
Renal Endothelial Single-Cell Transcriptomics Reveals Spatiotemporal Regulation and Divergent Roles of Differential Gene Transcription and Alternative Splicing in Murine Diabetic Nephropathy
 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
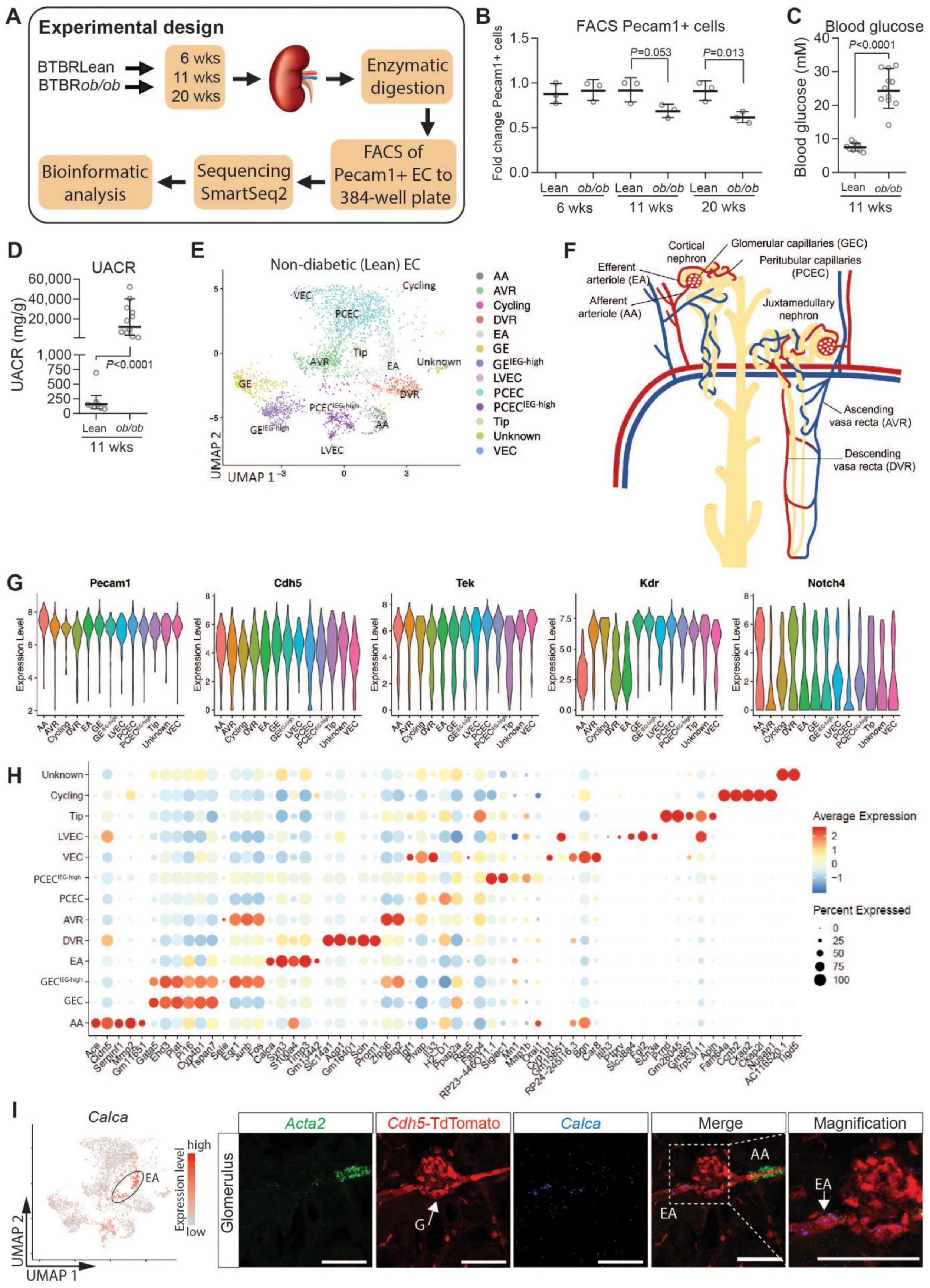
2.1. Molecular Profiling and EC Populations
2.2. Results from Renal EC Heterogeneity, Annotations, and Validation
2.3. Functional Zonation of the Healthy Renal Vasculature
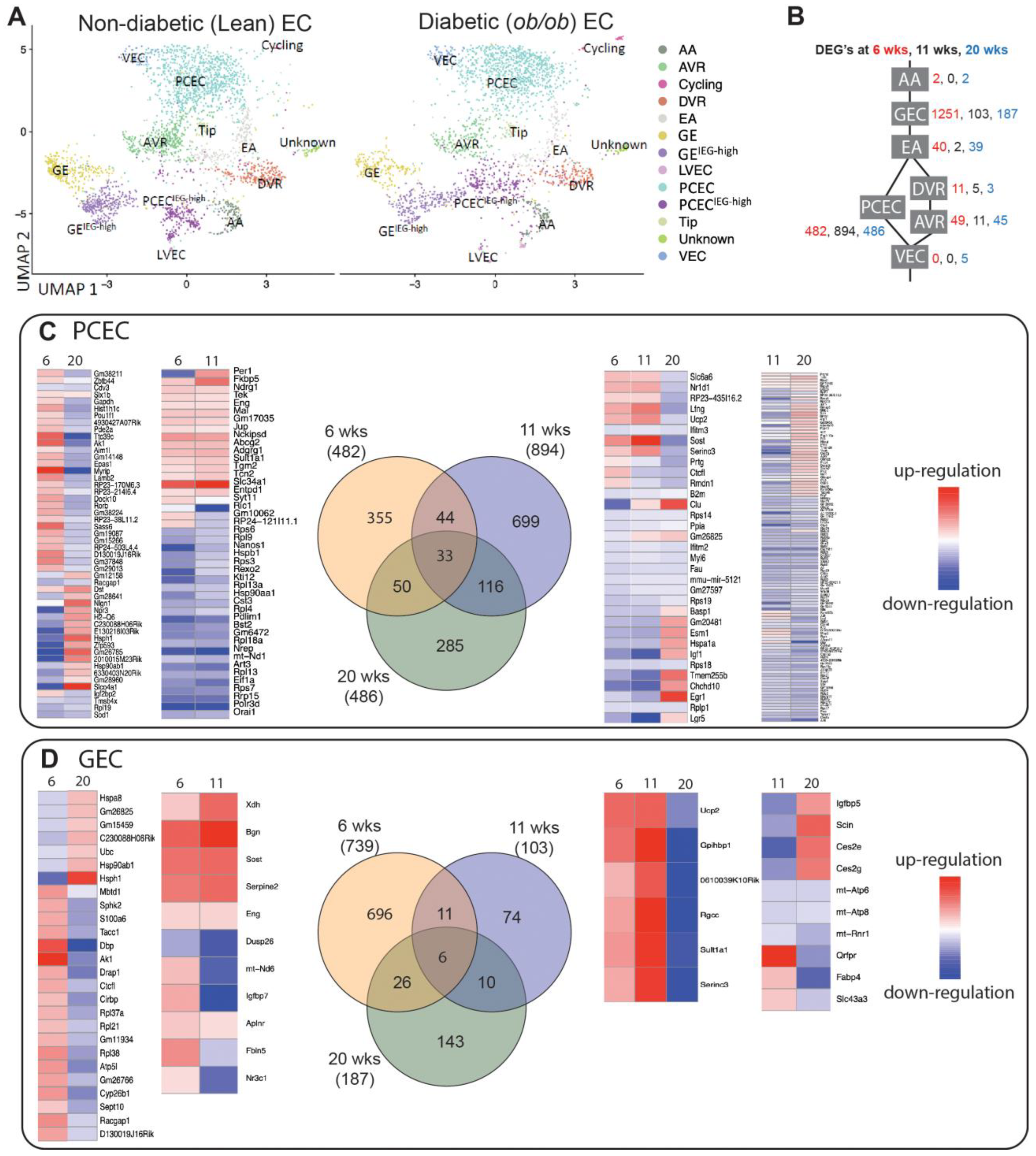
2.4. Transcriptional Responses in Progression of DKD
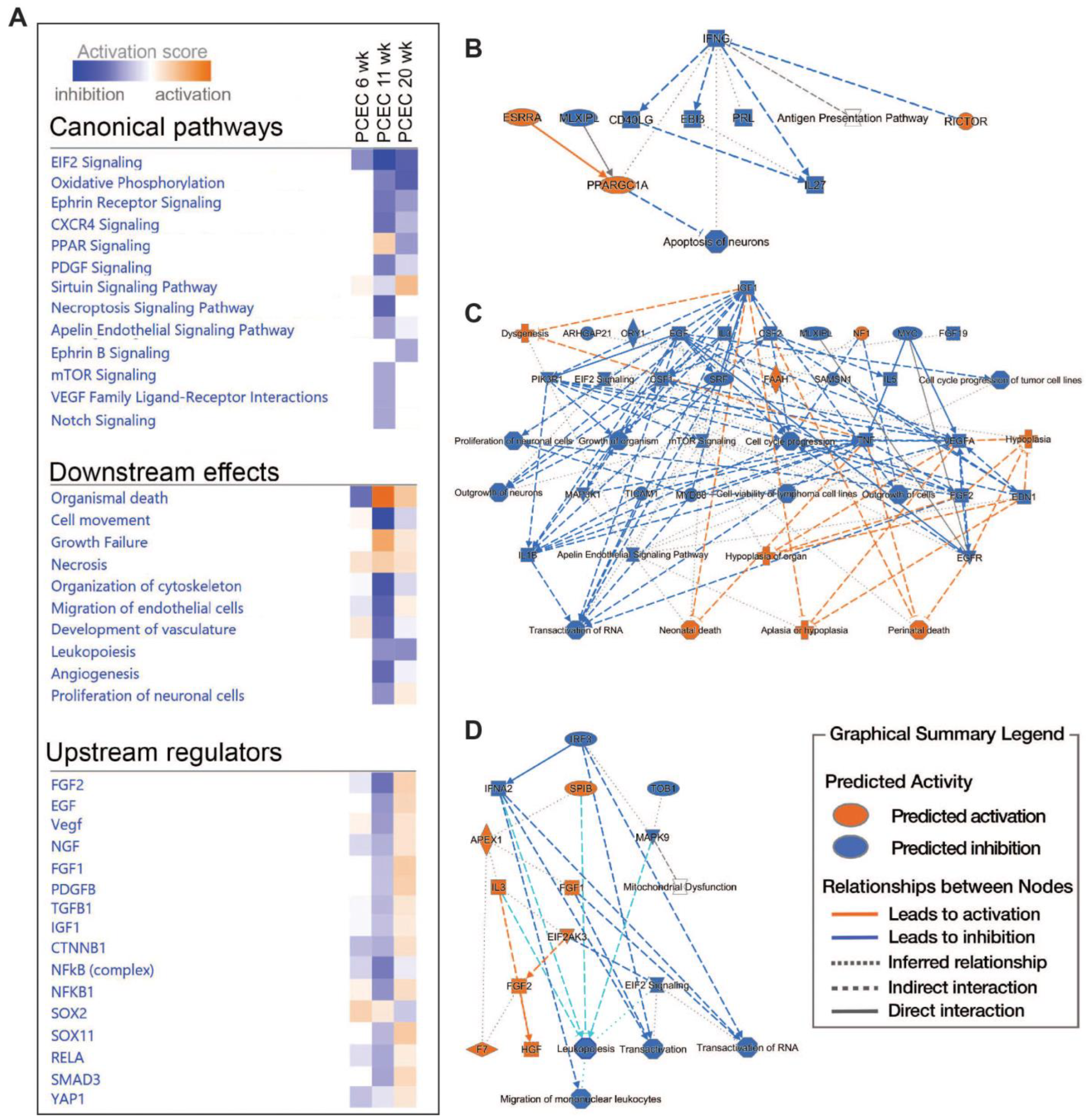
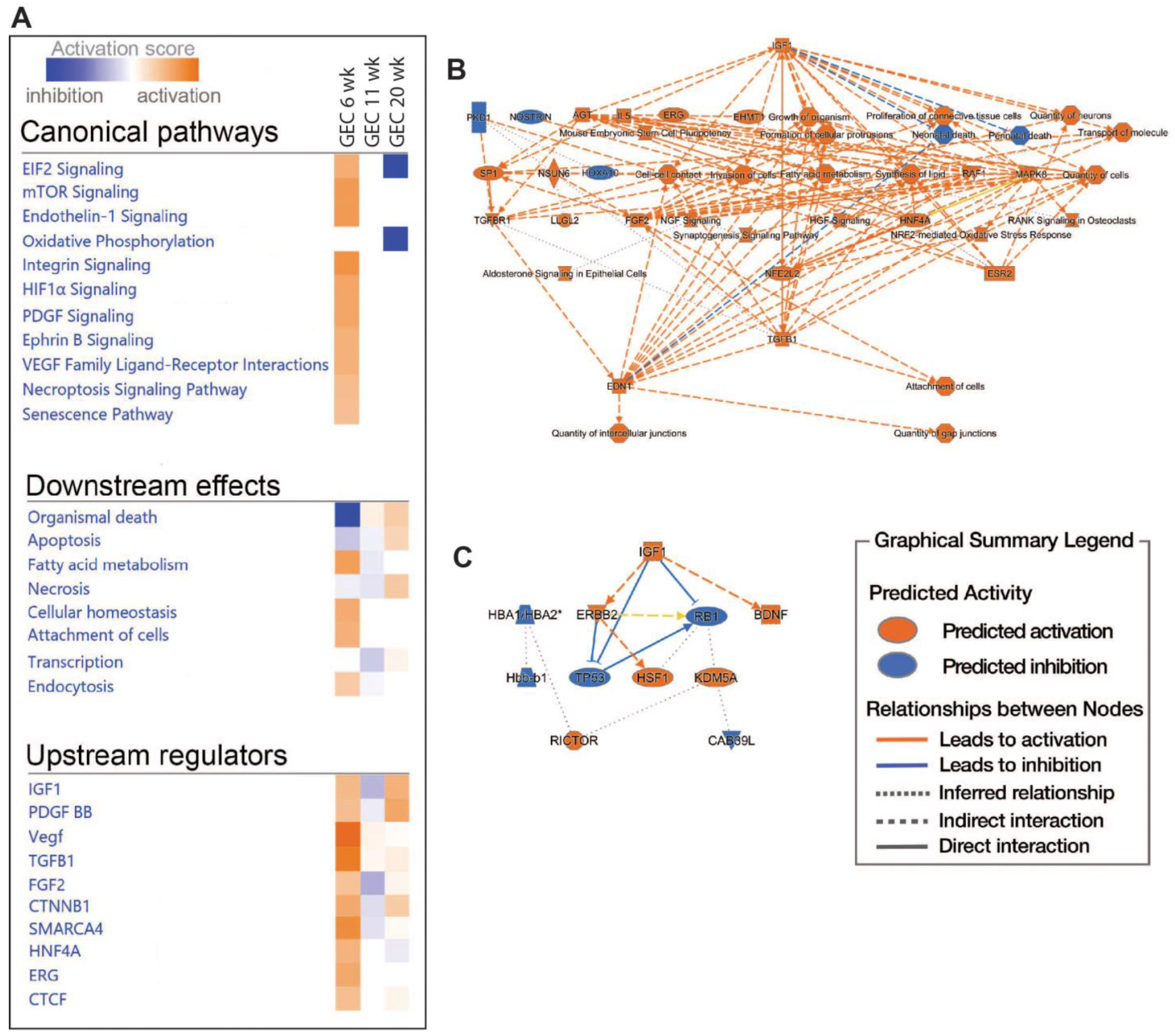
2.5. Pathway and Network Analyses of EC Responses in DKD
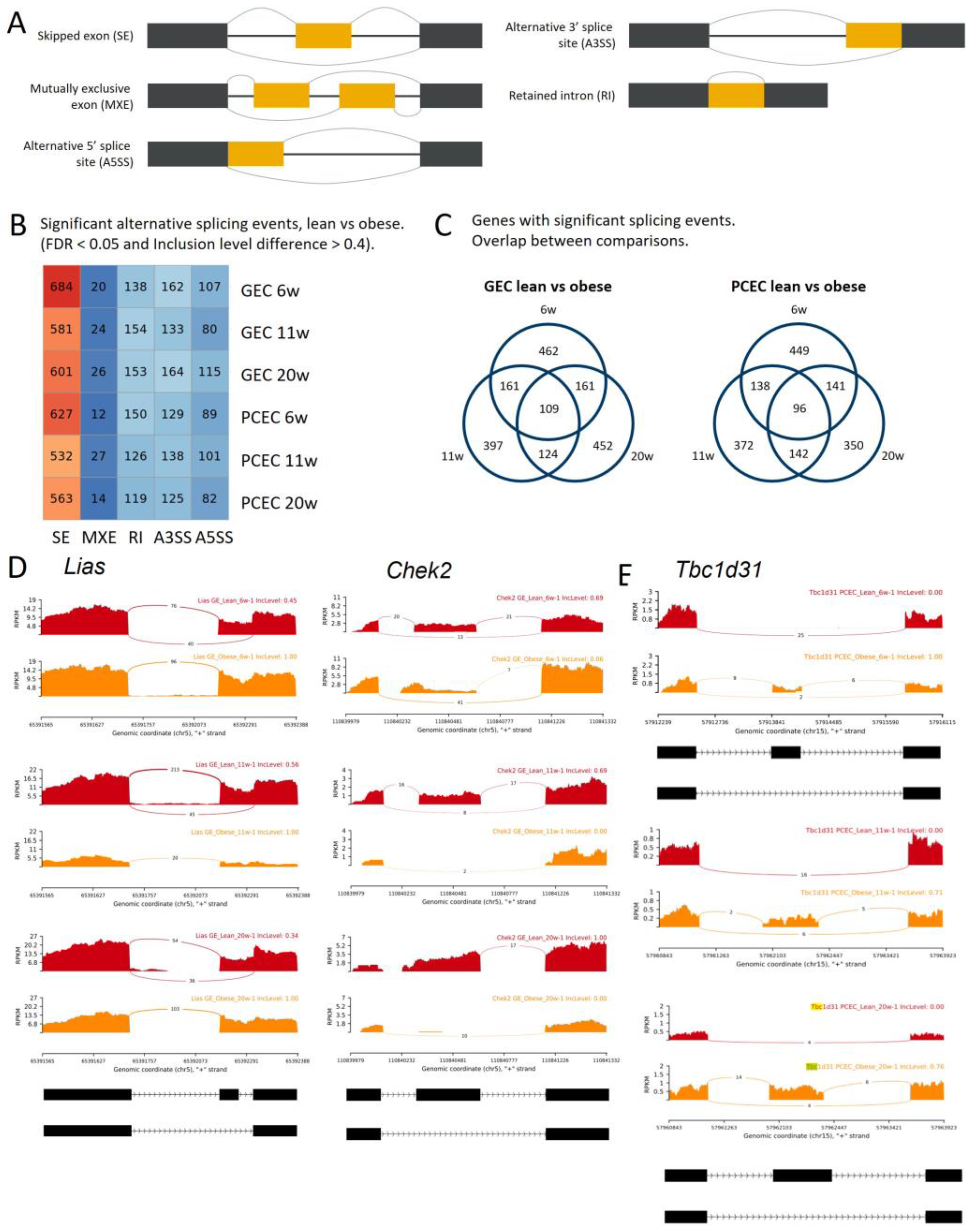
2.6. Alternative Splicing in Progression of DKD
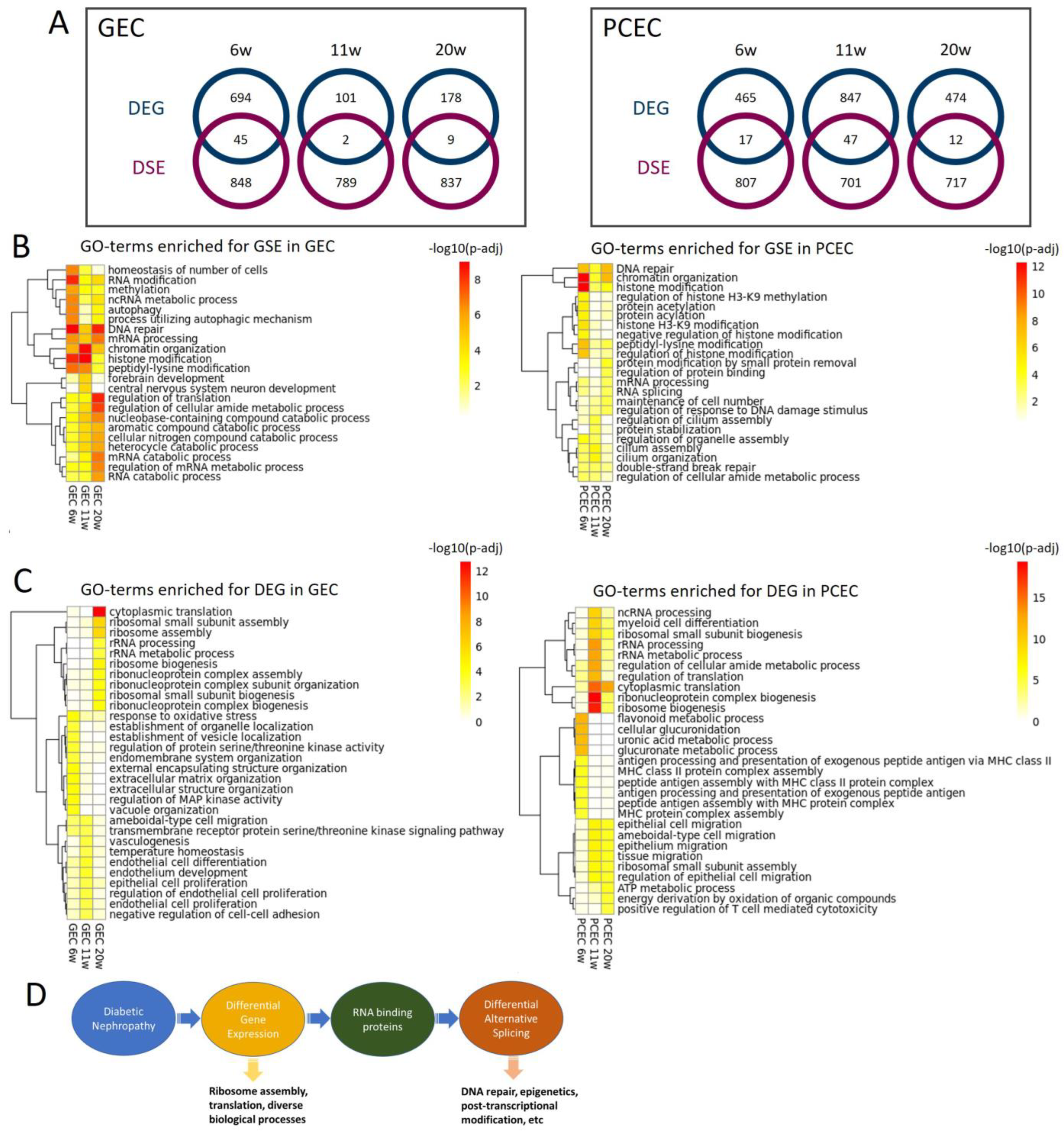
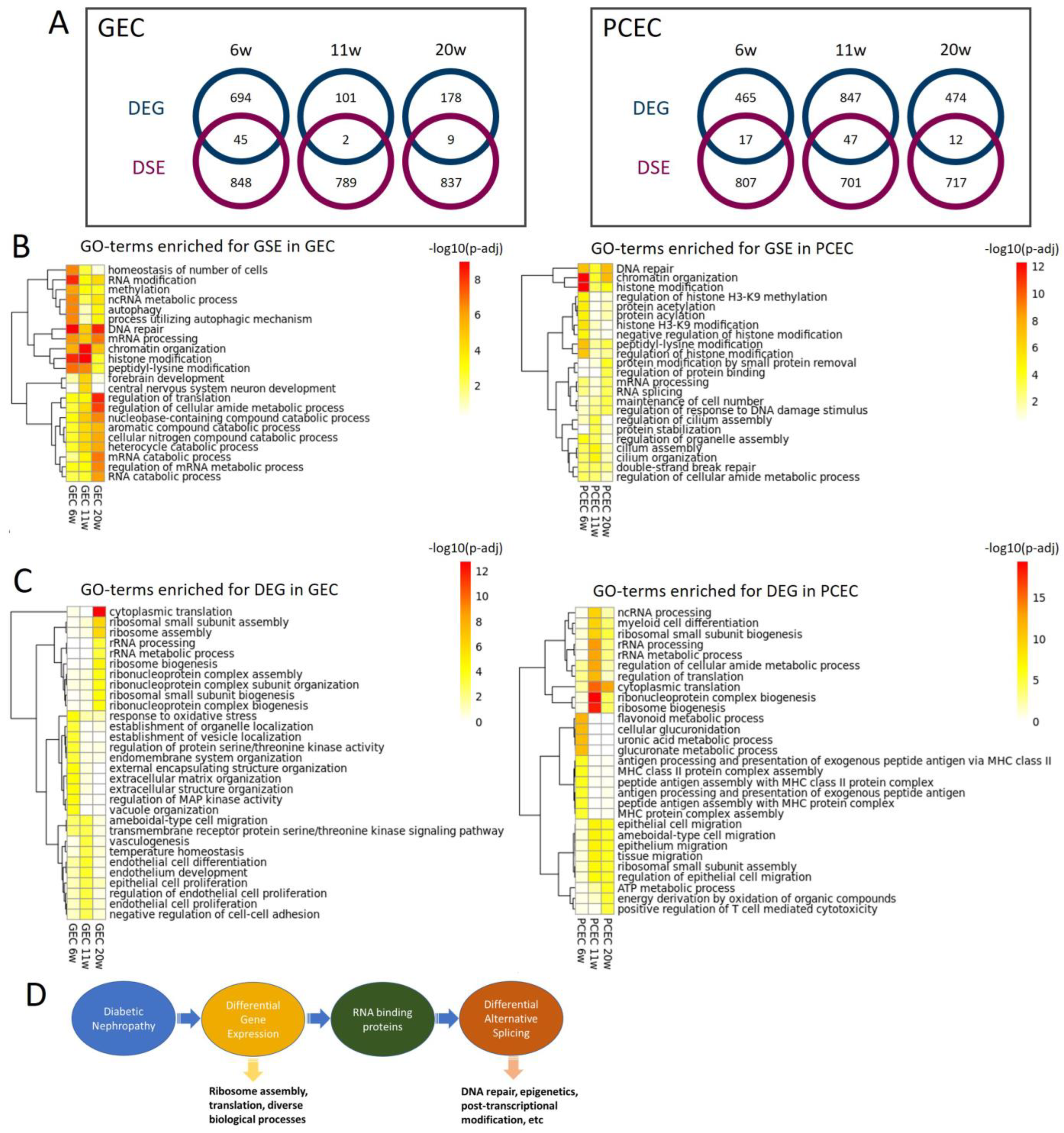
2.7. DEG and DSE Linked to Divergent Biological Processes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Renal Single Cells
4.3. Fluorescence-Activated Cell Sorting (FACS)
4.4. SmartSeq2 Library Preparation and Sequencing
4.5. Sequence Data Processing
4.6. Ingenuity Pathway Analysis
4.7. Alternative Splicing Analysis
4.8. Gene Ontology Enrichment Analysis
4.9. Identification of RNA Binding Proteins
4.10. Validation of EC Cluster Markers with RNA In Situ Hybridization (RNA-ISH)
4.11. Immunofluorescence Staining
4.12. Blood and Urine Analysis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of Ckd Progression. Int. J. Mol. Sci. 2020, 21, 8255. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.M.; McMillan, E.A.; Kunar, B.; Lis, R.; Zhang, T.; Lu, T.; Daniel, E.; Yokoyama, M.; Gomez-Salinero, J.M.; Sureshbabu, A.; et al. Molecular Determinants of Nephron Vascular Specialization in the Kidney. Nat. Commun. 2019, 10, 5705. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Meta, E.; Borri, M.; Goveia, J.; Rohlenova, K.; Conchinha, N.V.; Falkenberg, K.; Teuwen, L.-A.; de Rooij, L.; Kalucka, J.; et al. Single-Cell Rna Sequencing Reveals Renal Endothelium Heterogeneity and Metabolic Adaptation to Water Deprivation. J. Am. Soc. Nephrol. 2020, 31, 118–138. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, P.; Zambrano, S.; Dabaghie, D.; Hu, Y.; Möller-Hackbarth, K.; Unnersjö-Jess, D.; Korkut, G.G.; Charrin, E.; Jeansson, M.; et al. Single-cell RNA Sequencing Reveals the Mesangial Identity and Species Diversity of Glomerular Cell Transcriptomes. Nat. Commun. 2021, 12, 2141. [Google Scholar] [CrossRef]
- Zhang, K.; Kan, H.; Mao, A.; Yu, F.; Geng, L.; Zhou, T.; Feng, L.; Ma, X. Integrated Single-Cell Transcriptomic Atlas of Human Kidney Endothelial Cells. J. Am. Soc. Nephrol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.-J.; Goldstein, L.; Chen, Y.-J.J.; Lee, J.; Webster, J.D.; Roose-Girma, M.; Paudyal, S.C.; Modrusan, Z.; Dey, A.; Shaw, A.S. Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury. J. Am. Soc. Nephrol. 2020, 31, 2341–2354. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Vernon, K.; Zhou, Y.; Marshall, J.L.; Alimova, M.; Zhang, F.; Slyper, M.; Waldman, J.; Montesinos, M.S.; Dionne, D.; et al. Obesity-Instructed Trem2High Macrophages Identified by Comparative Analysis of Diabetic Mouse and Human Kidney at Single Cell Resolution. bioRxiv 2021, bioRxiv:2021.05.30.446342. [Google Scholar]
- Wu, H.; Villalobos, R.G.; Yao, X.; Reilly, D.; Chen, T.; Rankin, M.; Myshkin, E.; Breyer, M.D.; Humphreys, B.D. Mapping the Single-Cell Transcriptomic Response of Murine Diabetic Kidney Disease to Therapies. Cell Metab. 2022, 34, 1064–1078.e6. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.C.; Wu, H.; Kirita, Y.; Uchimura, K.; Ledru, N.; Rennke, H.G.; Welling, P.A.; Waikar, S.S.; Humphreys, B.D. The Single-Cell Transcriptomic Landscape of Early Human Diabetic Nephropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 19619–19625. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep Surveying of Alternative Splicing Complexity in the Human Transcriptome by High-Throughput Sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wineberg, Y.; Bar-Lev, T.H.; Futorian, A.; Ben-Haim, N.; Armon, L.; Ickowicz, D.; Oriel, S.; Bucris, E.; Yehuda, Y.; Pode-Shakked, N.; et al. Single-Cell RNA Sequencing Reveals mRNA Splice Isoform Switching during Kidney Development. J. Am. Soc. Nephrol. 2020, 31, 2278–2291. [Google Scholar] [CrossRef] [PubMed]
- Stoehr, J.P.; Nadler, S.T.; Schueler, K.L.; Rabaglia, M.E.; Yandell, B.S.; Metz, S.A.; Attie, A.D. Genetic Obesity Unmasks Nonlinear Interactions between Murine Type 2 Diabetes Susceptibility Loci. Diabetes 2000, 49, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Hudkins, K.L.; Pichaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Mühlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. Btbr Ob/Ob Mutant Mice Model Progressive Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Schueler, K.M.; Keller, M.P.; Mitok, K.A.; Simonett, S.P.; Hudkins, K.L.; Mehrotra, K.; Graham, M.J.; Lee, R.G.; Alpers, C.E. Reversal of Hypertriglyceridemia in Diabetic Btbr Ob/Ob Mice Does Not Prevent Nephropathy. Mod. Pathol. 2021, 101, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Gembardt, F.; Bartaun, C.; Jarzebska, N.; Mayoux, E.; Todorov, V.T.; Hohenstein, B.; Hugo, C. The Sglt2 Inhibitor Empagliflozin Ameliorates Early Features of Diabetic Nephropathy in Btbr Ob/Ob Type 2 Diabetic Mice with and without Hypertension. Am. J. Physiol. Physiol. 2014, 307, F317–F325. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell. Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef]
- Vestweber, D. Ve-Cadherin: The Major Endothelial Adhesion Molecule Controlling Cellular Junctions and Blood Vessel Formation. Arter. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef]
- Park, J.; Shrestha, R.; Qiu, C.; Kondo, A.; Huang, S.; Werth, M.; Li, M.; Barasch, J.; Suszták, K. Single-Cell Transcriptomics of the Mouse Kidney Reveals Potential Cellular Targets of Kidney Disease. Science 2018, 360, 758–763. [Google Scholar] [CrossRef]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular Localization and Expression of Angiotensin-Converting Enzyme 2 and Angiotensin-Converting Enzyme: Implications for Albuminuria in Diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef]
- Muris, T. Consortium, coordination Overall, coordination Logistical, collection Organ, processing, preparation Library, sequencing, analysis Computational data, annotation Cell type, group Writing, group Supplemental text writing, and investigators Principal. Single-Cell Transcriptomics of 20 Mouse Organs Creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar]
- Moffat, D.B. The Fine Structure of the Blood Vessels of the Renal Medulla with Particular Reference to the Control of the Medullary Circulation. J. Ultrastruct. Res. 1967, 19, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W. Fenestrated Glomerular Capillaries Are Unique. J. Am. Soc. Nephrol. 2008, 19, 1439–1440. [Google Scholar] [CrossRef]
- Stolz, D.B.; Sims-Lucas, S. Unwrapping the Origins and Roles of the Renal Endothelium. Pediatr. Nephrol. 2014, 30, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.-V.; Kubitza, M.; Palade, G.E. Pv-1 Is a Component of the Fenestral and Stomatal Diaphragms in Fenestrated Endothelia. Proc. Natl. Acad. Sci. USA 1999, 96, 13203–13207. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Horita, S.; Takahashi, T.; Tanabe, K.; Fuchinoue, S.; Teraoka, S.; Hattori, M.; Yamaguchi, Y. Glomerular Expression of Plasmalemmal Vesicle-Associated Protein-1 in Patients with Transplant Glomerulopathy. Am. J. Transplant. 2007, 7, 1954–1960. [Google Scholar] [CrossRef]
- Young, M.D.; Mitchell, T.; Braga, F.A.V.; Tran, M.G.; Stewart, B.J.; Ferdinand, J.; Collord, G.; Botting, R.A.; Popescu, D.-M.; Loudon, K.W.; et al. Single-Cell Transcriptomes from Human Kidneys Reveal the Cellular Identity of Renal Tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Pattaro, C.; Teumer, A.; Gorski, M.; Chu, A.Y.; Li, M.; Mijatovic, V.; Garnaas, M.; Tin, A.; Sorice, R.; Li, Y.; et al. Genetic Associations at 53 Loci Highlight Cell Types and Biological Pathways Relevant for Kidney Function. Nat. Commun. 2016, 7, 10023. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.; Otto, E.A.; Hoover, P.; Eddy, S.; Mariani, L.; Godfrey, B.; Berthier, C.C.; Eichinger, F.; Subramanian, L.; Harder, J.; et al. Single Cell Transcriptomics Identifies Focal Segmental Glomerulosclerosis Remission Endothelial Biomarker. J. Clin. Investig. 2020, 5, e133267. [Google Scholar] [CrossRef] [PubMed]
- Herschman, H.R. Primary Response Genes Induced by Growth Factors and Tumor Promoters. Annu. Rev. Biochem. 1991, 60, 281–319. [Google Scholar] [CrossRef]
- Fowler, T.; Sen, R.; Roy, A.L. Regulation of Primary Response Genes. Mol. Cell 2011, 44, 348–360. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; Betsholtz, C. Single-Cell Mrna Sequencing of the Mouse Brain Vasculature. Methods Mol. Biol. 2018, 1846, 309–324. [Google Scholar] [PubMed]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar]
- Stewart, B.J.; Ferdinand, J.; Young, M.D.; Mitchell, T.; Loudon, K.W.; Riding, A.M.; Richoz, N.; Frazer, G.L.; Staniforth, J.U.; Braga, F.A.V.; et al. Spatiotemporal Immune Zonation of the Human Kidney. Science 2019, 365, 1461–1466. [Google Scholar] [CrossRef]
- Taylor, M.S.; Francis, M. Decoding Dynamic Ca2+ Signaling in the Vascular Endothelium. Front. Physiol. 2014, 5, 447. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.A.; Tare, M.; Parkington, H.C. Endothelial Potassium Channels, Endothelium-Dependent Hyperpolarization and the Regulation of Vascular Tone in Health and Disease. Clin. Exp. Pharmacol. Physiol. 2004, 31, 641–649. [Google Scholar] [CrossRef]
- Schmidt, K.; de Wit, C. Endothelium-Derived Hyperpolarizing Factor and Myoendothelial Coupling: The In Vivo Perspective. Front. Physiol. 2020, 11, 602930. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Kurtz, A. Distribution and functional relevance of connexins in renin-producing cells. Pflugers Arch. Eur. J. Physiol. 2013, 465, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Marsen, T.A.; Simonson, M.S.; Dunn, M.J. Roles of calcium and kinases in regulation of thrombin-stimulated preproendothelin-1 transcription. Am. J. Physiol. 1996, 271, H1918–H1925. [Google Scholar] [CrossRef]
- Altmann, J.B.; Yan, G.; Meeks, J.F.; Abood, M.E.; Brailoiu, E.; Brailoiu, G.C. G protein-coupled estrogen receptor-mediated effects on cytosolic calcium and nanomechanics in brain microvascular endothelial cells. J. Neurochem. 2015, 133, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Vallon, V. Reduced Renal Calcium Excretion in the Absence of Sclerostin Expression: Evidence for a Novel Calcium-Regulating Bone Kidney Axis. J. Am. Soc. Nephrol. 2014, 25, 2159–2168. [Google Scholar] [CrossRef]
- Bach, L.A. The insulin-like growth factor system in kidney disease and hypertension. Curr. Opin. Nephrol. Hypertens. 2012, 21, 86–91. [Google Scholar] [CrossRef]
- Bossolasco, M.; Veillette, F.; Bertrand, R.; Mes-Masson, A.-M. Human TDE1, a TDE1/TMS family member, inhibits apoptosis in vitro and stimulates in vivo tumorigenesis. Oncogene 2006, 25, 4549–4558. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Xu, L.; Hiller, S.; Kim, H.-S.; Nickeleit, V.; James, L.R.; Maeda, N. Reduced Expression of Lipoic Acid Synthase Accelerates Diabetic Nephropathy. J. Am. Soc. Nephrol. 2012, 23, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yan, T.; Xiong, C.; Chang, M.; Gao, Q.; Yao, S.; Wu, W.; Yi, X.; Xu, G. Overexpression of Lipoic Acid Synthase Gene Alleviates Diabetic Nephropathy of LeprDb/Db Mice. BMJ Open Diabetes Res. Care 2021, 9, e002260. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Oltean, S. Alternative Splicing in CKD. J. Am. Soc. Nephrol. 2016, 27, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jeong, K.; Li, J.; Murphy, J.M.; Vukadin, L.; Stone, J.K.; Richard, A.; Tran, J.; Gillespie, G.Y.; Flemington, E.K.; et al. Son Drives Oncogenic Rna Splicing in Glioblastoma by Regulating Ptbp1/Ptbp2 Switching and Rbfox2 Activity. Nat. Commun. 2021, 12, 5551. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wu, L.; Wang, Q.; Zhao, X.; Chen, T.; Yin, C.; Yan, L.; Yang, X. Insulin-Like Growth Factor 2 Mrna-Binding Protein 2-Regulated Alternative Splicing of Nuclear Factor 1 C-Type Causes Excessive Granulosa Cell Proliferation in Polycystic Ovary Syndrome. Cell Prolif. 2022, 55, e13216. [Google Scholar] [CrossRef] [PubMed]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V. and Segerstolpe, Å. Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef]
- Ericsson, A.; Tonelius, P.; Lal, M.; Sabirsh, A.; Böttcher, G.; William-Olsson, L.; Strömstedt, M.; Johansson, C.; Hyberg, G.; Tapani, S.; et al. The Effects of Dual Pparalpha/Gamma Agonism Compared with Ace Inhibition in the Btbrob/Ob Mouse Model of Diabetes and Diabetic Nephropathy. Physiol. Rep. 2017, 5, e13186. [Google Scholar] [CrossRef]
- Granqvist, A.B.; Ericsson, A.; Sanchez, J.; Tonelius, P.; William-Olsson, L.; Dahlqvist, U.; Andersson, A.-K.; Tomic, T.T.; Hudkins, K.L.; Alpers, C.E.; et al. High-Protein Diet Accelerates Diabetes and Kidney Disease in the Btbrob/Ob Mouse. Am. J. Physiol. Physiol. 2020, 318, F763–F771. [Google Scholar] [CrossRef]
- Fan, Y.; Yi, Z.; D’agati, V.D.; Sun, Z.; Zhong, F.; Zhang, W.; Wen, J.; Zhou, T.; Li, Z.; He, L.; et al. Comparison of Kidney Transcriptomic Profiles of Early and Advanced Diabetic Nephropathy Reveals Potential New Mechanisms for Disease Progression. Diabetes 2019, 68, 2301–2314. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Kim, Y.; Caramori, M.L.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Walker, P.C.; Mauer, M. Diabetic Nephropathy Is Associated with Gene Expression Levels of Oxidative Phosphorylation and Related Pathways. Diabetes 2006, 55, 1826–1831. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.B.; Zhi, H.; Zhang, X.Y.; Liang, J.W.; He, J.; Su, C.; Xia, W.H.; Zhang, G.X.; Tao, J. Mitochondrial Dysfunction-Mediated Decline in Angiogenic Capacity of Endothelial Progenitor Cells Is Associated with Capillary Rarefaction in Patients with Hypertension Via Downregulation of Cxcr4/Jak2/Sirt5 Signaling. EBioMedicine 2019, 42, 64–75. [Google Scholar] [CrossRef]
- Kasinath, B.S.; Mariappan, M.M.; Sataranatarajan, K.; Lee, M.J.; Choudhury, G.G.; Feliers, D. Novel Mechanisms of Protein Synthesis in Diabetic Nephropathy—Role of mRNA Translation. Rev. Endocr. Metab. Disord. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.B.; Duan, C. Igf-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, Z.; Mokoena, F.; Hull, R. Abnormalities in Alternative Splicing in Diabetes: Therapeutic Targets. J. Mol. Endocrinol. 2017, 59, R93–R107. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, V.A.; Fulton, J.R.; Margariti, A. Alternative Splicing: A Key Mediator of Diabetic Vasculopathy. Genes 2021, 12, 1332. [Google Scholar] [CrossRef]
- Zhao, H.; Kong, H.; Wang, B.; Wu, S.; Chen, T.; Cui, Y. RNA-Binding Proteins and Alternative Splicing Genes Are Coregulated in Human Retinal Endothelial Cells Treated with High Glucose. J. Diabetes Res. 2022, 2022, 7680513. [Google Scholar] [CrossRef]
- Hishikawa, A.; Hayashi, K.; Yoshimoto, N.; Nakamichi, R.; Homma, K.; Itoh, H. DNA Damage and Expression of DNA Methylation Modulators in Urine-Derived Cells of Patients with Hypertension and Diabetes. Sci. Rep. 2020, 10, 3377. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, A.; Hayashi, K.; Abe, T.; Kaneko, M.; Yokoi, H.; Azegami, T.; Nakamura, M.; Yoshimoto, N.; Kanda, T.; Sakamaki, Y.; et al. Decreased KAT5 Expression Impairs DNA Repair and Induces Altered DNA Methylation in Kidney Podocytes. Cell Rep. 2019, 26, 1318–1332.e4. [Google Scholar] [CrossRef] [PubMed]
- Arzalluz-Luque, A.; Conesa, A. Single-Cell Rnaseq for the Study of Isoforms-How Is That Possible? Genome Biol. 2018, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Pitulescu, M.E.; Schmidt, I.; Benedito, R.; Adams, R.H. Inducible Gene Targeting in the Neonatal Vasculature and Analysis of Retinal Angiogenesis in Mice. Nat. Protoc. 2010, 5, 1518–1534. [Google Scholar] [CrossRef]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; A Zariwala, H.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A Robust and High-Throughput Cre Reporting and Characterization System for the Whole Mouse Brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Faridani, O.R.; Björklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length Rna-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. Tophat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using Umap. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Kaminow, B.; Yunusov, D.; Dobin, A. Starsolo: Accurate, Fast and Versatile Mapping/Quantification of Single-Cell and Single-Nucleus Rna-Seq Data. bioRxiv 2021, bioRxiv:2021.05.05.442755. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.-X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, A.-X.; Jeansson, M.; He, L.; Wigge, L.; Tonelius, P.; Tati, R.; Cederblad, L.; Muhl, L.; Uhrbom, M.; Liu, J.; et al. Renal Endothelial Single-Cell Transcriptomics Reveals Spatiotemporal Regulation and Divergent Roles of Differential Gene Transcription and Alternative Splicing in Murine Diabetic Nephropathy. Int. J. Mol. Sci. 2024, 25, 4320. https://doi.org/10.3390/ijms25084320
Zhou A-X, Jeansson M, He L, Wigge L, Tonelius P, Tati R, Cederblad L, Muhl L, Uhrbom M, Liu J, et al. Renal Endothelial Single-Cell Transcriptomics Reveals Spatiotemporal Regulation and Divergent Roles of Differential Gene Transcription and Alternative Splicing in Murine Diabetic Nephropathy. International Journal of Molecular Sciences. 2024; 25(8):4320. https://doi.org/10.3390/ijms25084320
Chicago/Turabian StyleZhou, Alex-Xianghua, Marie Jeansson, Liqun He, Leif Wigge, Pernilla Tonelius, Ramesh Tati, Linda Cederblad, Lars Muhl, Martin Uhrbom, Jianping Liu, and et al. 2024. "Renal Endothelial Single-Cell Transcriptomics Reveals Spatiotemporal Regulation and Divergent Roles of Differential Gene Transcription and Alternative Splicing in Murine Diabetic Nephropathy" International Journal of Molecular Sciences 25, no. 8: 4320. https://doi.org/10.3390/ijms25084320