
Evolutionary Changes in Primate Glutamate Dehydrogenases 1 and 2 Influence the Protein Regulation by Ligands, Targeting and Posttranslational Modifications


Abstract
1. Introduction
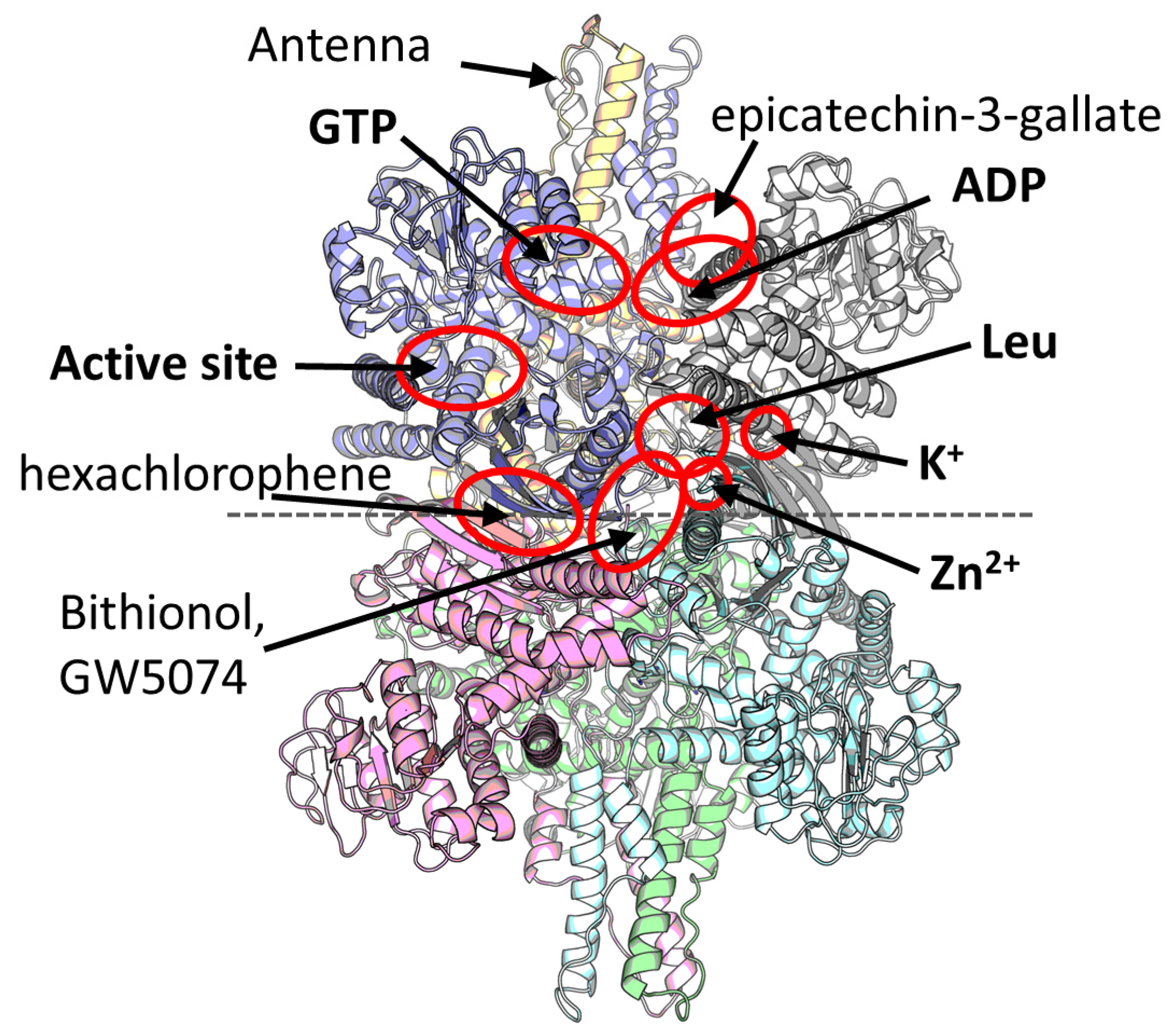
2. Multiple Ligand Binding Sites of the Mammalian GDH

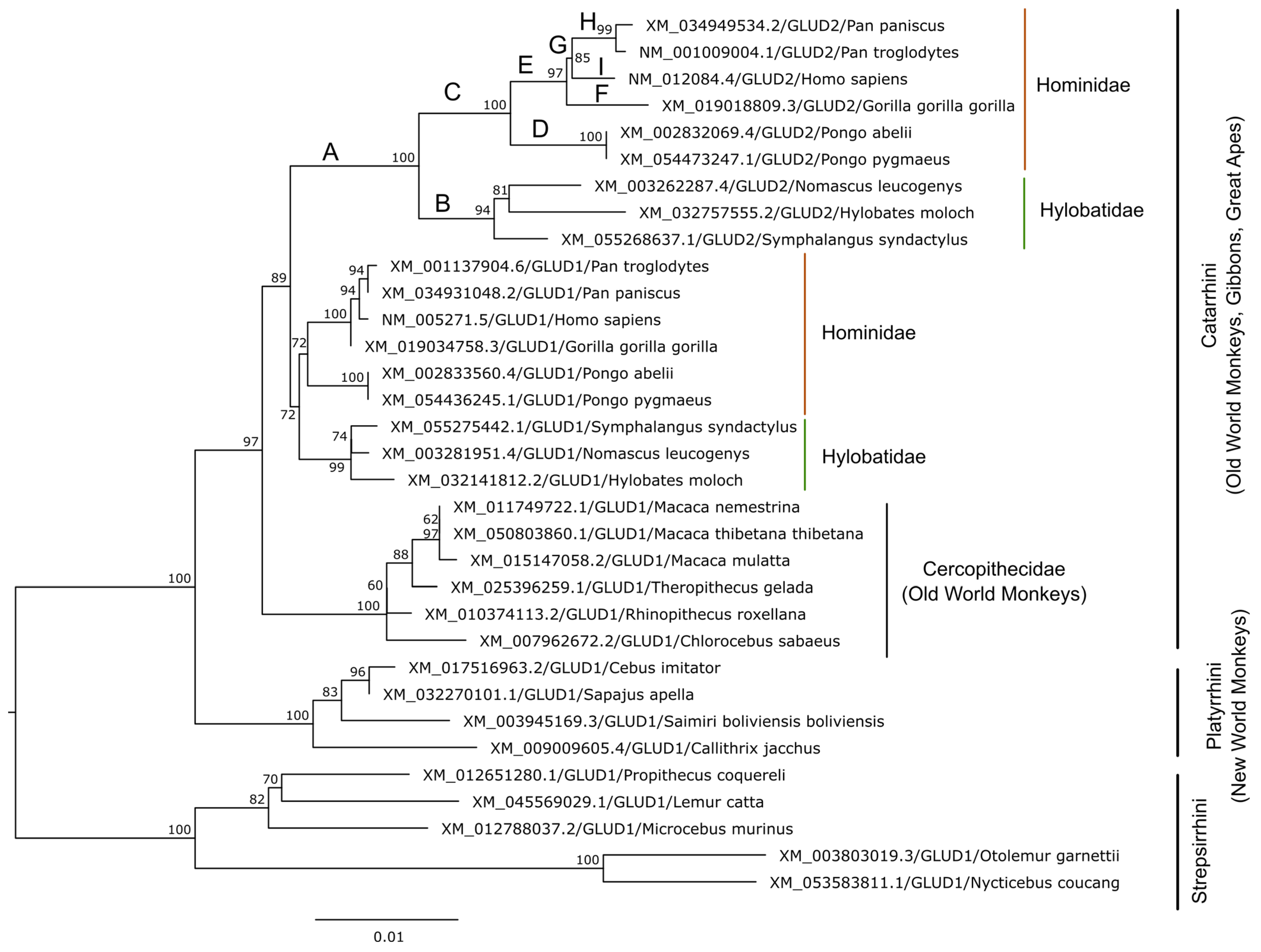
3. Evolution of Glutamate Dehydrogenases in Hominoids
3.1. GDH Duplication in Primates
3.2. Specific Properties and Expression of GLUD2
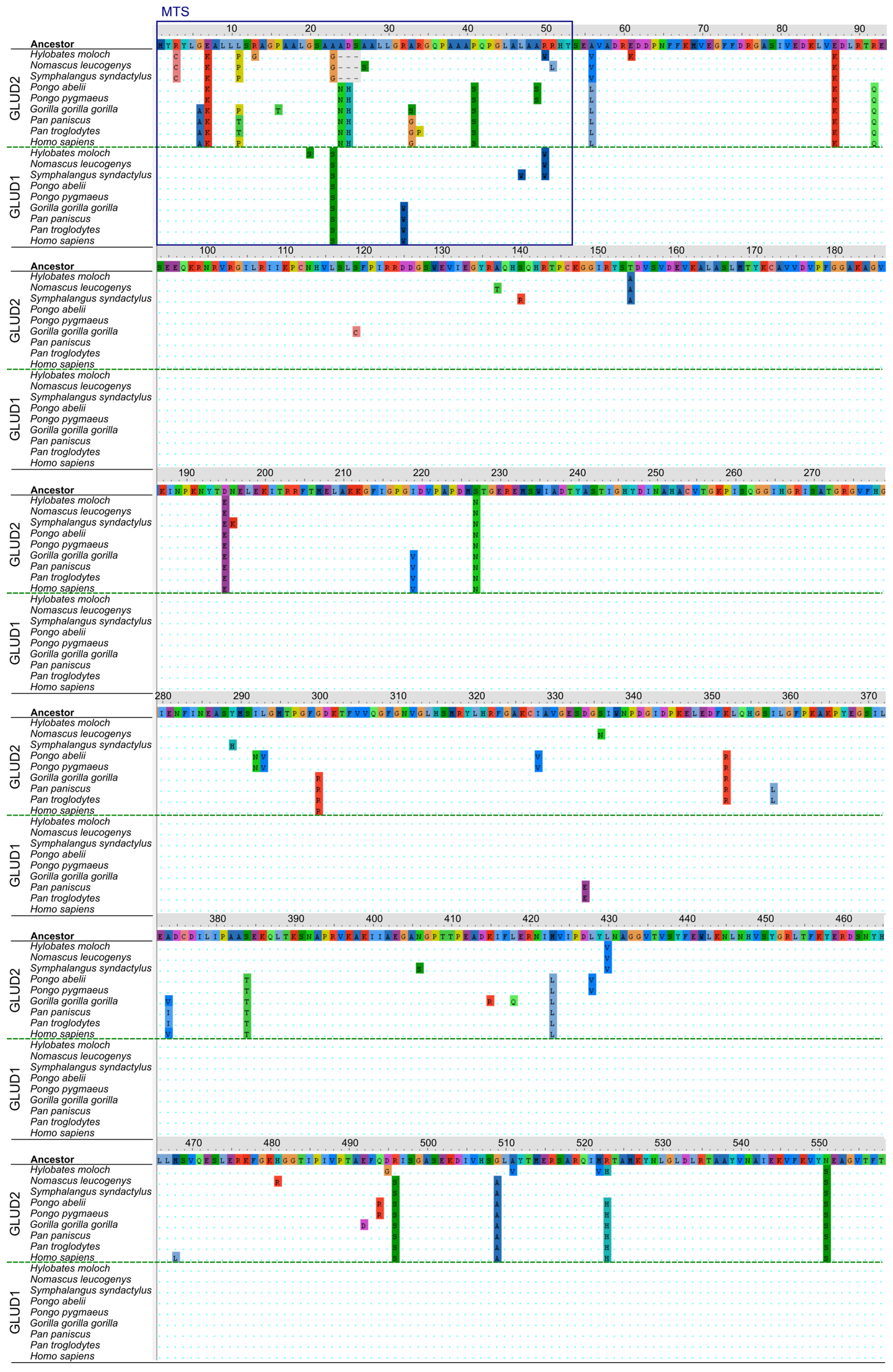
3.3. Structural Analysis of Mutation Sites Occuring upon Evolution of “Mature” GLUD2
3.4. Analysis of Mutation Sites Occuring in the MTS of Ape GDH
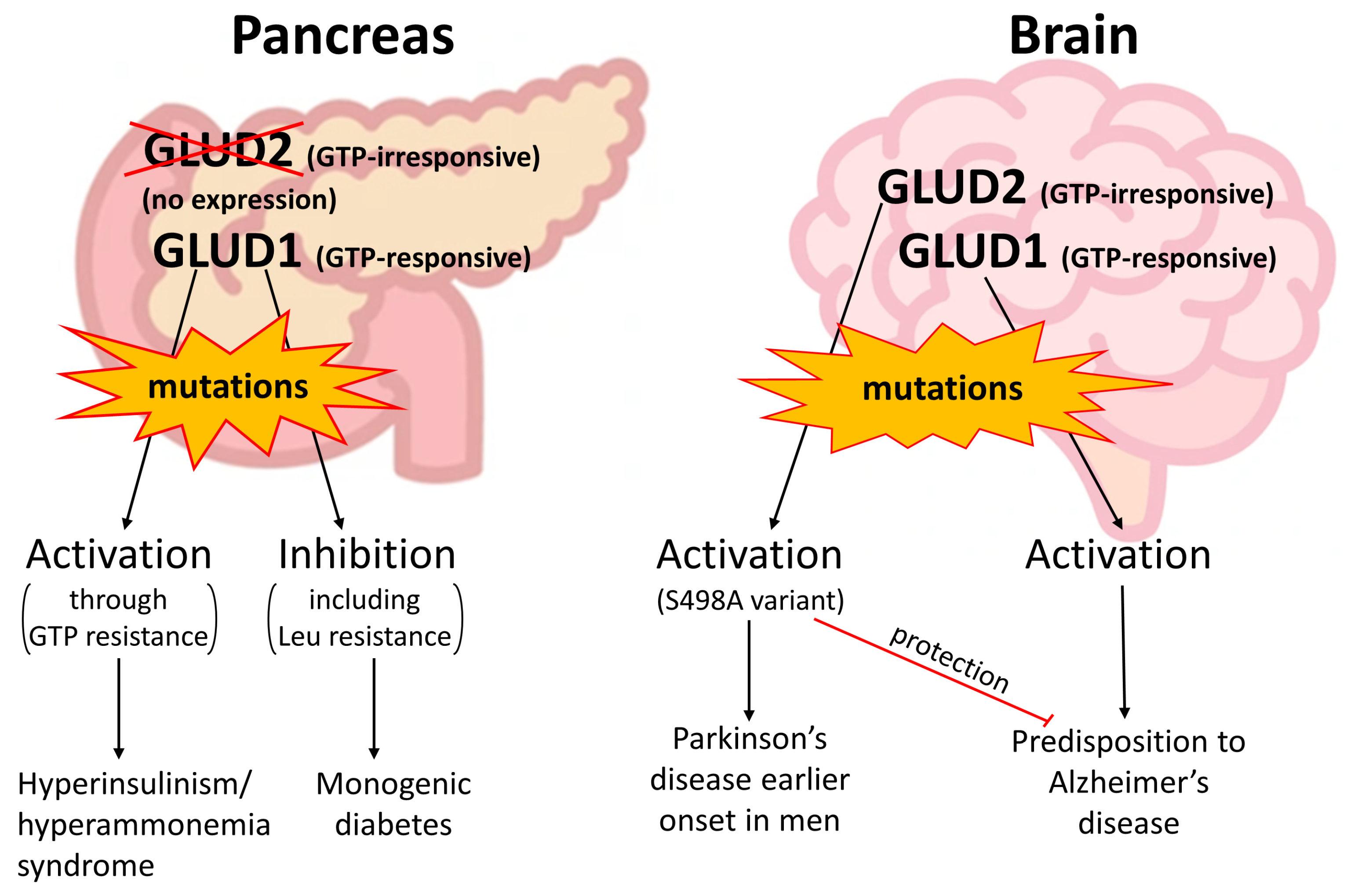
4. Clinically Relevant Genetic Variants of GDH Revealed in Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Allele Frequency | Submitted Condition | Structural Data | References |
|---|---|---|---|---|
| GLUD1 | ||||
| A18V | – | HI/HA | Modified MTS sequence | – |
| G35E | – | HI/HA; MD | Modified MTS sequence | – |
| Q36R | 0.00280 | HI/HA | Modified MTS sequence | – |
| H52N | 0.00002 | HI/HA | Modified MTS sequence | – |
| D126N | 0.00839 | HI/HA; MD | Mutation at the entrance to the Leu site, hexachlorophene and GW5074/bithionol sites. May affect regulation by Leu. | – |
| R274C | – | HI/HA | A GTP-binding residue. Decreases GTP inhibition. | [115,116,117,118,119] |
| R322H | – | HI/HA | A GTP-binding residue. Decreases GTP inhibition. | [115,116,117] |
| S357F | – | HI/HA | A phosphorylatable [120] residue at the entrance to the NADPH site. | – |
| D375N | 0.00003 | Uncertain | Mutation at the surface of the NAD domain. | – |
| P489R | – | HI/HA | A residue between two helices of the antenna. Should affect allosteric regulation of GDH. | – |
| S498L | – | HI/HA | A residue of the antenna, small helix. Should affect allosteric regulation of GDH (see S498A below). | [102,107,116,118,121,122,123,124] |
| H507Y | – | HI/HA; FH | A GTP-binding residue. Decreases GTP inhibition. | [102,125] |
| R523H | – | HI/HA | A pivot helix residue, substituted in GLUD2. May slightly affect ADP activation [39]. | – |
| N551S | – | HI/HA | A residue in proximity to the C-terminal/Leu site, substituted in GLUD2. May influence Leu activation and hexachlorophene and inhibition by GW5074 or bithionol. | – |
| GLUD2 | ||||
| S498A | 0.03285 | PD | A residue of the antenna, small helix. Activity and allosteric regulation by estrogens are affected [112]. | [112] |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GDH | glutamate dehydrogenase, |
| MTS | mitochondrial targeting sequence, |
| HI/HA | hyperinsulinism/hyperammonemia syndrome. |
References
- Hohnholt, M.C.; Andersen, V.H.; Andersen, J.V.; Christensen, S.K.; Karaca, M.; Maechler, P.; Waagepetersen, H.S. Glutamate dehydrogenase is essential to sustain neuronal oxidative energy metabolism during stimulation. J. Cereb. Blood Flow. Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 1754–1768. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Plaitakis, A. The role of glutamate dehydrogenase in mammalian ammonia metabolism. Neurotox. Res. 2012, 21, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Marin-Hernandez, A.; Gallardo-Perez, J.C.; Pacheco-Velazquez, S.C.; Robledo-Cadena, D.X.; Padilla-Flores, J.A.; Saavedra, E.; Rodriguez-Enriquez, S. Physiological Role of Glutamate Dehydrogenase in Cancer Cells. Front. Oncol. 2020, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Stanley, C.A. Untangling the glutamate dehydrogenase allosteric nightmare. Trends Biochem. Sci. 2008, 33, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Minambres, B.; Olivera, E.R.; Jensen, R.A.; Luengo, J.M. A new class of glutamate dehydrogenases (GDH). Biochemical and genetic characterization of the first member, the AMP-requiring NAD-specific GDH of Streptomyces clavuligerus. J. Biol. Chem. 2000, 275, 39529–39542. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, M.; Melero, R.; Huet, C.; López-Alonso, J.P.; Delgado, S.; Dodu, A.; Bruch, E.M.; Abriata, L.A.; Alzari, P.M.; Valle, M.; et al. 3D architecture and structural flexibility revealed in the subfamily of large glutamate dehydrogenases by a mycobacterial enzyme. Commun. Biol. 2021, 4, 684. [Google Scholar] [CrossRef] [PubMed]
- Munn, E.A. Structure of oligomeric and polymeric forms of ox liver glutamate dehydrogenase examined by electron microscopy. Biochim. Biophys. Acta 1972, 285, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Schmidt, T.; Fang, J.; Stanley, C.A.; Smith, T.J. Structural studies on ADP activation of mammalian glutamate dehydrogenase and the evolution of regulation. Biochemistry 2003, 42, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Talal, N.; Tomkins, G.M. Allosteric Properties of Glutamate Dehydrogenases from Different Sources. Science 1964, 146, 1309–1311. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Bunik, V.I.; Bruch, E.M.; Bellinzoni, M. Structural Basis for the Binding of Allosteric Activators Leucine and ADP to Mammalian Glutamate Dehydrogenase. Int. J. Mol. Sci. 2022, 23, 11306. [Google Scholar] [CrossRef]
- Shashidharan, P.; Michaelidis, T.M.; Robakis, N.K.; Kresovali, A.; Papamatheakis, J.; Plaitakis, A. Novel human glutamate dehydrogenase expressed in neural and testicular tissues and encoded by an X-linked intronless gene. J. Biol. Chem. 1994, 269, 16971–16976. [Google Scholar] [CrossRef]
- Burki, F.; Kaessmann, H. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat. Genet. 2004, 36, 1061–1063. [Google Scholar] [CrossRef]
- Zhang, Z.; Carriero, N.; Gerstein, M. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet 2004, 20, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Deloukas, P.; Dauwerse, J.G.; Moschonas, N.K.; van Ommen, G.J.; van Loon, A.P. Three human glutamate dehydrogenase genes (GLUD1, GLUDP2, and GLUDP3) are located on chromosome 10q, but are not closely physically linked. Genomics 1993, 17, 676–681. [Google Scholar] [CrossRef]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Litso, I.; Plaitakis, A.; Fadouloglou, V.E.; Providaki, M.; Kokkinidis, M.; Zaganas, I. Structural Evolution of Primate Glutamate Dehydrogenase 2 as Revealed by In Silico Predictions and Experimentally Determined Structures. Biomolecules 2023, 14, 22. [Google Scholar] [CrossRef]
- Kotzamani, D.; Plaitakis, A. Alpha helical structures in the leader sequence of human GLUD2 glutamate dehydrogenase responsible for mitochondrial import. Neurochem. Int. 2012, 61, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Katrakili, N.; Plaitakis, A.; Tokatlidis, K. Import of a major mitochondrial enzyme depends on synergy between two distinct helices of its presequence. Biochem. J. 2016, 473, 2813–2829. [Google Scholar] [CrossRef]
- Rosso, L.; Marques, A.C.; Reichert, A.S.; Kaessmann, H. Mitochondrial targeting adaptation of the hominoid-specific glutamate dehydrogenase driven by positive Darwinian selection. PLoS Genet 2008, 4, e1000150. [Google Scholar] [CrossRef]
- Peterson, P.E.; Smith, T.J. The structure of bovine glutamate dehydrogenase provides insights into the mechanism of allostery. Structure 1999, 7, 769–782. [Google Scholar] [CrossRef]
- Smith, T.J.; Peterson, P.E.; Schmidt, T.; Fang, J.; Stanley, C.A. Structures of bovine glutamate dehydrogenase complexes elucidate the mechanism of purine regulation. J. Mol. Biol. 2001, 307, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Yielding, K.L.; Tomkins, G.M. An Effect of L-Leucine and Other Essential Amino Acids on the Structure and Activity of Glutamic Dehydrogenase. Proc. Natl. Acad. Sci. USA 1961, 47, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; Kmiotek, E. Regulation of glutamate dehydrogenase by palmitoyl-coenzyme A. Arch. Biochem. Biophys. 1981, 212, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.F.; Foster, D.S. The absence of zinc in bovine liver glutamate dehydrogenase. J. Biol. Chem. 1970, 245, 6190–6195. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.; Powell, L.; Sinanan, L.; Neal, J.; Li, M.; Smith, T.; Bell, E. A novel mechanism of V-type zinc inhibition of glutamate dehydrogenase results from disruption of subunit interactions necessary for efficient catalysis. FEBS J. 2011, 278, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Smith, C.J.; Walker, M.T.; Smith, T.J. Novel Inhibitors Complexed with Glutamate Dehydrogenase. J. Biol. Chem. 2009, 284, 22988–23000. [Google Scholar] [CrossRef]
- Li, C.; Li, M.; Chen, P.; Narayan, S.; Matschinsky, F.M.; Bennett, M.J.; Stanley, C.A.; Smith, T.J. Green tea polyphenols control dysregulated glutamate dehydrogenase in transgenic mice by hijacking the ADP activation site. J. Biol. Chem. 2011, 286, 34164–34174. [Google Scholar] [CrossRef] [PubMed]
- Borompokas, N.; Papachatzaki, M.M.; Kanavouras, K.; Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Plaitakis, A. Estrogen modification of human glutamate dehydrogenases is linked to enzyme activation state. J. Biol. Chem. 2010, 285, 31380–31387. [Google Scholar] [CrossRef]
- Spanaki, C.; Zaganas, I.; Kounoupa, Z.; Plaitakis, A. The complex regulation of human glud1 and glud2 glutamate dehydrogenases and its implications in nerve tissue biology. Neurochem. Int. 2012, 61, 470–481. [Google Scholar] [CrossRef]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Di Salvo, M.L.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583. [Google Scholar] [CrossRef]
- Li, M.; Allen, A.; Smith, T.J. High Throughput Screening Reveals Several New Classes of Glutamate Dehydrogenase Inhibitors. Biochemistry 2007, 46, 15089–15102. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.; Kwagh, J.; Fang, J.; Stanley, C.A.; Smith, T.J. Evolution of glutamate dehydrogenase regulation of insulin homeostasis is an example of molecular exaptation. Biochemistry 2004, 43, 14431–14443. [Google Scholar] [CrossRef] [PubMed]
- Hoffpauir, Z.A.; Sherman, E.; Smith, T.J. Dissecting the Antenna in Human Glutamate Dehydrogenase: Understanding Its Role in Subunit Communication and Allosteric Regulation. Biochemistry 2019, 58, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Hamza, M.A.; Engel, P.C. Homotropic allosteric control in clostridial glutamate dehydrogenase: Different mechanisms for glutamate and NAD+? FEBS Lett. 2008, 582, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.E.; Engel, P.C.; Parker, D.M. Functional studies of a glutamate dehydrogenase with known three-dimensional structure: Steady-state kinetics of the forward and reverse reactions catalysed by the NAD(+)-dependent glutamate dehydrogenase of Clostridium symbiosum. Biochim. Biophys. Acta 1991, 1115, 123–130. [Google Scholar] [CrossRef]
- Tomita, T.; Kuzuyama, T.; Nishiyama, M. Structural basis for leucine-induced allosteric activation of glutamate dehydrogenase. J. Biol. Chem. 2011, 286, 37406–37413. [Google Scholar] [CrossRef]
- LeJohn, H.B.; Cameron, L.E.; Yang, B.; Rennie, S.L. Molecular characterization of an NAD-specific glutamate dehydrogenase gene inducible by L-glutamine. Antisense gene pair arrangement with L-glutamine-inducible heat shock 70-like protein gene. J. Biol. Chem. 1994, 269, 4523–4531. [Google Scholar] [CrossRef] [PubMed]
- Dimovasili, C.; Fadouloglou, V.E.; Kefala, A.; Providaki, M.; Kotsifaki, D.; Kanavouras, K.; Sarrou, I.; Plaitakis, A.; Zaganas, I.; Kokkinidis, M. Crystal structure of glutamate dehydrogenase 2, a positively selected novel human enzyme involved in brain biology and cancer pathophysiology. J. Neurochem. 2021, 157, 802–815. [Google Scholar] [CrossRef]
- Plaitakis, A.; Metaxari, M.; Shashidharan, P. Nerve tissue-specific (GLUD2) and housekeeping (GLUD1) human glutamate dehydrogenases are regulated by distinct allosteric mechanisms: Implications for biologic function. J. Neurochem. 2000, 75, 1862–1869. [Google Scholar] [CrossRef]
- Shashidharan, P.; Clarke, D.D.; Ahmed, N.; Moschonas, N.; Plaitakis, A. Nerve tissue-specific human glutamate dehydrogenase that is thermolabile and highly regulated by ADP. J. Neurochem. 1997, 68, 1804–1811. [Google Scholar] [CrossRef]
- Dimovasili, C.; Aschner, M.; Plaitakis, A.; Zaganas, I. Differential interaction of hGDH1 and hGDH2 with manganese: Implications for metabolism and toxicity. Neurochem. Int. 2015, 88, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Cario, E.; Jung, S.; Harder D’Heureuse, J.; Schulte, C.; Sturm, A.; Wiedenmann, B.; Goebell, H.; Dignass, A.U. Effects of exogenous zinc supplementation on intestinal epithelial repair in vitro. Eur. J. Clin. Investig. 2000, 30, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.M.; Kim, E.A.; Yang, S.J.; Choi, S.Y.; Cho, S.W.; Huh, J.W. Amino acid changes within antenna helix are responsible for different regulatory preferences of human glutamate dehydrogenase isozymes. J. Biol. Chem. 2007, 282, 19510–19517. [Google Scholar] [CrossRef] [PubMed]
- Kanavouras, K.; Mastorodemos, V.; Borompokas, N.; Spanaki, C.; Plaitakis, A. Properties and molecular evolution of human GLUD2 (neural and testicular tissue-specific) glutamate dehydrogenase. J. Neurosci. Res. 2007, 85, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, E.A. Human intronless genes: Functional groups, associated diseases, evolution, and mRNA processing in absence of splicing. Biochem. Biophys. Res. Commun. 2012, 424, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vockel, M.; Riera-Escamilla, A.; Tuttelmann, F.; Krausz, C. The X chromosome and male infertility. Hum. Genet. 2021, 140, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Zaganas, I.; Kleopa, K.A.; Plaitakis, A. Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J. Biol. Chem. 2010, 285, 16748–16756. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Expression of human GLUD1 and GLUD2 glutamate dehydrogenases in steroid producing tissues. Mol. Cell. Endocrinol. 2015, 415, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Mear, L.; Zhong, W.; Digre, A.; Katona, B.; Sjostedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Barisciano, G.; Colangelo, T.; Rosato, V.; Muccillo, L.; Taddei, M.L.; Ippolito, L.; Chiarugi, P.; Galgani, M.; Bruzzaniti, S.; Matarese, G.; et al. miR-27a is a master regulator of metabolic reprogramming and chemoresistance in colorectal cancer. Br. J. Cancer 2020, 122, 1354–1366. [Google Scholar] [CrossRef]
- Rosato, V.; Barisciano, G.; Muccillo, L.; Colangelo, T.; Bianchi, F.; Mazzoccoli, G.; Colantuoni, V.; Sabatino, L. miR-27a-3p Is a Master Regulator of Metabolic Reprogramming in Colorectal Cancer. Available online: https://www.nanaonlus.org/wp-content/uploads/2018/07/Poster-Rosato-Valeria.pdf (accessed on 7 March 2024).
- Mateus, I.; Feijo, M.; Espinola, L.M.; Vaz, C.V.; Correia, S.; Socorro, S. Glucose and glutamine handling in the Sertoli cells of transgenic rats overexpressing regucalcin: Plasticity towards lactate production. Sci. Rep. 2018, 8, 10321. [Google Scholar] [CrossRef]
- Broeder, J.A.; Smith, C.H.; Moe, A.J. Glutamate oxidation by trophoblasts in vitro. Am. J. Physiol. 1994, 267, C189–C194. [Google Scholar] [CrossRef] [PubMed]
- Vedelek, V.; Vedelek, B.; Lorincz, P.; Juhasz, G.; Sinka, R. A comparative analysis of fruit fly and human glutamate dehydrogenases in Drosophila melanogaster sperm development. Front. Cell Dev. Biol. 2023, 11, 1281487. [Google Scholar] [CrossRef]
- Descalzi, G.; Gao, V.; Steinman, M.Q.; Suzuki, A.; Alberini, C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019, 2, 247. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Kharbanda, S.; Peale, F.; Deng, Y.; Daemen, A.; Forrest, W.F.; Kwong, M.; Hedehus, M.; Hatzivassiliou, G.; et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc. Natl. Acad. Sci. USA 2014, 111, 14217–14222. [Google Scholar] [CrossRef] [PubMed]
- Tiburcio, P.D.B.; Gillespie, D.L.; Jensen, R.L.; Huang, L.E. Extracellular glutamate and IDH1(R132H) inhibitor promote glioma growth by boosting redox potential. J. Neuro-Oncol. 2020, 146, 427–437. [Google Scholar] [CrossRef]
- Franceschi, S.; Corsinovi, D.; Lessi, F.; Tantillo, E.; Aretini, P.; Menicagli, M.; Scopelliti, C.; Civita, P.; Pasqualetti, F.; Naccarato, A.G.; et al. Mitochondrial enzyme GLUD2 plays a critical role in glioblastoma progression. EBioMedicine 2018, 37, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.D.; Lykke, K.; Bryk, J.; Stridh, M.H.; Zaganas, I.; Skytt, D.M.; Schousboe, A.; Bak, L.K.; Enard, W.; Paabo, S.; et al. Expression of the human isoform of glutamate dehydrogenase, hGDH2, augments TCA cycle capacity and oxidative metabolism of glutamate during glucose deprivation in astrocytes. Glia 2017, 65, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, S.; Jiang, X.; Bryk, J.; Naumann, R.; Enard, W.; Tomita, M.; Sugimoto, M.; Khaitovich, P.; Paabo, S. Mice carrying a human GLUD2 gene recapitulate aspects of human transcriptome and metabolome development. Proc. Natl. Acad. Sci. USA 2016, 113, 5358–5363. [Google Scholar] [CrossRef]
- Spanaki, C.; Kotzamani, D.; Kleopa, K.; Plaitakis, A. Evolution of GLUD2 Glutamate Dehydrogenase Allows Expression in Human Cortical Neurons. Mol. Neurobiol. 2016, 53, 5140–5148. [Google Scholar] [CrossRef]
- Bao, X.; Pal, R.; Hascup, K.N.; Wang, Y.; Wang, W.T.; Xu, W.; Hui, D.; Agbas, A.; Wang, X.; Michaelis, M.L.; et al. Transgenic expression of Glud1 (glutamate dehydrogenase 1) in neurons: In vivo model of enhanced glutamate release, altered synaptic plasticity, and selective neuronal vulnerability. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 13929–13944. [Google Scholar] [CrossRef]
- Bunik, V.; Artiukhov, A.; Aleshin, V.; Mkrtchyan, G. Multiple Forms of Glutamate Dehydrogenase in Animals: Structural Determinants and Physiological Implications. Biology 2016, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press (OUP): Oxford, UK, 2000. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Wang, Y.T.; Yen, H.Y.; Tsou, C.C.; Ku, W.C.; Lin, P.Y.; Chen, H.Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y.J. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Plaitakis, A. Single amino acid substitution (G456A) in the vicinity of the GTP binding domain of human housekeeping glutamate dehydrogenase markedly attenuates GTP inhibition and abolishes the cooperative behavior of the enzyme. J. Biol. Chem. 2002, 277, 26422–26428. [Google Scholar] [CrossRef]
- Zaganas, I.; Spanaki, C.; Karpusas, M.; Plaitakis, A. Substitution of Ser for Arg-443 in the regulatory domain of human housekeeping (GLUD1) glutamate dehydrogenase virtually abolishes basal activity and markedly alters the activation of the enzyme by ADP and L-leucine. J. Biol. Chem. 2002, 277, 46552–46558. [Google Scholar] [CrossRef]
- Plaitakis, A.; Spanaki, C.; Mastorodemos, V.; Zaganas, I. Study of structure-function relationships in human glutamate dehydrogenases reveals novel molecular mechanisms for the regulation of the nerve tissue-specific (GLUD2) isoenzyme. Neurochem. Int. 2003, 43, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Mastorodemos, V.; Kanavouras, K.; Sundaram, S.; Providaki, M.; Petraki, Z.; Kokkinidis, M.; Zaganas, I.; Logothetis, D.E.; Plaitakis, A. Side-chain interactions in the regulatory domain of human glutamate dehydrogenase determine basal activity and regulation. J. Neurochem. 2015, 133, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol. Cell. Proteom. 2015, 14, 1599–1615. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, R.; Qu, X.; Yu, H.; Chu, H.; Zhang, Y.; Zhu, W.; Wu, X.; Gao, H.; Tao, B.; et al. alpha-Ketoglutarate-Activated NF-kappaB Signaling Promotes Compensatory Glucose Uptake and Brain Tumor Development. Mol. Cell 2019, 76, 148–162 e147. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Eckersdorff, M.; Li, Y.; Silva, J.C.; Fermin, D.; Kurtev, M.V.; Giallourakis, C.; Comb, M.J.; Alt, F.W.; Lombard, D.B. Calorie restriction alters mitochondrial protein acetylation. Aging Cell 2009, 8, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.M.; Cheng, J.; Gordon, J.I. Quantitative assessment of the impact of the gut microbiota on lysine epsilon-acetylation of host proteins using gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2012, 109, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef]
- Kanavouras, K.; Borompokas, N.; Latsoudis, H.; Stagourakis, A.; Zaganas, I.; Plaitakis, A. Mutations in human GLUD2 glutamate dehydrogenase affecting basal activity and regulation. J. Neurochem. 2009, 109 (Suppl. 1), 167–173. [Google Scholar] [CrossRef]
- Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Bessa, M.; Plaitakis, A. Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J. Neurosci. Res. 2005, 79, 65–73. [Google Scholar] [CrossRef]
- Pajecka, K.; Nielsen, C.W.; Hauge, A.; Zaganas, I.; Bak, L.K.; Schousboe, A.; Plaitakis, A.; Waagepetersen, H.S. Glutamate dehydrogenase isoforms with N-terminal (His)6- or FLAG-tag retain their kinetic properties and cellular localization. Neurochem. Res. 2014, 39, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Islam, M.M.; Zaganas, I. Interaction between glutamate dehydrogenase (GDH) and L-leucine catabolic enzymes: Intersecting metabolic pathways. Neurochem. Int. 2011, 59, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Purohit, J.S.; Tomar, R.S.; Panigrahi, A.K.; Pandey, S.M.; Singh, D.; Chaturvedi, M.M. Chicken liver glutamate dehydrogenase (GDH) demonstrates a histone H3 specific protease (H3ase) activity in vitro. Biochimie 2013, 95, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Rajas, F.; Gire, V.; Rousset, B. Involvement of a membrane-bound form of glutamate dehydrogenase in the association of lysosomes to microtubules. J. Biol. Chem. 1996, 271, 29882–29890. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Crail, J.P.; Laffoon, M.M.; Fernandez, W.G.; Menze, M.A.; Konkle, M.E. Identification of disulfide bond formation between MitoNEET and glutamate dehydrogenase 1. Biochemistry 2013, 52, 8969–8971. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nnatubeugo, C.; Johnson, E.; Gisondi, S.; Roland, F.; Geldenhuys, W.J.; Menze, M.A.; Konkle, M.E. The Mitochondrial Protein MitoNEET as a Probe for the Allostery of Glutamate Dehydrogenase. Molecules 2022, 27, 8314. [Google Scholar] [CrossRef] [PubMed]
- Mathioudakis, L.; Bourbouli, M.; Daklada, E.; Kargatzi, S.; Michaelidou, K.; Zaganas, I. Localization of Human Glutamate Dehydrogenases Provides Insights into Their Metabolic Role and Their Involvement in Disease Processes. Neurochem. Res. 2019, 44, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Miyazaki, T.; Miyazaki, J.; Kuzuyama, T.; Nishiyama, M. Hetero-oligomeric glutamate dehydrogenase from Thermus thermophilus. Microbiology 2010, 156, 3801–3813. [Google Scholar] [CrossRef] [PubMed]
- Mastorodemos, V.; Kotzamani, D.; Zaganas, I.; Arianoglou, G.; Latsoudis, H.; Plaitakis, A. Human GLUD1 and GLUD2 glutamate dehydrogenase localize to mitochondria and endoplasmic reticulum. Biochem. Cell Biol. 2009, 87, 505–516. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Stanley, C.A.; Lieu, Y.K.; Hsu, B.Y.; Burlina, A.B.; Greenberg, C.R.; Hopwood, N.J.; Perlman, K.; Rich, B.H.; Zammarchi, E.; Poncz, M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N. Engl. J. Med. 1998, 338, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A. Two genetic forms of hyperinsulinemic hypoglycemia caused by dysregulation of glutamate dehydrogenase. Neurochem. Int. 2011, 59, 465–472. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. The structure and allosteric regulation of glutamate dehydrogenase. Neurochem. Int. 2011, 59, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; Macdonald, M.J. The complex mechanism of glutamate dehydrogenase in insulin secretion. Diabetes 2011, 60, 2450–2454. [Google Scholar] [CrossRef] [PubMed]
- Raizen, D.M.; Brooks-Kayal, A.; Steinkrauss, L.; Tennekoon, G.I.; Stanley, C.A.; Kelly, A. Central nervous system hyperexcitability associated with glutamate dehydrogenase gain of function mutations. J. Pediatr. 2005, 146, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Roze, E.; Dionisi, C.; Escande, F.; Valayannopoulos, V.; Feillet, F.; Heinrichs, C.; Chadefaux-Vekemans, B.; Dan, B.; de Lonlay, P. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev. Med. Child. Neurol. 2008, 50, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Goto, S.; Sako, W.; Miyashiro, A.; Kim, I.; Escande, F.; Harada, M.; Morigaki, R.; Asanuma, K.; Mizobuchi, Y.; et al. Generalized dystonia in a patient with a novel mutation in the GLUD1 gene. Mov. Disord. 2012, 27, 1198–1199. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Targa Dias Anastacio, H.; Matosin, N.; Ooi, L. Neuronal hyperexcitability in Alzheimer’s disease: What are the drivers behind this aberrant phenotype? Transl. Psychiatry 2022, 12, 257. [Google Scholar] [CrossRef]
- Plaitakis, A.; Latsoudis, H.; Kanavouras, K.; Ritz, B.; Bronstein, J.M.; Skoula, I.; Mastorodemos, V.; Papapetropoulos, S.; Borompokas, N.; Zaganas, I.; et al. Gain-of-function variant in GLUD2 glutamate dehydrogenase modifies Parkinson’s disease onset. Eur. J. Hum. Genet. 2010, 18, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gong, J.; Ding, L.; Zhang, Z.; Pan, X.; Chen, X.; Guo, W.; Zhang, X.; Yang, X.; Peng, G.; et al. Functional validation of a human GLUD2 variant in a murine model of Parkinson’s disease. Cell Death Dis. 2020, 11, 897. [Google Scholar] [CrossRef] [PubMed]
- Mathioudakis, L.; Dimovasili, C.; Bourbouli, M.; Latsoudis, H.; Kokosali, E.; Gouna, G.; Vogiatzi, E.; Basta, M.; Kapetanaki, S.; Panagiotakis, S.; et al. Study of Alzheimer’s disease- and frontotemporal dementia-associated genes in the Cretan Aging Cohort. Neurobiol. Aging 2023, 123, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Liang, X.J.; Li, W.J.; Wu, D.; Liu, M.; Cao, B.Y.; Chen, J.J.; Qin, M.; Meng, X.; Gong, C.X. Clinical and Molecular Spectrum of Glutamate Dehydrogenase Gene Defects in 26 Chinese Congenital Hyperinsulinemia Patients. J. Diabetes Res. 2018, 2018, 2802540. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Fernandez-Ramos, C.; Vela, A.; Velayos, T.; Aguayo, A.; Urrutia, I.; Rica, I.; Castano, L.; Spanish Congenital Hyperinsulinism, G. Clinical and genetic characterization of congenital hyperinsulinism in Spain. Eur. J. Endocrinol. 2016, 174, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.; Kinner, M.; Passarge, M.; Superti-Furga, A.; Mayatepek, E.; Meissner, T.; Schneppenheim, R.; Schaub, J. Novel missense mutations outside the allosteric domain of glutamate dehydrogenase are prevalent in European patients with the congenital hyperinsulinism-hyperammonemia syndrome. Hum. Genet. 2001, 108, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Sarajlija, A.; Milenkovic, T.; Djordjevic, M.; Mitrovic, K.; Todorovic, S.; Kecman, B.; Hussain, K. Early Presentation of Hyperinsulinism/Hyperammonemia Syndrome in Three Serbian Patients. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 228–231. [Google Scholar] [CrossRef]
- Strajnar, A.; Tansek, M.Z.; Podkrajsek, K.T.; Battelino, T.; Groselj, U. Hyperinsulinism-hyperammonemia Syndrome in an Infant with Seizures. Balk. J. Med. Genet. 2018, 21, 77–81. [Google Scholar] [CrossRef]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J. Proteome Res. 2013, 12, 2414–2421. [Google Scholar] [CrossRef]
- Barrosse-Antle, M.; Su, C.; Chen, P.; Boodhansingh, K.E.; Smith, T.J.; Stanley, C.A.; De Leon, D.D.; Li, C. A severe case of hyperinsulinism due to hemizygous activating mutation of glutamate dehydrogenase. Pediatr. Diabetes 2017, 18, 911–916. [Google Scholar] [CrossRef]
- Sang, Y.; Xu, Z.; Liu, M.; Yan, J.; Wu, Y.; Zhu, C.; Ni, G. Mutational analysis of ABCC8, KCNJ11, GLUD1, HNF4A and GCK genes in 30 Chinese patients with congenital hyperinsulinism. Endocr. J. 2014, 61, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Proverbio, M.C.; Mangano, E.; Gessi, A.; Bordoni, R.; Spinelli, R.; Asselta, R.; Valin, P.S.; Di Candia, S.; Zamproni, I.; Diceglie, C.; et al. Whole genome SNP genotyping and exome sequencing reveal novel genetic variants and putative causative genes in congenital hyperinsulinism. PLoS ONE 2013, 8, e68740. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Taki, T.; Ohura, T.; Kato, H.; Yanagisawa, M.; Hayashi, Y. Novel missense mutations in the glutamate dehydrogenase gene in the congenital hyperinsulinism-hyperammonemia syndrome. J. Pediatr. 2000, 136, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Odom, J.; Gieron-Korthals, M.; Shulman, D.; Newkirk, P.; Prijoles, E.; Sanchez-Valle, A. A novel mutation in GLUD1 causing hyperinsulinism-hyperammonemia in a patient with high density of homozygosity on microarray: A case report. J. Med. Case Rep. 2016, 10, 25. [Google Scholar] [CrossRef] [PubMed][Green Version]



| Clade (Figure 2) | Group/Organism | Substitution | Structural Area | Unique Novel Feature | References |
|---|---|---|---|---|---|
| A | Hominoidea | A56V a | Proximity to NAD(P)(H) site | – | |
| Hominoidea | S227N | Proximity to NAD(P)(H) site | Phospho-Ser site removal | [78,79] | |
| Hominoidea | E87K | Proximity to C-terminal site | – | ||
| Hominoidea | D195E | Proximity to C-terminal site | – | ||
| Hominoidea | N551S | Proximity to C-terminal/Leu site | – | [80] | |
| Hominoidea | R496S b | Within/close to antenna | Altered allosteric regulation | [45,81,82,83] | |
| Hominoidea | G509A c | Within/close to antenna | Altered stability | [45,80,82] | |
| B | Hylobatidae | T154A # | Proximity to NAD(P)(H) site | Phospho-Tre site removal | [78] |
| Hylobatidae | L430V # | Hydrophobic core of NAD domain | – | ||
| Hylobates | E61K # | Proximity to NAD(P)(H) site | – | ||
| Hylobates moloch | D495G | Within/close to antenna | Altered regulation (predicted) | This work | |
| Hylobates moloch | S496R b | Within/close to antenna | Reversed mutation | ||
| Hylobates moloch | A509G c | Within/close to antenna | Reversed mutation | ||
| Hylobates moloch | A511V | Within/close to antenna | Altered regulation (predicted) | This work | |
| Hylobates moloch | M522V | Close to antenna/Hydrophobic core of NAD domain | Altered regulation/activity (predicted) | This work | |
| Hylobates moloch | R523H d,** | Proximity to ADP site | – | [80] | |
| Symphalangus | S140R | K+ site | Altered Leu-K-ADP interaction | [10] | |
| Symphalangus | N196K | Proximity to C-terminal site | – | ||
| Symphalangus | Y289H | Hydrophobic core/surface of NAD domain | – | ||
| Symphalangus | N406S | NAD(P)(H) site | – | ||
| Nomascus | A137T | Protein hydrophobic core/surface | – | ||
| Nomascus | S336N | Surface of NAD domain | Phospho-Ser site removal | [79] | |
| Nomascus | H481R | Within/close to antenna | – | ||
| C | Hominidae | V56L a | Proximity to NAD(P)(H) site | – | |
| Hominidae | S384T | Proximity to NAD(P)(H) site | Phospho-Ser/Tre site modification | [79,84,85,86] | |
| Hominidae | K352R e | Proximity to NAD(P)(H) site | Ac-Lys site removal | [87,88,89] | |
| Hominidae | R92Q | Proximity to C-terminal site | – | ||
| Hominidae | M423L | Hydrophobic core of NAD domain | – | [81] | |
| Hominidae | R523H d,** | Proximity to ADP site | – | [80] | |
| D | Pongo | I292N | Surface of NAD domain | – | |
| Pongo | L293V | Hydrophobic core of NAD domain | – | ||
| Pongo | I328V | Hydrophobic core of NAD domain | – | ||
| Pongo | L428V | Hydrophobic core of NAD domain | – | ||
| Pongo | Q494R | Within/close to antenna | – | [90] | |
| E | Homininae | A374V f | Hydrophobic core of NAD domain | – | |
| Homininae | I219V | Hydrophobic core of NAD domain/hexachlorophene site | – | ||
| Homininae | G300R | Surface of NAD domain | – | ||
| F | Gorilla | S119C | Subunit interface | Novel S-S bond formation | |
| Gorilla | K415R | Surface of NAD domain | Ac-Lys site removal | [88,89] | |
| Gorilla | L418Q | Surface of NAD domain | – | ||
| Gorilla | E492D | Within/close to antenna | – | ||
| G | Hominini | – | – | – | |
| H | Pan | I358L | Hydrophobic core of NAD domain | – | |
| Pan | V374I f | Hydrophobic core of NAD domain | – | ||
| I | Homo | R352K e | Proximity to NAD(P)(H) site | Ac-Lys site return | [87,88,89] |
| Homo | M468L | Within/close to antenna | – | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleshina, Y.A.; Aleshin, V.A. Evolutionary Changes in Primate Glutamate Dehydrogenases 1 and 2 Influence the Protein Regulation by Ligands, Targeting and Posttranslational Modifications. Int. J. Mol. Sci. 2024, 25, 4341. https://doi.org/10.3390/ijms25084341
Aleshina YA, Aleshin VA. Evolutionary Changes in Primate Glutamate Dehydrogenases 1 and 2 Influence the Protein Regulation by Ligands, Targeting and Posttranslational Modifications. International Journal of Molecular Sciences. 2024; 25(8):4341. https://doi.org/10.3390/ijms25084341
Chicago/Turabian StyleAleshina, Yulia A., and Vasily A. Aleshin. 2024. "Evolutionary Changes in Primate Glutamate Dehydrogenases 1 and 2 Influence the Protein Regulation by Ligands, Targeting and Posttranslational Modifications" International Journal of Molecular Sciences 25, no. 8: 4341. https://doi.org/10.3390/ijms25084341
APA StyleAleshina, Y. A., & Aleshin, V. A. (2024). Evolutionary Changes in Primate Glutamate Dehydrogenases 1 and 2 Influence the Protein Regulation by Ligands, Targeting and Posttranslational Modifications. International Journal of Molecular Sciences, 25(8), 4341. https://doi.org/10.3390/ijms25084341