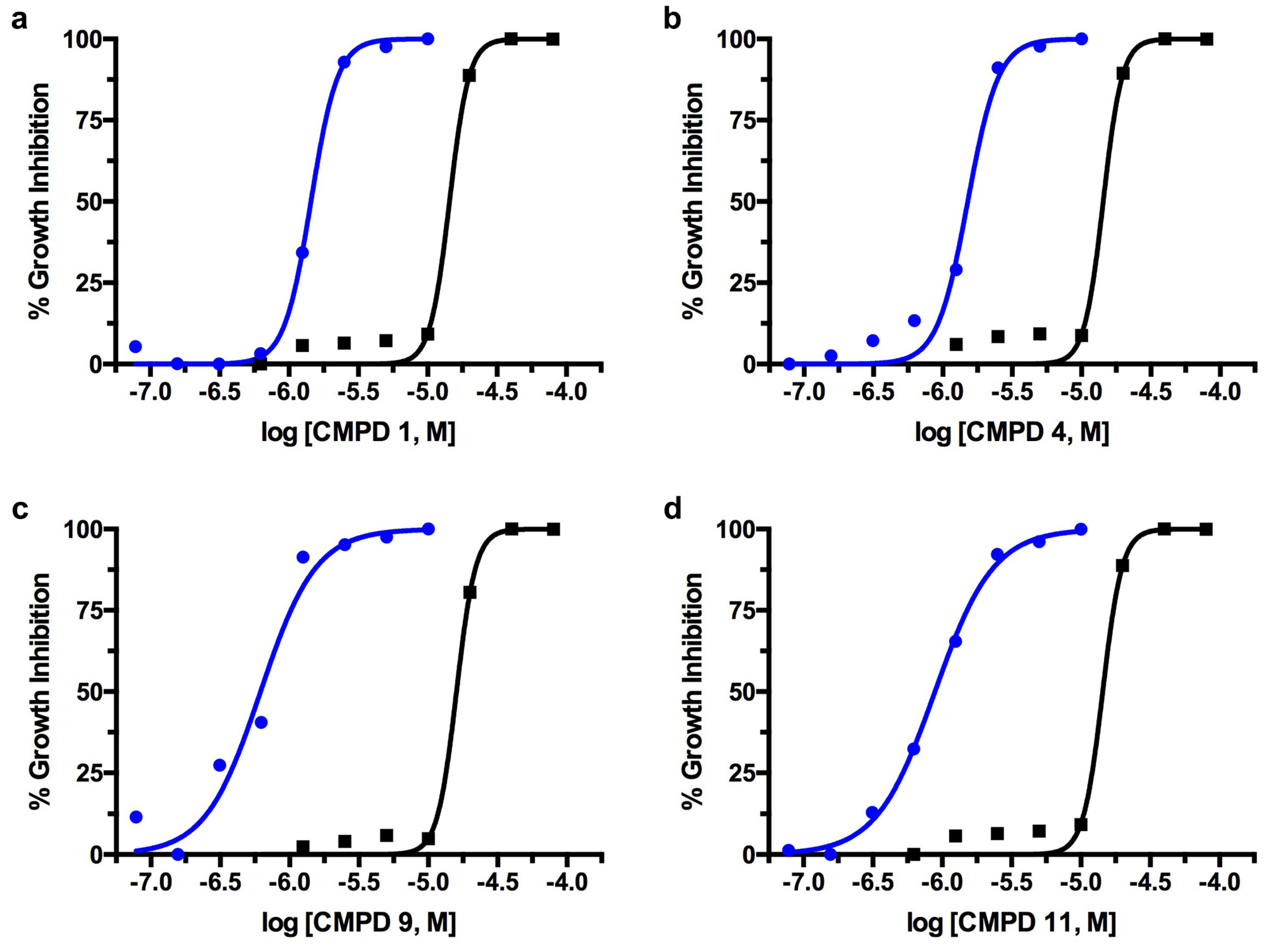
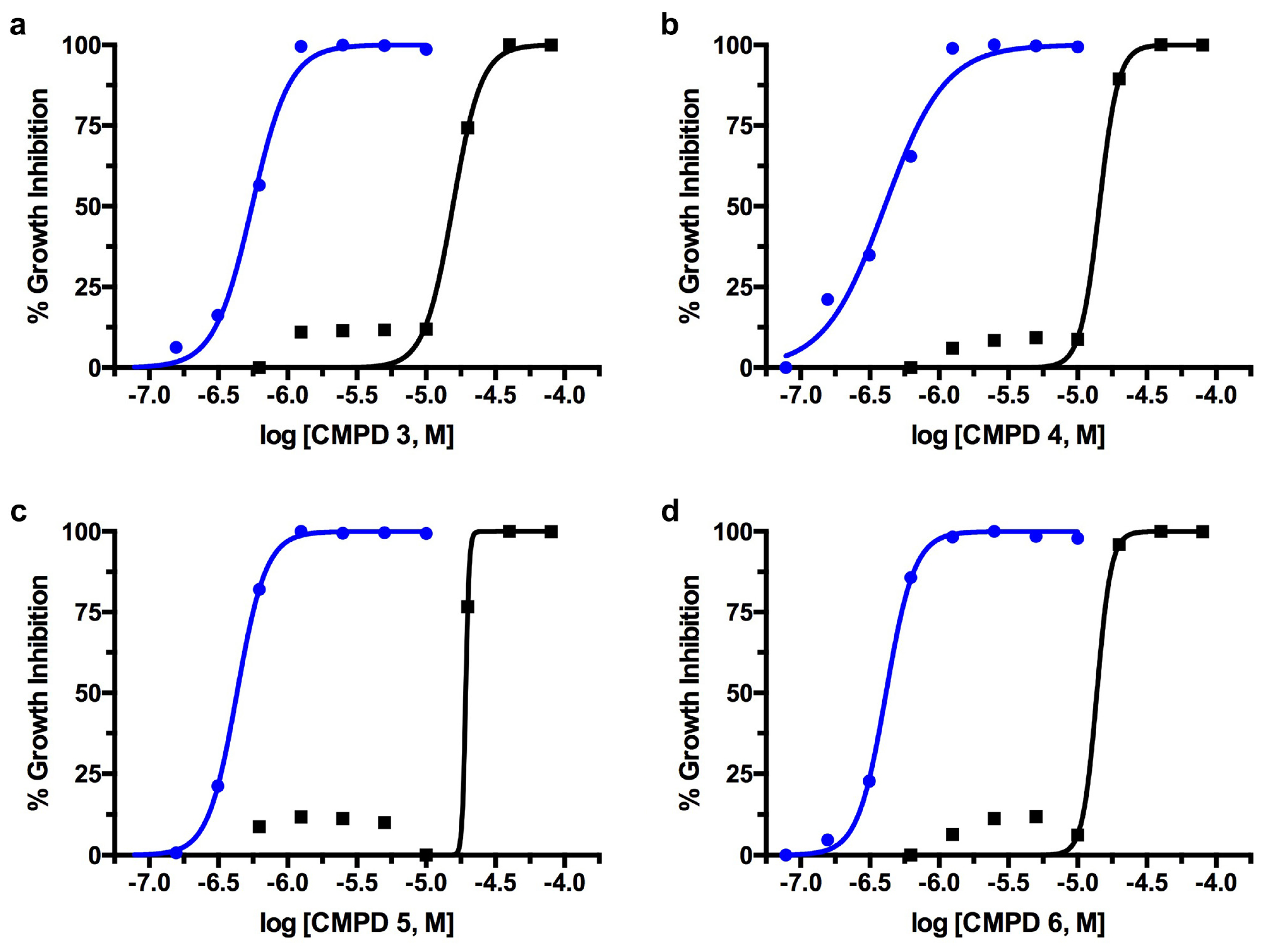
2.1. TcGlcK Primary Screen, Confirmatory Assay, and T. cruzi Biological Assay
2.2. T. brucei Biological Assay
2.3. Mammalian Cytotoxicity Assay
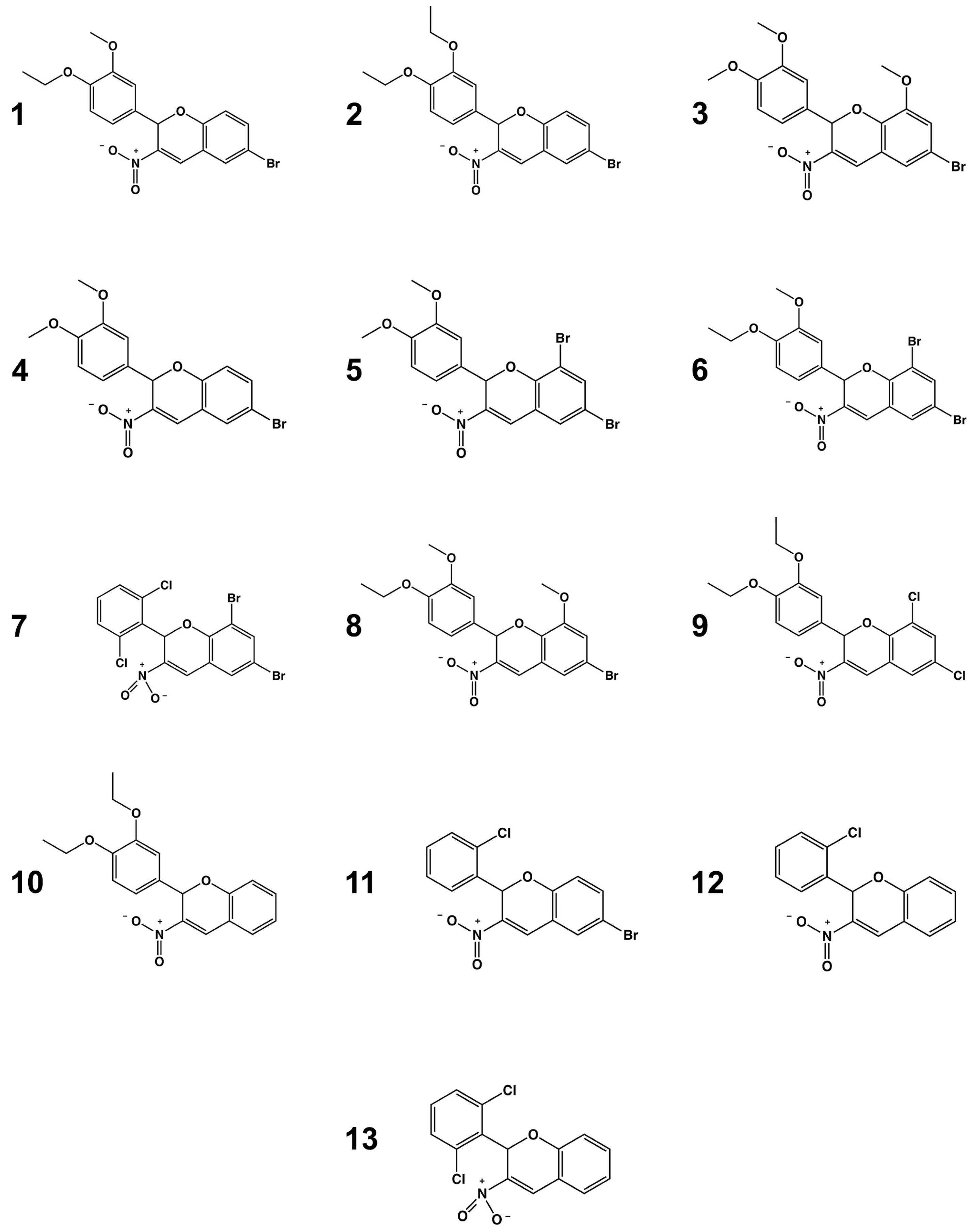
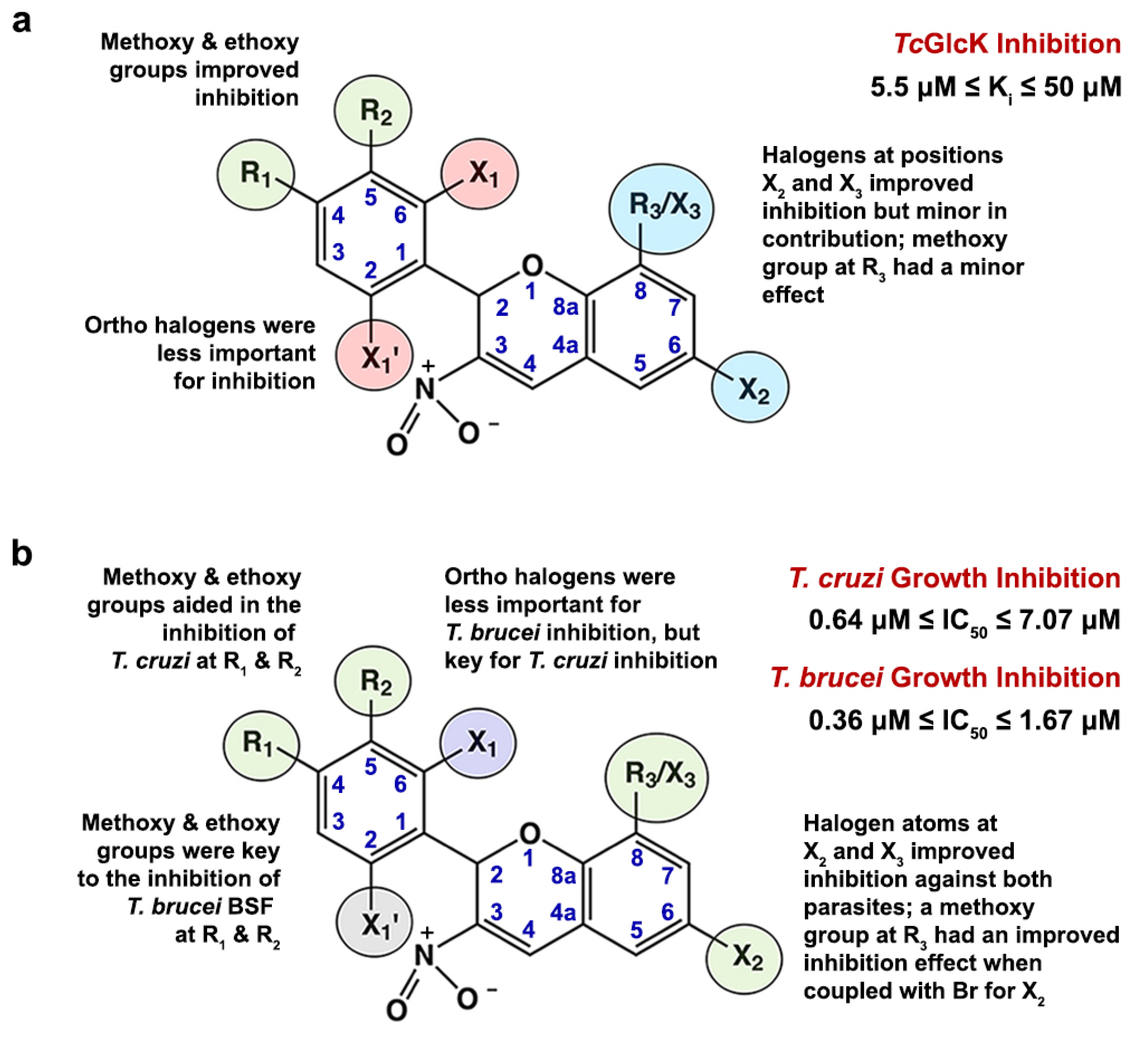
2.4. Structure–Activity Relationship Analysis
3. Materials and Methods
3.1. Chemicals
3.2. Recombinant Overexpression and Purification of TcGlcK
3.3. Primary Screening Assay of TcGlcK and 3-Nitro-2-Phenyl-2H-Chromene Analogues
3.4. Confirmatory Assay
3.5. In Vitro T. cruzi Biological Assay
3.6. In Vitro T. brucei Biological Assay
3.7. In Vitro Biological Assay for the Cytotoxicity of Mammalian NIH-3T3 Fibroblasts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kjos, S.A.; Snowden, K.F.; Olson, J.K. Biogeography and Trypanosoma cruzi infection prevalence of Chagas disease vectors in Texas, USA. Vector-Borne Zoonotic Dis. 2009, 9, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization, Media Centre Fact Sheets Homepage. Updated 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 10 March 2024).
- Coura, J.R.; Borges-Pereira, J. Chagas disease: 100 years after its discovery. A systematic review. Acta Trop. 2010, 115, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention, Parasites Homepage. Updated February 2019. Available online: http://www.cdc.gov/parasites/chagas/ (accessed on 10 March 2024).
- Bern, C. Chagas’ disease. N. Engl. J. Med. 2015, 373, 456–466. [Google Scholar] [CrossRef]
- Wang, T.; Wang, L.; He, J.; Chang, L.; Shi, J. Recent research progress on small molecule compounds and its derivatives of antiparasitic drugs. Chin. Chem. Lett. 2023, 34, 108359. [Google Scholar] [CrossRef]
- Rassi, A.J.; Dias, J.C.P.; Marin-Neto, J.A.; Rassi, A. Challenges and opportunities for primary, secondary, and tertiary prevention of Chagas’ disease. Heart 2009, 95, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Docampo, R. Specific chemotherapy of Chagas disease: Controversies and advances. Trends Parasitol. 2003, 19, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, C.; García-Vázquez, E.; Carrilero, B.; Simón, M.; Franco, F.; Iborra, M.A.; Gil-Gallardo, L.J.; Segovia, M. Tolerance and adherence of patients with chronic Chagas disease treated with benznidazole. Rev. Soc. Bras. Med. Trop. 2023, 56, e0384-2022. [Google Scholar] [CrossRef]
- Castro, J.A.; Montalto de Mecca, M.; Bartel, L.C. Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef]
- Sánchez-Valdéz, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R.L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. eLife 2018, 7, e34039. [Google Scholar] [CrossRef]
- Duschak, V.G.; Couto, A.S. An insight on targets and patented drugs for chemotherapy of Chagas disease. Recent Pat. Anti-Infect. Drug Discov. 2007, 2, 19–51. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Cáceres, A.J.; Portillo, R.; Acosta, H.; Rosales, D.; Quiñones, W.; Avilán, L.; Salazar, L.; Dubourdieu, M.; Michels, P.A.M.; Concepción, J.L. Molecular and biochemical characterization of hexokinase from Trypanosoma cruzi. Mol. Biochem. Parasitol. 2003, 126, 251–262. [Google Scholar] [CrossRef]
- Cáceres, A.J.; Quiñones, W.; Gualdrón, M.; Cordeiro, A.; Avilán, L.; Michels, P.A.M.; Concepción, J.L. Molecular and biochemical characterization of novel glucokinases from Trypanosoma cruzi and Leishmania spp. Mol. Biochem. Parasitol. 2007, 156, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Mercaldi, G.F.; D’Antonio, E.L.; Aguessi, A.; Rodriguez, A.; Cordeiro, A.T. Discovery of antichagasic inhibitors by high-throughput screening with Trypanosoma cruzi glucokinase. Bioorg. Med. Chem. Lett. 2019, 29, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Acosta, H.; Burchmore, R.; Naula, C.; Gualdrón-López, M.; Quintero-Troconis, E.; Cáceres, A.J.; Michels, P.A.M.; Concepción, J.L.; Quiñones, W. Proteomic analysis of glycosomes from Trypanosoma cruzi epimastigotes. Mol. Biochem. Parasitol. 2019, 229, 62–74. [Google Scholar] [CrossRef]
- Jäger, T.; Koch, O.; Flohé, L. (Eds.) Trypanosomatid Diseases: Molecular Routes to Drug Discovery; Wiley-Blackwell: Weinheim, Germany, 2013; Volume 4, ISBN 978-3-527-33255-7. [Google Scholar] [CrossRef]
- Mercaldi, G.F.; Dawson, A.; Hunter, W.N.; Cordeiro, A.T. The structure of a Trypanosoma cruzi glucose-6-phosphate dehydrogenase reveals differences from the mammalian enzyme. FEBS Lett. 2016, 590, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P. The pentose phosphate pathway and parasitic protozoa. Parasitol. Today 1997, 13, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, D.A.; Cazzulo, J.J. The pentose phosphate pathway in Trypanosoma cruzi. FEMS Microbiol. Lett. 2004, 234, 117–123. [Google Scholar] [CrossRef]
- Shaw-Simpson, S.; Lentini, G.; Dumoulin, P.C.; Burleigh, B.A. Modulation of host central carbon metabolism and in situ glucose uptake by intracellular Trypanosoma cruzi amastigotes. PLoS Pathog. 2017, 13, e1006747. [Google Scholar] [CrossRef]
- Bringaud, F.; Rivière, L.; Coustou, V. Energy metabolism of trypanosomatids: Adaptation to available carbon sources. Mol. Biochem. Parasitol. 2006, 149, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.M.; Kearns, S.P.; Millington, M.E.; Buechner, G.S.; Alvarez, B.E.J.; Daneshian, L.; Abiskaroon, B.; Chruszcz, M.; D’Antonio, E.L. At the outer part of the active site in Trypanosoma cruzi glucokinase: The role of phenylalanine 337. Biochimie 2024, 218, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Green, S.B.; Lanier, R.J., Jr.; Carey, S.M.; Morgan, D.R.; Gracz, H.; Sherman, J.; Rodriguez, A.; D’Antonio, E.L. Synthesis, biochemical, and biological evaluation of C2 linkage derivatives of amino sugars, inhibitors of glucokinase from Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 2021, 47, 128227. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, E.L.; Deinema, M.S.; Kearns, S.P.; Frey, T.A.; Tanghe, S.; Perry, K.; Roy, T.A.; Gracz, H.S.; Rodriguez, A.; D’Antonio, J. Structure-based approach to the identification of a novel group of selective glucosamine analogue inhibitors of Trypanosoma cruzi glucokinase. Mol. Biochem. Parasitol. 2015, 204, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Gabaldón-Figueira, J.C.; Martinez-Peinado, N.; Escabia, E.; Ros-Lucas, A.; Chatelain, E.; Scandale, I.; Gascon, J.; Pinazo, M.J.; Alonso-Padilla, J. State-of-the-art in the drug discovery pathway for Chagas disease: A framework for drug development and target validation. Res. Rep. Trop. Med. 2023, 14, 1–19. [Google Scholar] [CrossRef]
- Chatelain, E. Chagas disease drug discovery: Toward a new era. J. Biomol. Screening 2015, 20, 22–35. [Google Scholar] [CrossRef]
- Omolabi, K.F.; Odeniran, P.O.; Olotu, F.A.; Soliman, M.E.S. A mechanistic probe into the dual inhibition of T. cruzi glucokinase and hexokinase in Chagas disease treatment—A stone killing two birds? Chem. Biodivers. 2021, 18, e2000863. [Google Scholar] [CrossRef] [PubMed]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef]
- Cordeiro, A.T.; Cáceres, A.J.; Vertommen, D.; Concepción, J.L.; Michels, P.A.M.; Versées, W. The crystal structure of Trypanosoma cruzi glucokinase reveals features determining oligomerization and anomer specificity of hexose-phosphorylating enzymes. J. Mol. Biol. 2007, 372, 1215–1226. [Google Scholar] [CrossRef]
- Srivastava, S.S.; Darling, J.E.; Suryadi, J.; Morris, J.C.; Drew, M.E.; Subramaniama, S. Plasmodium vivax and human hexokinases share similar active sites but display distinct quaternary architectures. IUCrJ 2020, 7, 453–461. [Google Scholar] [CrossRef]
- Dillenberger, M.; Werner, A.-D.; Velten, A.-S.; Rahlfs, S.; Becker, K.; Fritz-Wolf, K. Structural analysis of Plasmodium falciparum hexokinase provides novel information about catalysis due to a Plasmodium-specific insertion. Int. J. Mol. Sci. 2023, 24, 12739. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Fowler, M.L.; Morris, M.T.; Morris, J.C. The anti-trypanosomal agent lonidamine inhibits Trypanosoma brucei hexokinase 1. Mol. Biochem. Parasitol. 2008, 158, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Mercaldi, G.F.; Ranzani, A.T.; Cordeiro, A.T. Discovery of new uncompetitive inhibitors of glucose-6-phosphate dehydrogenase. J. Biomol. Screen. 2014, 19, 1362–1371. [Google Scholar] [CrossRef]
- Andriani, G.; Chessler, A.-D.C.; Courtemanche, G.; Burleigh, B.A.; Rodriguez, A. Activity in vivo of anti-Trypanosoma cruzi compounds selected from a high throughput screening. PLoS Neglected Trop. Dis. 2011, 5, e1298. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, O.; Metibemu, D.S.; Crown, O.; Adeyinka, O.S.; Kaiser, M.; Shoji, N.; Silva, M.; Rodriguez, A.; Ogungbe, I.V. Discovery of an orally active nitrothiophene-based antitrypanosomal agent. Eur. J. Med. Chem. 2024, 263, 115954. [Google Scholar] [CrossRef]
- Lowe, D.M.; Corbett, P.T.; Murray-Rust, P.; Glen, R.C. Chemical name to structure: OPSIN, an open source solution. J. Chem. Inf. Model 2011, 51, 739–753. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | TcGlcK Ki (µM) a | LmG6PDH Inh20 (%) c,d | T. cruzi IC50 (µM) b,e | T. brucei IC50 (µM) b,f | NIH-3T3 TC50 (µM) b,g |
|---|---|---|---|---|---|
| 1 | 6.2 ± 1.2 b | 15.4 ± 0.1 | 1.49 ± 0.07 | 0.61 ± 0.03 | 16.0 ± 1.84 |
| 2 | 46 | 0.0 ± 0.0 | 1.44 ± 0.07 | 1.19 ± 0.04 | 22.2 ± 0.33 |
| 3 | 24 | 0.0 ± 0.0 | 0.96 ± 0.02 | 0.52 ± 0.05 | 17.5 ± 1.90 |
| 4 | 5.5 ± 1.8 b | 12.1 ± 0.0 | 1.31 ± 0.15 | 0.44 ± 0.03 | 16.4 ± 1.91 |
| 5 | 35 | 0.0 ± 0.0 | 0.64 ± 0.23 | 0.42 ± 0.04 | 19.5 ± 0.51 |
| 6 | 6.1 | 16.1 ± 0.0 | 2.56 ± 0.26 | 0.36 ± 0.06 | 18.1 ± 0.24 |
| 7 | 50 | 0.0 ± 0.0 | 0.64 ± 0.02 | 0.90 ± 0.33 | 11.9 ± 0.51 |
| 8 | 30 | 0.0 ± 0.0 | 1.85 ± 0.15 | 0.74 ± 0.14 | 26.1 ± 0.57 |
| 9 | 12.3 ± 5.5 b | 10.1 ± 0.1 | 0.64 ± 0.17 | 0.66 ± 0.05 | 17.5 ± 1.60 |
| 10 | 23 | 0.0 ± 0.0 | 2.56 ± 0.26 | 1.12 ± 0.30 | 9.45 ± 2.20 |
| 11 | 20.0 ± 4.3 b | 15.0 ± 0.1 | 0.77 ± 0.02 | 0.86 ± 0.20 | 4.12 ± 1.95 |
| 12 | 14 | 8.6 ± 0.1 | 7.07 ± 1.19 | 1.67 ± 0.06 | 10.9 ± 1.55 |
| 13 | 25 | 0.0 ± 0.0 | 3.08 ± 0.08 | 1.28 ± 0.10 | nd h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carey, S.M.; O’Neill, D.M.; Conner, G.B.; Sherman, J.; Rodriguez, A.; D’Antonio, E.L. Discovery of Strong 3-Nitro-2-Phenyl-2H-Chromene Analogues as Antitrypanosomal Agents and Inhibitors of Trypanosoma cruzi Glucokinase. Int. J. Mol. Sci. 2024, 25, 4319. https://doi.org/10.3390/ijms25084319
Carey SM, O’Neill DM, Conner GB, Sherman J, Rodriguez A, D’Antonio EL. Discovery of Strong 3-Nitro-2-Phenyl-2H-Chromene Analogues as Antitrypanosomal Agents and Inhibitors of Trypanosoma cruzi Glucokinase. International Journal of Molecular Sciences. 2024; 25(8):4319. https://doi.org/10.3390/ijms25084319
Chicago/Turabian StyleCarey, Shane M., Destiny M. O’Neill, Garrett B. Conner, Julian Sherman, Ana Rodriguez, and Edward L. D’Antonio. 2024. "Discovery of Strong 3-Nitro-2-Phenyl-2H-Chromene Analogues as Antitrypanosomal Agents and Inhibitors of Trypanosoma cruzi Glucokinase" International Journal of Molecular Sciences 25, no. 8: 4319. https://doi.org/10.3390/ijms25084319
APA StyleCarey, S. M., O’Neill, D. M., Conner, G. B., Sherman, J., Rodriguez, A., & D’Antonio, E. L. (2024). Discovery of Strong 3-Nitro-2-Phenyl-2H-Chromene Analogues as Antitrypanosomal Agents and Inhibitors of Trypanosoma cruzi Glucokinase. International Journal of Molecular Sciences, 25(8), 4319. https://doi.org/10.3390/ijms25084319