
Effect of Regulation of Chemerin/Chemokine-like Receptor 1/Stimulator of Interferon Genes Pathway on Astrocyte Recruitment to Aβ Plaques

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
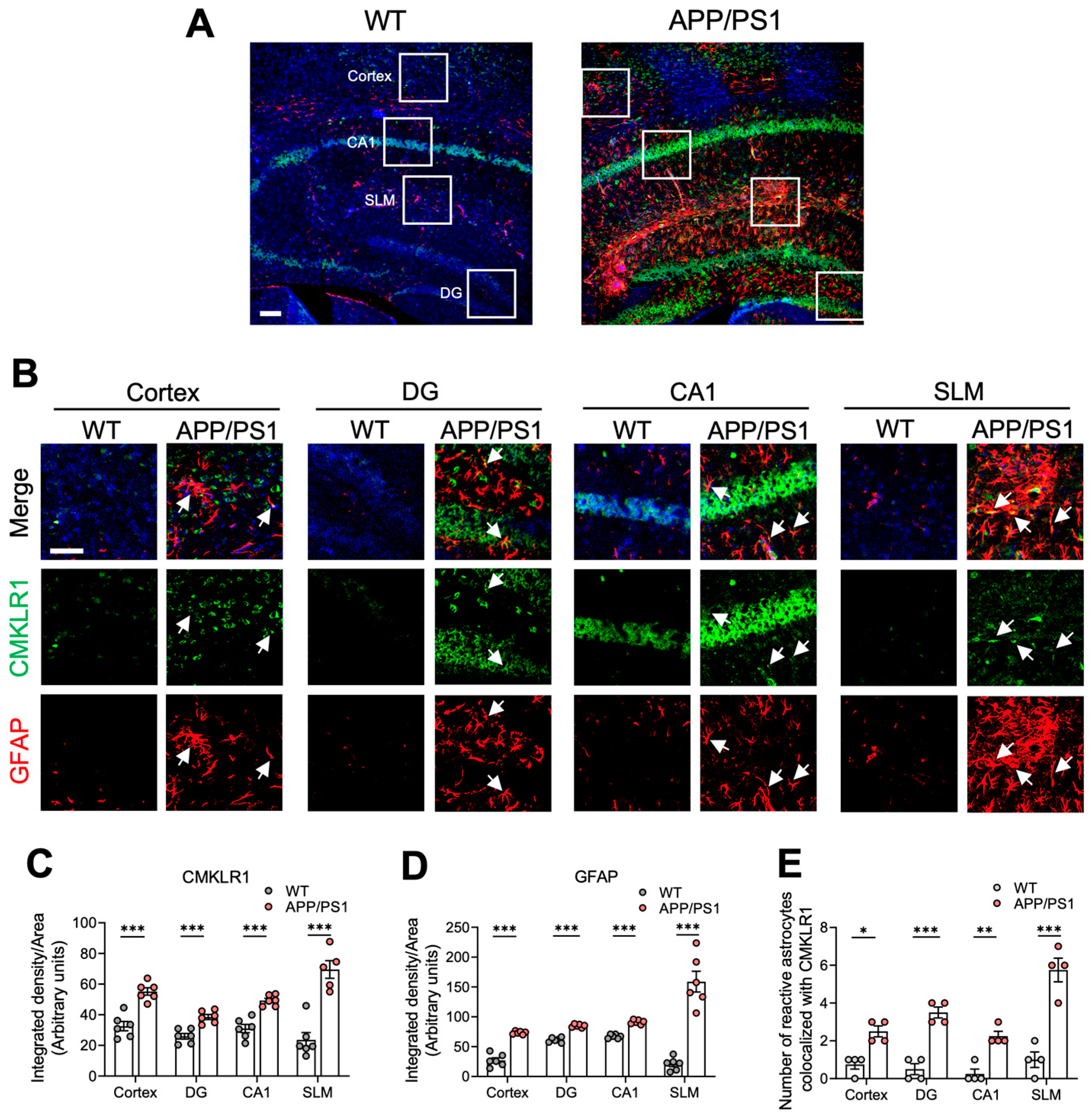
2.1. Elevated CMKLR1-Expressed Astrocytes in the Brain of APP/PS1 Mice
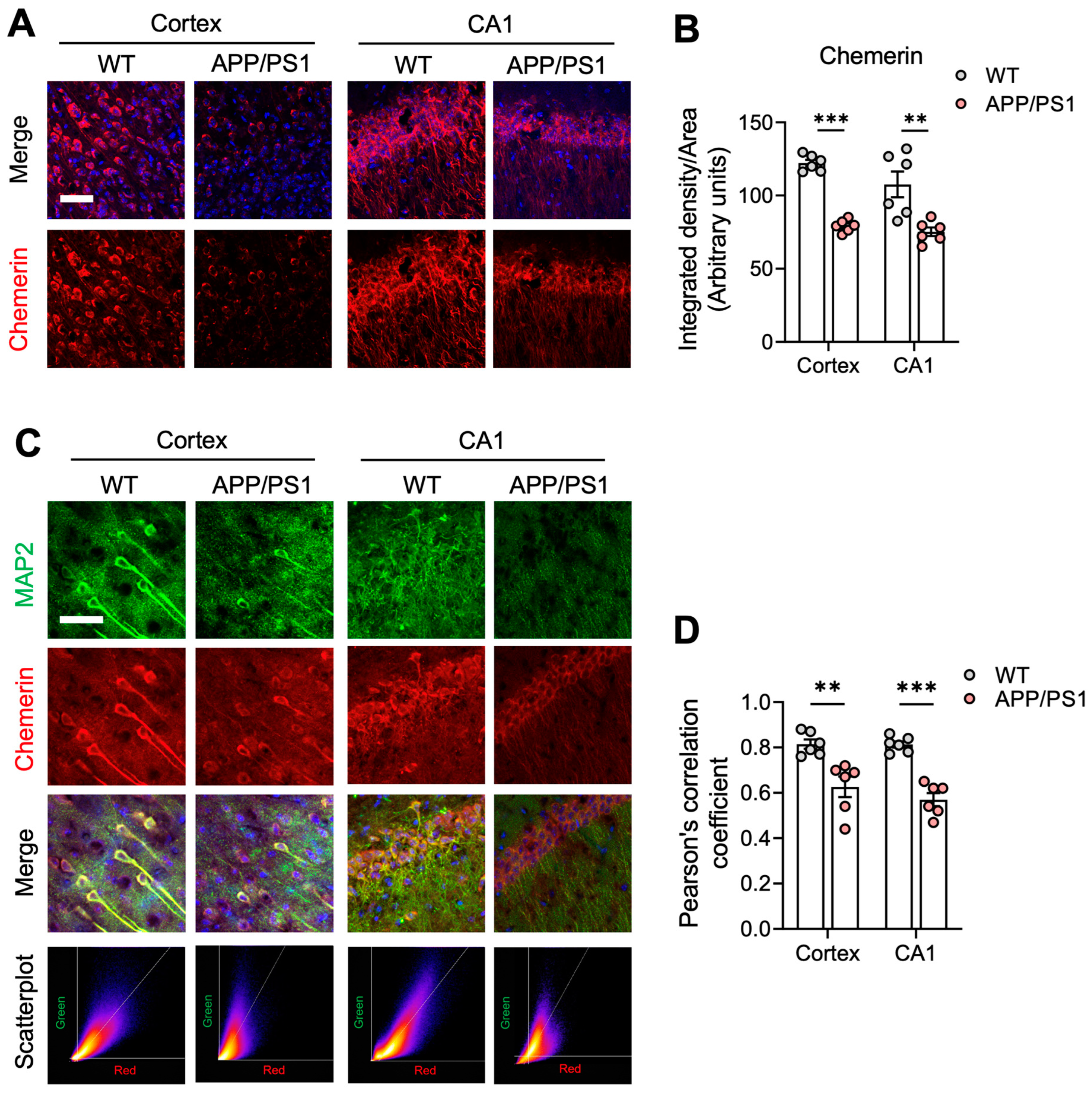
2.2. The Chemerin/CMKLR1 Axis Is Weakened in APP/PS1 Mice
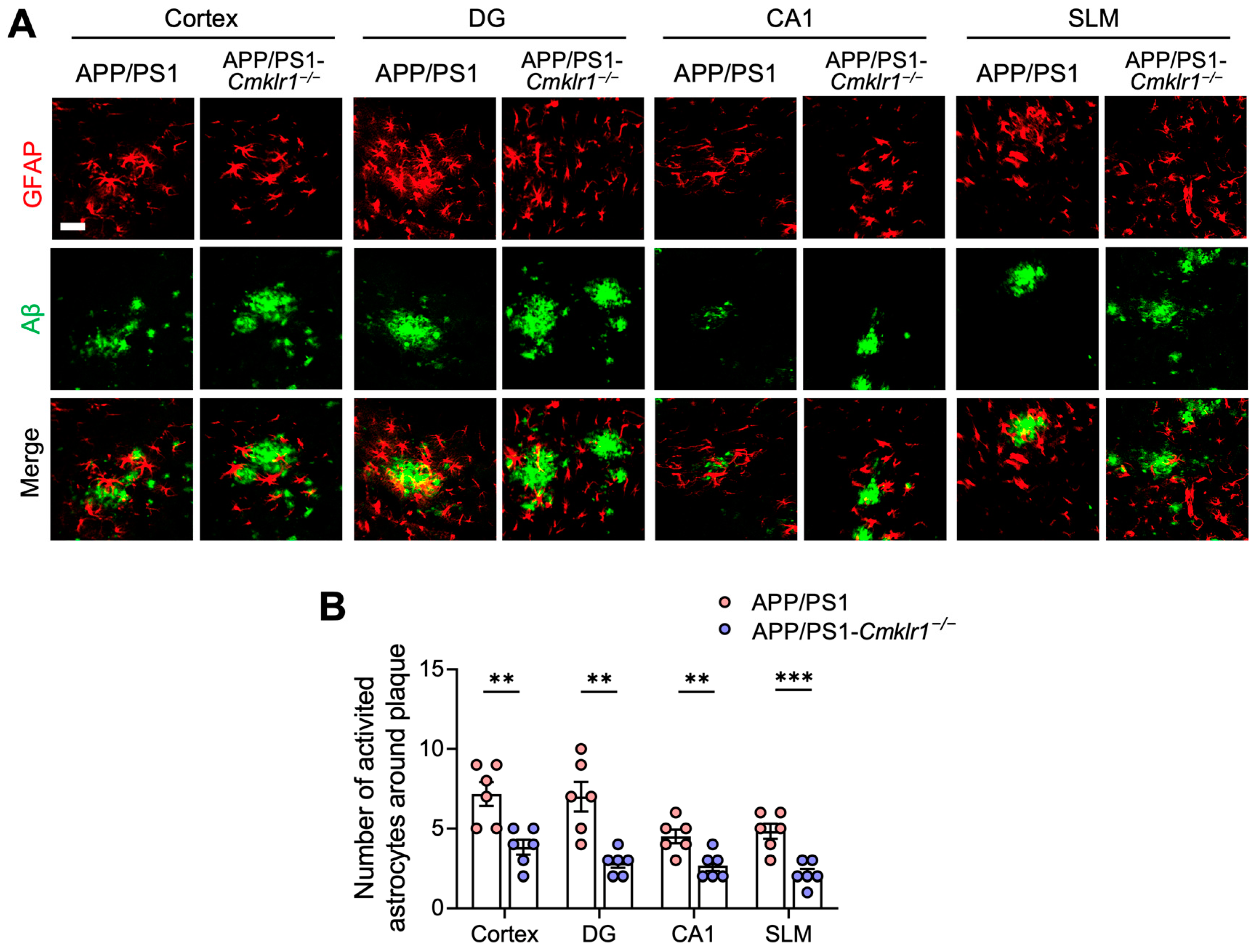
2.3. CMKLR1 Deficiency Reduces the Colocalization of Astrocytes with Aβ Plaques in the Brain of APP/PS1 Mice
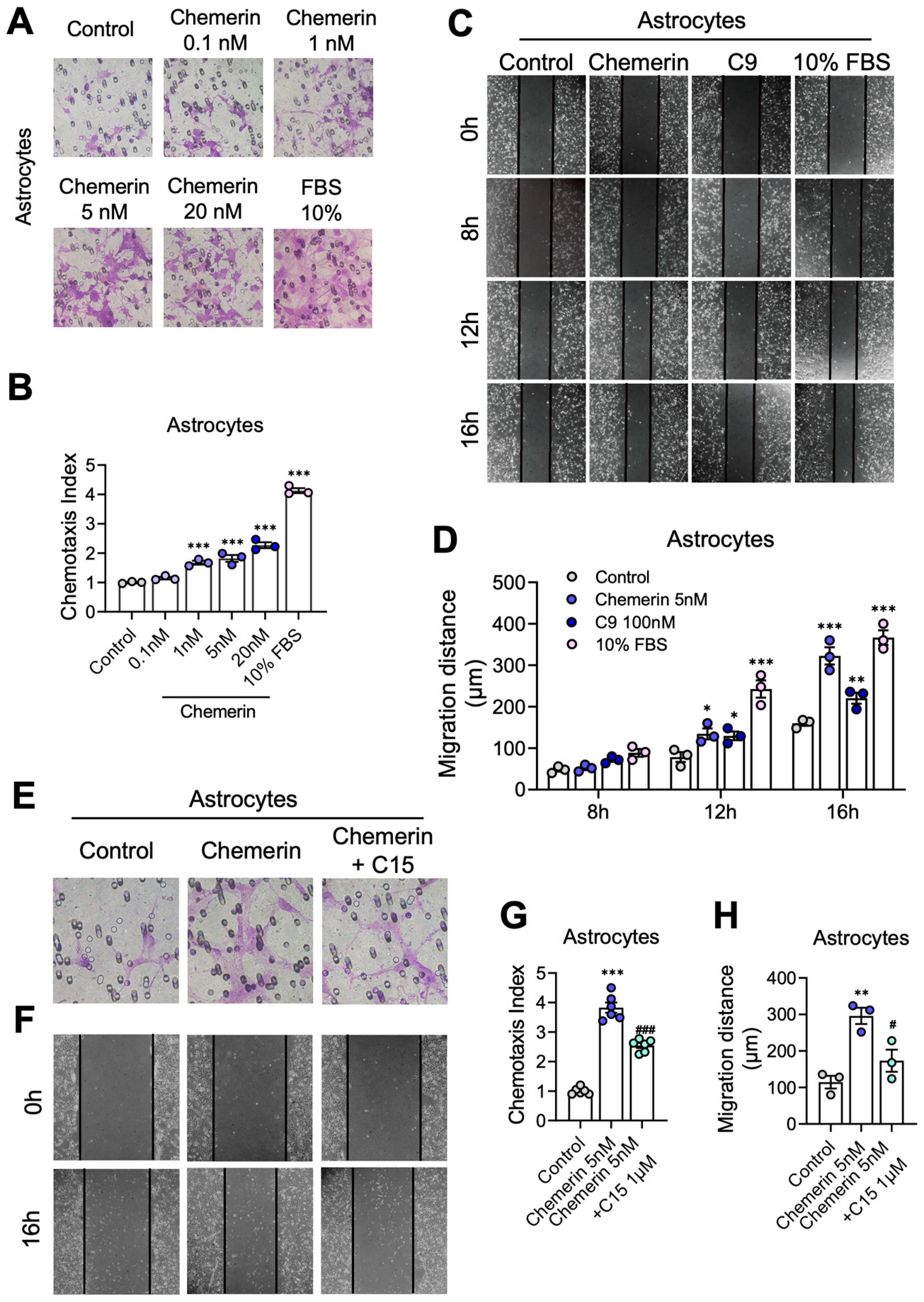
2.4. Activation of the Chemerin/CMKLR1 Axis Induces Astrocyte Migration
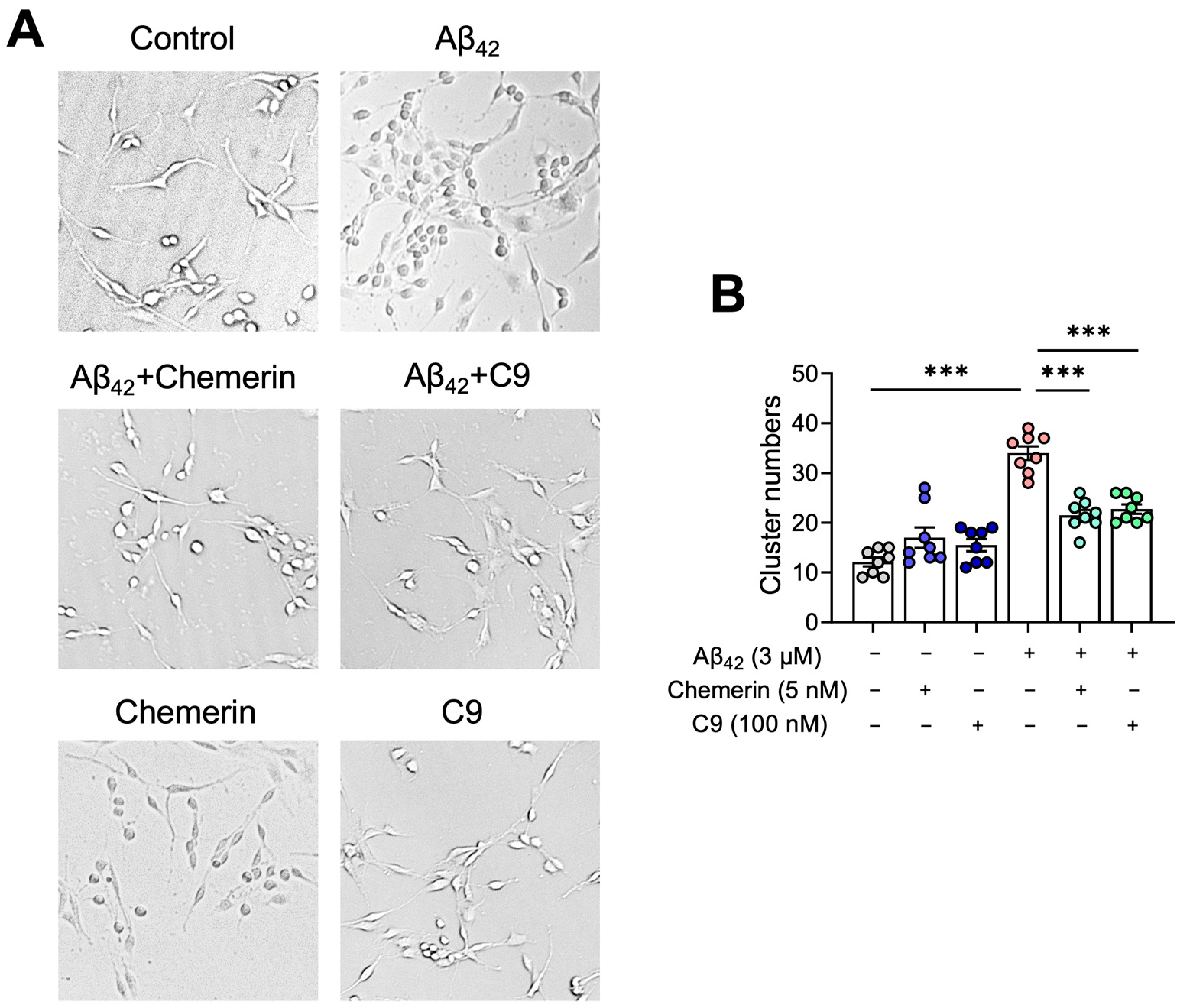
2.5. Chemerin Suppresses Aβ-Induced Aggregation of Astrocytes
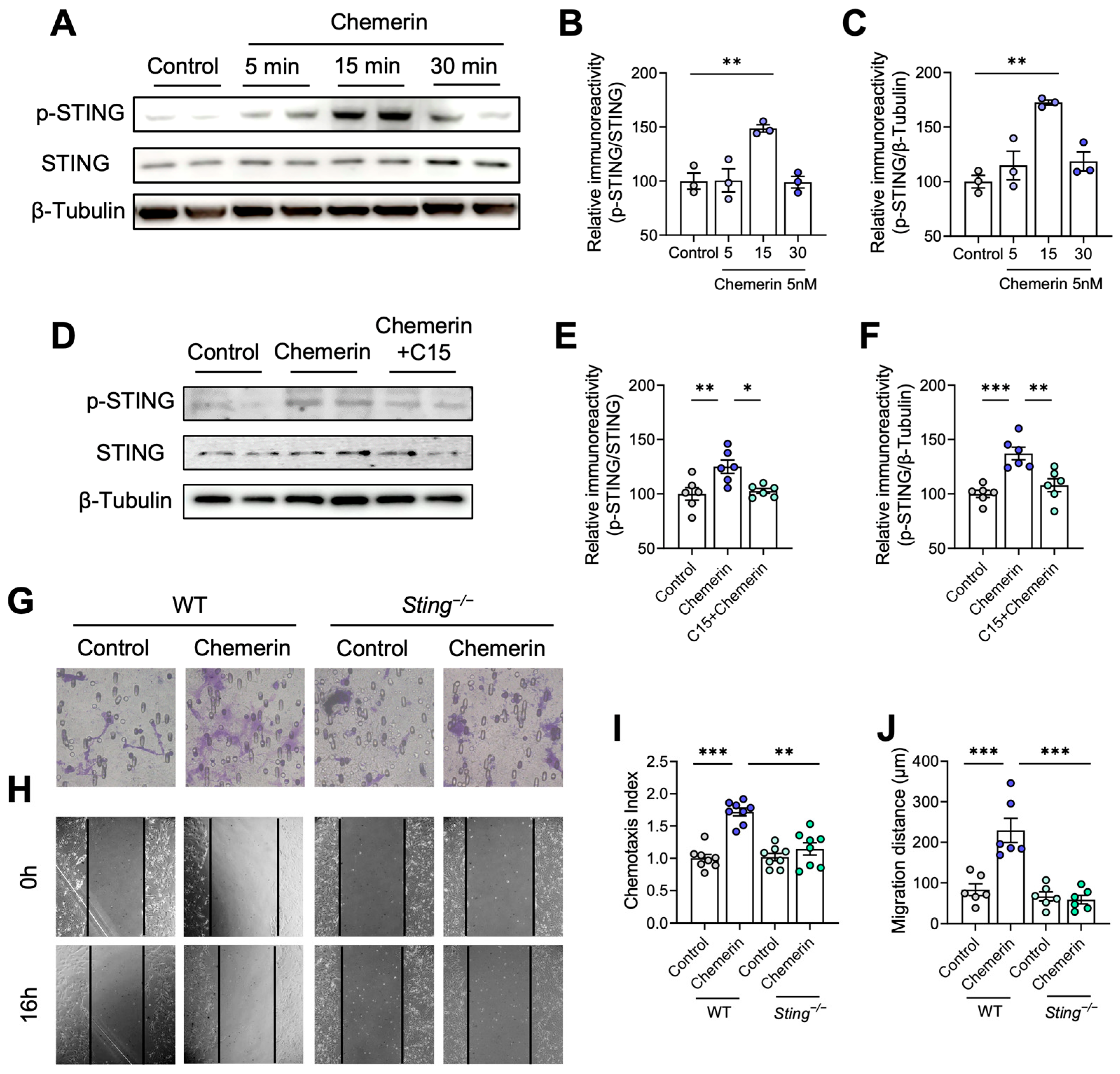
2.6. STING Contributes to Astrocyte Migration Induced by Chemerin/CMKLR1 Axis
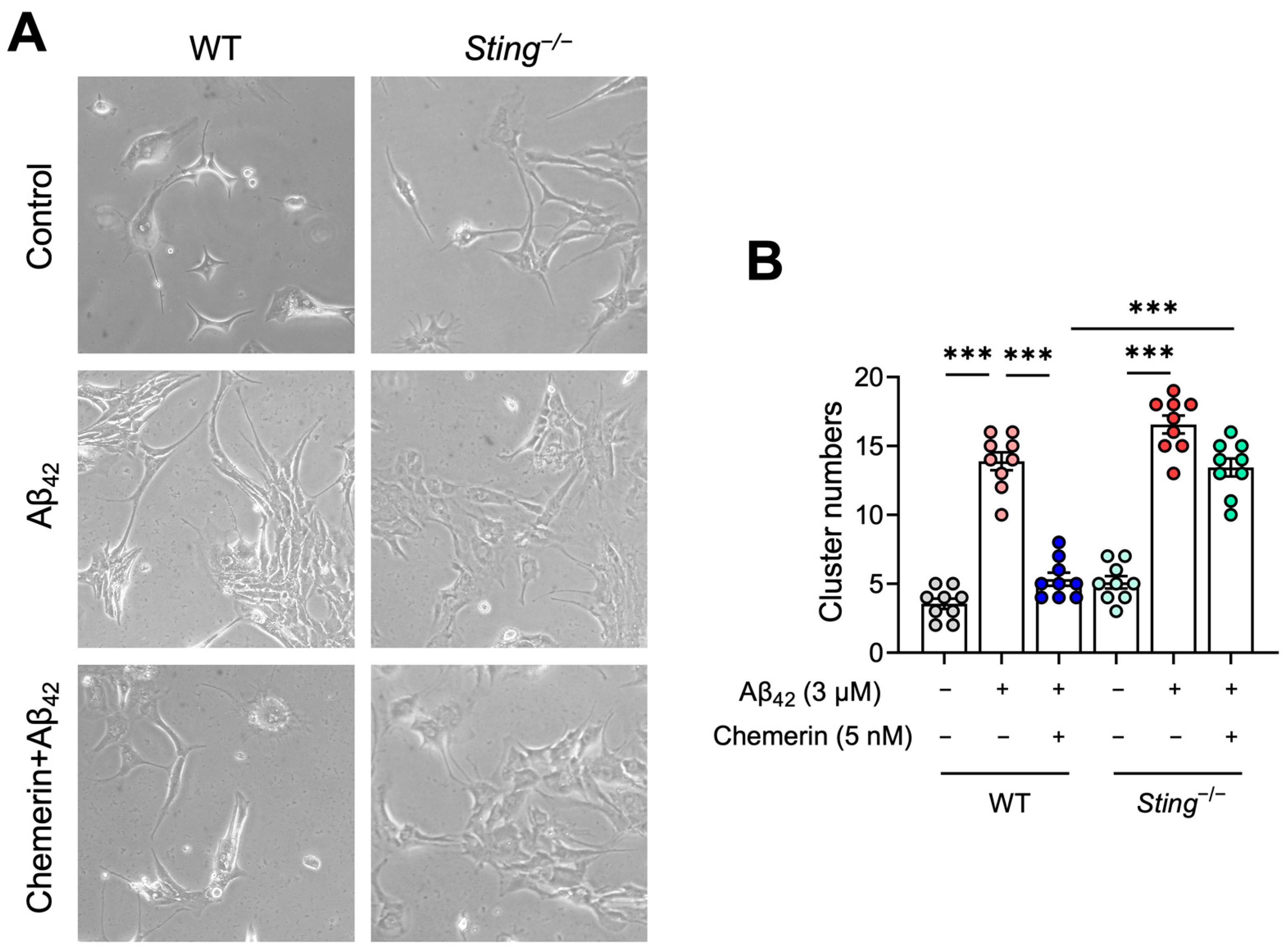
2.7. STING Contributes to the Inhibitory Effect of Chemerin on Aβ-Induced Aggregation of Astrocytes
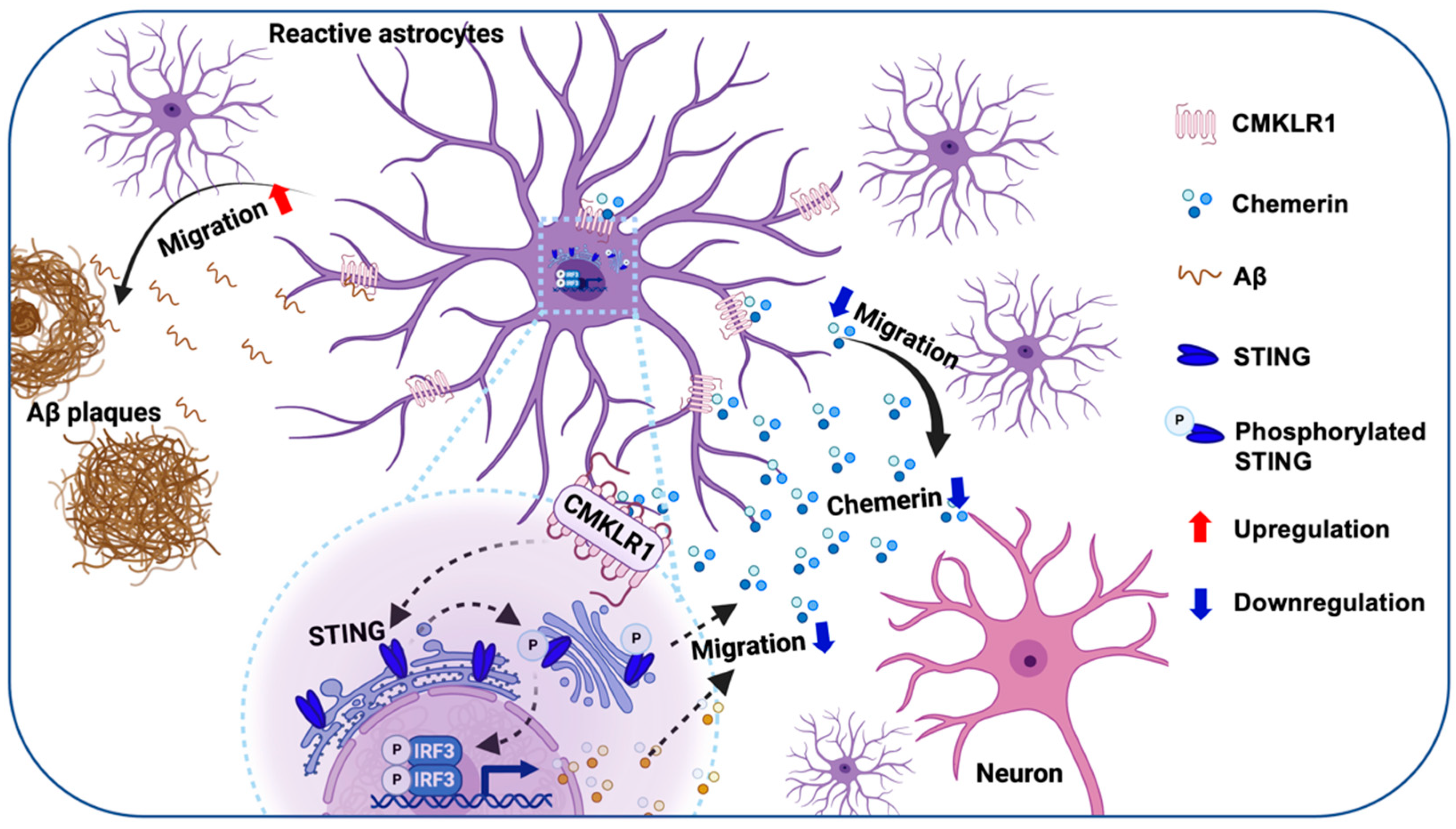
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Animals
4.3. Astrocyte Cultures
4.4. Immunofluorescence Staining and Image Analysis
4.5. Boyden Chamber Assay
4.6. Scratch-Wound Assay
4.7. Assessment of Astrocyte Aggregation
4.8. Western Blot
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, K. 2021 marks a new era for Alzheimer’s therapeutics. Lancet Neurol. 2022, 21, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approval for Biogen’s aducanumab sparks Alzheimer disease firestorm. Nat. Rev. Drug Discov. 2021, 20, 496. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ye, R.D. Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Zhuang, J.; Zhang, H.; Gao, C. beta-Amyloid activates reactive astrocytes by enhancing glycolysis of astrocytes. Mol. Biol. Rep. 2022, 49, 4699–4707. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, A.B.; Hennessy, E.; Murray, C.L.; Nazmi, A.; Delaney, H.J.; Healy, D.; Fagan, S.G.; Rooney, M.; Stewart, E.; Lewis, A.; et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer’s disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 1735–1755. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; Le Poul, E.; Migeotte, I.; Brézillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Silverio, A.M.; Butcher, E.C. Chemokine-like receptor 1 expression and chemerin-directed chemotaxis distinguish plasmacytoid from myeloid dendritic cells in human blood. J. Immunol. 2005, 174, 244–251. [Google Scholar] [CrossRef]
- Kennedy, A.J.; Davenport, A.P. International Union of Basic and Clinical Pharmacology CIII: Chemerin Receptors CMKLR1 (Chemerin(1)) and GPR1 (Chemerin(2)) Nomenclature, Pharmacology, and Function. Pharmacol. Rev. 2018, 70, 174–196. [Google Scholar] [CrossRef]
- Bufano, M.; Laffranchi, M.; Sozzani, S.; Raimondo, D.; Silvestri, R.; Coluccia, A. Exploring CCRL2 chemerin binding using accelerated molecular dynamics. Proteins 2022, 90, 1714–1720. [Google Scholar] [CrossRef]
- Zabel, B.A.; Nakae, S.; Zúñiga, L.; Kim, J.Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef]
- Meder, W.; Wendland, M.; Busmann, A.; Kutzleb, C.; Spodsberg, N.; John, H.; Richter, R.; Schleuder, D.; Meyer, M.; Forssmann, W.G. Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett. 2003, 555, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Oppenheim, J.J. Chemokine-like receptor 1 (CMKLR1) and chemokine (C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp. Cell Res. 2011, 317, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Huang, Y.; Ling, X.; Qin, H.; Wang, M.; Luo, B. Chemerin/CMKLR1 Axis Promotes Inflammation and Pyroptosis by Activating NLRP3 Inflammasome in Diabetic Cardiomyopathy Rat. Front. Physiol. 2020, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yu, Y.; Liu, J.; Li, S.; He, H.; Cheng, N.; Ye, R.D. The chemerin receptor CMKLR1 is a functional receptor for amyloid-β peptide. J. Alzheimers Dis. 2015, 43, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, A.; Gong, P.; Chen, Y.; Ye, R.D.; Qian, F.; Zhang, Y.; Yu, Y. The Chemokine-like Receptor 1 Deficiency Improves Cognitive Deficits of AD Mice and Attenuates Tau Hyperphosphorylation via Regulating Tau Seeding. J. Neurosci. 2020, 40, 6991–7007. [Google Scholar] [CrossRef] [PubMed]
- Emre, C.; Hjorth, E.; Bharani, K.; Carroll, S.; Granholm, A.C.; Schultzberg, M. Receptors for pro-resolving mediators are increased in Alzheimer’s disease brain. Brain Pathol. 2020, 30, 614–640. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Gong, P.; Zhang, H.; Chen, Y.; Yao, S.; Li, W.; Zhang, Y.; Yu, Y. The Chemerin/CMKLR1 Axis Is Involved in the Recruitment of Microglia to Abeta Deposition through p38 MAPK Pathway. Int. J. Mol. Sci. 2022, 23, 9041. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, L.; Zhang, Y.; Yuan, Y.; Miao, Z.; Lu, K.; Zhang, X.; Ni, R.; Zhang, H.; Zhao, Y.; et al. ChemR23 signaling ameliorates cognitive impairments in diabetic mice via dampening oxidative stress and NLRP3 inflammasome activation. Redox Biol. 2022, 58, 102554. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, N.; Ding, Y.; Zhang, Y.; Li, Q.; Flores, J.; Haghighiabyaneh, M.; Doycheva, D.; Tang, J.; Zhang, J.H. Chemerin suppresses neuroinflammation and improves neurological recovery via CaMKK2/AMPK/Nrf2 pathway after germinal matrix hemorrhage in neonatal rats. Brain Behav. Immunol. 2018, 70, 179–193. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, N.; Ding, Y.; Doycheva, D.M.; Zhang, Y.; Li, Q.; Flores, J.; Haghighiabyaneh, M.; Tang, J.; Zhang, J.H. Chemerin reverses neurological impairments and ameliorates neuronal apoptosis through ChemR23/CAMKK2/AMPK pathway in neonatal hypoxic-ischemic encephalopathy. Cell Death Dis. 2019, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Ma, G.; Li, X.; Zhao, J.; Zhao, Z.; Zeng, J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5xFAD mice. Nat. Aging 2023, 3, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, A.M.; Marriott, I. Human microglia and astrocytes express cGAS-STING viral sensing components. Neurosci. Lett. 2017, 658, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.N.; Lehallier, B.; Zardeneta, M.E.; Mistry, K.N.; Do, D.; Marsh, S.E.; Abud, E.M.; et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 2017, 96, 1290–1302.e6. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Getahun, A.; Knowles, H.M.; Mogan, J.; Akerlund, L.J.; Packard, T.A.; Perraud, A.L.; Cambier, J.C. STING/MPYS mediates host defense against Listeria monocytogenes infection by regulating Ly6C(hi) monocyte migration. J. Immunol. 2013, 190, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lin, W.; Kuang, Y.; Wang, J.; Xu, S.; Shen, C.; Qiu, Q.; Shi, M.; Xiao, Y.; Liang, L.; et al. cGAS/STING signaling in the regulation of rheumatoid synovial aggression. Ann. Transl. Med. 2022, 10, 431. [Google Scholar] [CrossRef]
- Yuan, M.; Guo, X.L.; Chen, J.H.; He, Y.; Liu, Z.Q.; Zhang, H.P.; Ren, J.; Xu, Q. Anlotinib suppresses proliferation, migration, and immune escape of gastric cancer cells by activating the cGAS-STING/IFN-β pathway. Neoplasma 2022, 69, 807–819. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Park, J.; Griciuc, A.; Kim, E.; Choi, S.H.; Iwamoto, Y.; Kiss, M.G.; Christie, K.A.; Vinegoni, C.; Poller, W.C.; et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 2021, 595, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.L.; Hart, R.; Russ, A.; Dixon, J.P.C.; Colledge, W.H.; Doran, J.; Hendrick, A.G.; Carlton, M.B.L.; Greaves, D.R. Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J. Exp. Med. 2008, 205, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yan, L.; Liu, N.; Xu, M.; Cai, H. IFI16 promotes cervical cancer progression by upregulating PD-L1 in immunomicroenvironment through STING-TBK1-NF-kB pathway. Biomed. Pharmacother. 2020, 123, 109790. [Google Scholar] [CrossRef] [PubMed]
- Parolini, S.; Santoro, A.; Marcenaro, E.; Luini, W.; Massardi, L.; Facchetti, F.; Communi, D.; Parmentier, M.; Majorana, A.; Sironi, M.; et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 2007, 109, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Riboldi, E.; Wittamer, V.; Gentili, F.; Luini, W.; Marrelli, S.; Vecchi, A.; Franssen, J.D.; Communi, D.; Massardi, L.; et al. Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 2005, 201, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Pachynski, R.K.; Zabel, B.A.; Kohrt, H.E.; Tejeda, N.M.; Monnier, J.; Swanson, C.D.; Holzer, A.K.; Gentles, A.J.; Sperinde, G.V.; Edalati, A.; et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med. 2012, 209, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, J.; Guo, L.; Cai, W.; Wu, Y.; Chen, W.; Tang, X. Chemerin stimulates aortic smooth muscle cell proliferation and migration via activation of autophagy in VSMCs of metabolic hypertension rats. Am. J. Transl. Res. 2019, 11, 1327–1342. [Google Scholar] [PubMed]
- Kumar, J.D.; Aolymat, I.; Tiszlavicz, L.; Reisz, Z.; Garalla, H.M.; Beynon, R.; Simpson, D.; Dockray, G.J.; Varro, A. Chemerin acts via CMKLR1 and GPR1 to stimulate migration and invasion of gastric cancer cells: Putative role of decreased TIMP-1 and TIMP-2. Oncotarget 2019, 10, 98–112. [Google Scholar] [CrossRef]
- Peng, L.; Chen, Y.; Li, Y.; Feng, P.; Zheng, Y.; Dong, Y.; Yang, Y.; Wang, R.; Li, A.; Yan, J.; et al. Chemerin Regulates the Proliferation and Migration of Pulmonary Arterial Smooth Muscle Cells via the ERK1/2 Signaling Pathway. Front. Pharmacol. 2022, 13, 767705. [Google Scholar] [CrossRef]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING mediates neurodegeneration and neuroinflammation in nigrostriatal alpha-synucleinopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2118819119. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Zhang, Y.; Tan, S.; Sun, J.; Ye, M.; Gao, H.; Pu, K.; Wu, M.; Wang, Q.; Zhai, Q. Therapeutic targeting of STING-TBK1-IRF3 signalling ameliorates chronic stress induced depression-like behaviours by modulating neuroinflammation and microglia phagocytosis. Neurobiol. Dis. 2022, 169, 105739. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Cheng, J.; Kong, X.; Li, S.; Li, X.; Zhang, M.; Zhang, H.; Yang, T.; Dong, Y.; Li, J.; et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 2020, 10, 9644–9662. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Zhang, M.; Frugier, T.; Bedoui, S.; Taylor, J.M.; Crack, P.J. STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J. Neuroinflamm. 2018, 15, 323. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cao, Y.; Dang, C.; Han, B.; Han, R.; Ma, H.; Hao, J.; Wang, L. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 2020, 12, e11002. [Google Scholar] [CrossRef]
- Li, S.Q.; Yu, Y.; Han, J.Z.; Wang, D.; Liu, J.; Qian, F.; Fan, G.H.; Bucala, R.; Ye, R.D. Deficiency of macrophage migration inhibitory factor attenuates tau hyperphosphorylation in mouse models of Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Maziuk, B.F.; Apicco, D.J.; Cruz, A.L.; Jiang, L.; Ash, P.E.A.; da Rocha, E.L.; Zhang, C.; Yu, W.H.; Leszyk, J.; Abisambra, J.F.; et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Liu, J.; Gong, P.; Chen, Y.; Zhang, H.; Zhang, Y.; Yu, Y. Serum amyloid A inhibits astrocyte migration via activating p38 MAPK. J. Neuroinflamm. 2020, 17, 254. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Q.; Su, N.; Gong, P.; Zhang, H.B.; Liu, J.; Wang, D.; Sun, Y.P.; Zhang, Y.; Qian, F.; Zhao, B.; et al. The Expression of Formyl Peptide Receptor 1 is Correlated with Tumor Invasion of Human Colorectal Cancer. Sci. Rep. 2017, 7, 5918. [Google Scholar] [CrossRef]
- Huang, W.C.; Yen, F.C.; Shie, F.S.; Pan, C.M.; Shiao, Y.J.; Yang, C.N.; Huang, F.L.; Sung, Y.J.; Tsay, H.J. TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J. Neuroinflamm. 2010, 7, 28. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Chen, Y.; Chen, Y.; Zheng, J.; Wu, W.; Wang, L.; Wang, H.; Yu, Y. Effect of Regulation of Chemerin/Chemokine-like Receptor 1/Stimulator of Interferon Genes Pathway on Astrocyte Recruitment to Aβ Plaques. Int. J. Mol. Sci. 2024, 25, 4324. https://doi.org/10.3390/ijms25084324
Liu Z, Chen Y, Chen Y, Zheng J, Wu W, Wang L, Wang H, Yu Y. Effect of Regulation of Chemerin/Chemokine-like Receptor 1/Stimulator of Interferon Genes Pathway on Astrocyte Recruitment to Aβ Plaques. International Journal of Molecular Sciences. 2024; 25(8):4324. https://doi.org/10.3390/ijms25084324
Chicago/Turabian StyleLiu, Zhen, Yijun Chen, Yanqing Chen, Jiayi Zheng, Wanning Wu, Linlin Wang, Hanqi Wang, and Yang Yu. 2024. "Effect of Regulation of Chemerin/Chemokine-like Receptor 1/Stimulator of Interferon Genes Pathway on Astrocyte Recruitment to Aβ Plaques" International Journal of Molecular Sciences 25, no. 8: 4324. https://doi.org/10.3390/ijms25084324
APA StyleLiu, Z., Chen, Y., Chen, Y., Zheng, J., Wu, W., Wang, L., Wang, H., & Yu, Y. (2024). Effect of Regulation of Chemerin/Chemokine-like Receptor 1/Stimulator of Interferon Genes Pathway on Astrocyte Recruitment to Aβ Plaques. International Journal of Molecular Sciences, 25(8), 4324. https://doi.org/10.3390/ijms25084324