
Targeting Macrophages: Therapeutic Approaches in Diabetic Kidney Disease

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Activation of Macrophages and Recruitment of Monocytes in Early Stage of Diabetic Kidney Disease
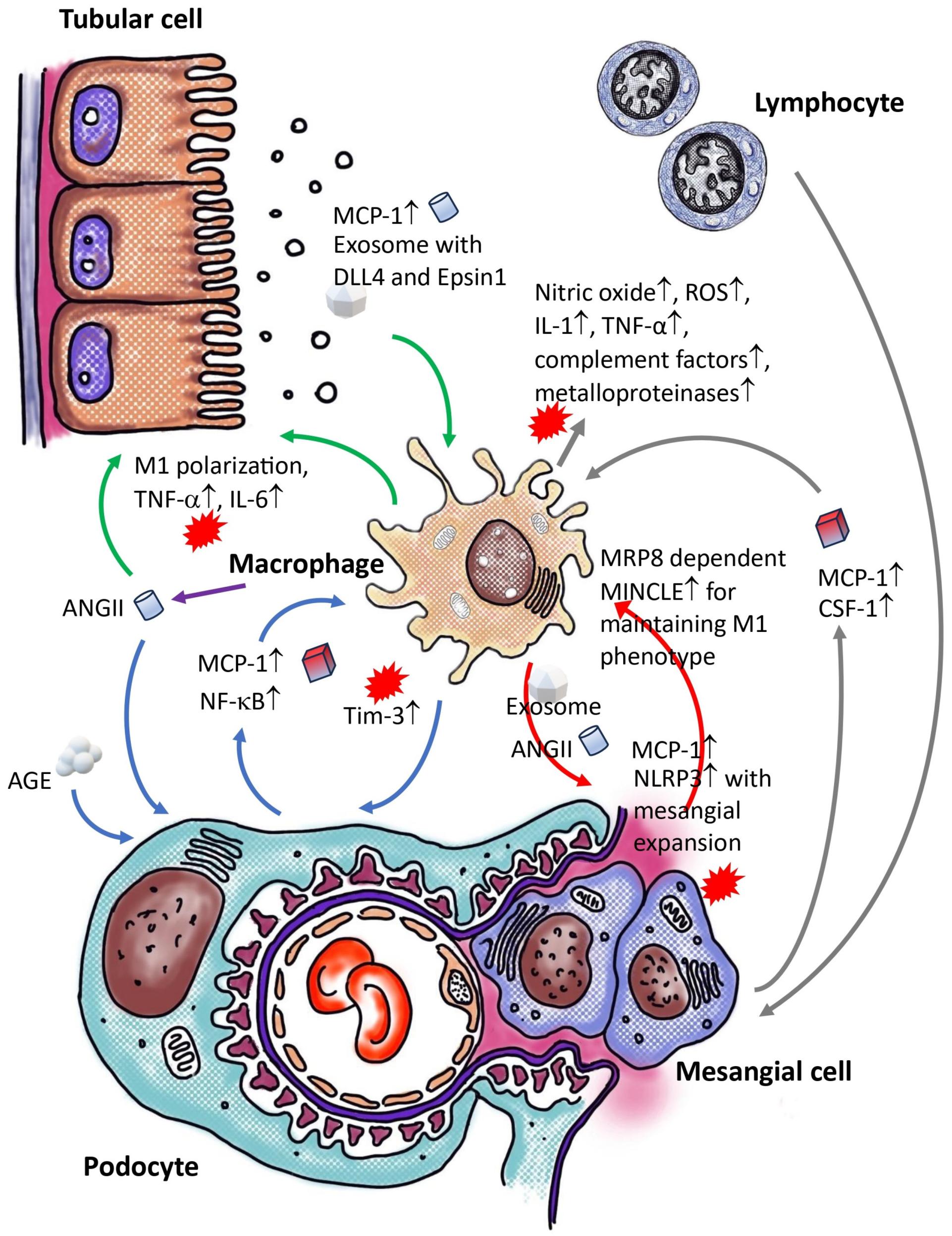
3. Crosstalk between Macrophages and Non-Myeloid Cells within the Renal Tissue of Diabetic Individuals
3.1. Crosstalk between Macrophage and Podocyte within Diabetic Kidneys
3.2. Crosstalk of Macrophage and Epithelial Cell/Tubular Cell within Diabetic Kidney
3.3. Crosstalk between Macrophage and Mesangial Cells within Diabetic Kidney
3.4. Crosstalk between Macrophage and Immune Cells within Diabetic Kidney
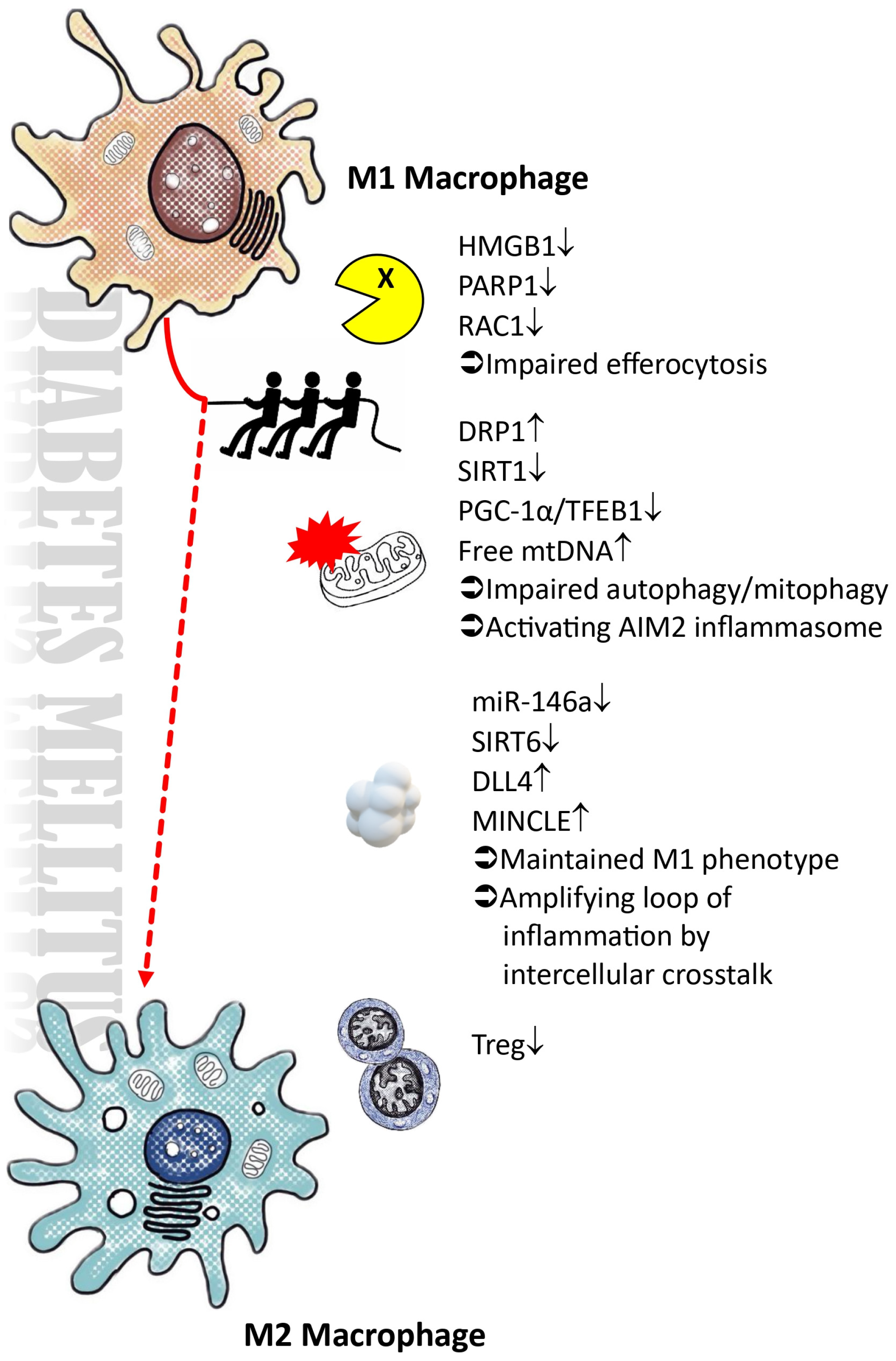
4. Polarization of Macrophages within Diabetic Microenvironment
4.1. Impairments in the Autophagic Process, Particularly Mitophagy, within a High-Glucose Microenvironment
4.2. Impaired Efferocytosis and Expression of AIM2 Inflammasomes Impede the Resolution of Inflammation
4.3. The Presence of Exosomes in DLL4 and the Disruption of microRNA Regulation Hinder the M2 Polarization Process
4.4. Elevated Blood Sugar Levels Lead to the Suppression of SIRT6, Thereby Impeding the Process of M2 Macrophage Polarization
5. The Participation of Macrophages in the Development of Renal Fibrosis in Diabetic Kidney Disease
5.1. Epithelial to Mesenchymal Transition vs. Macrophage–Myofibroblast Transition
5.2. M2 Macrophage, to Be or Not to Be? That Is a Question
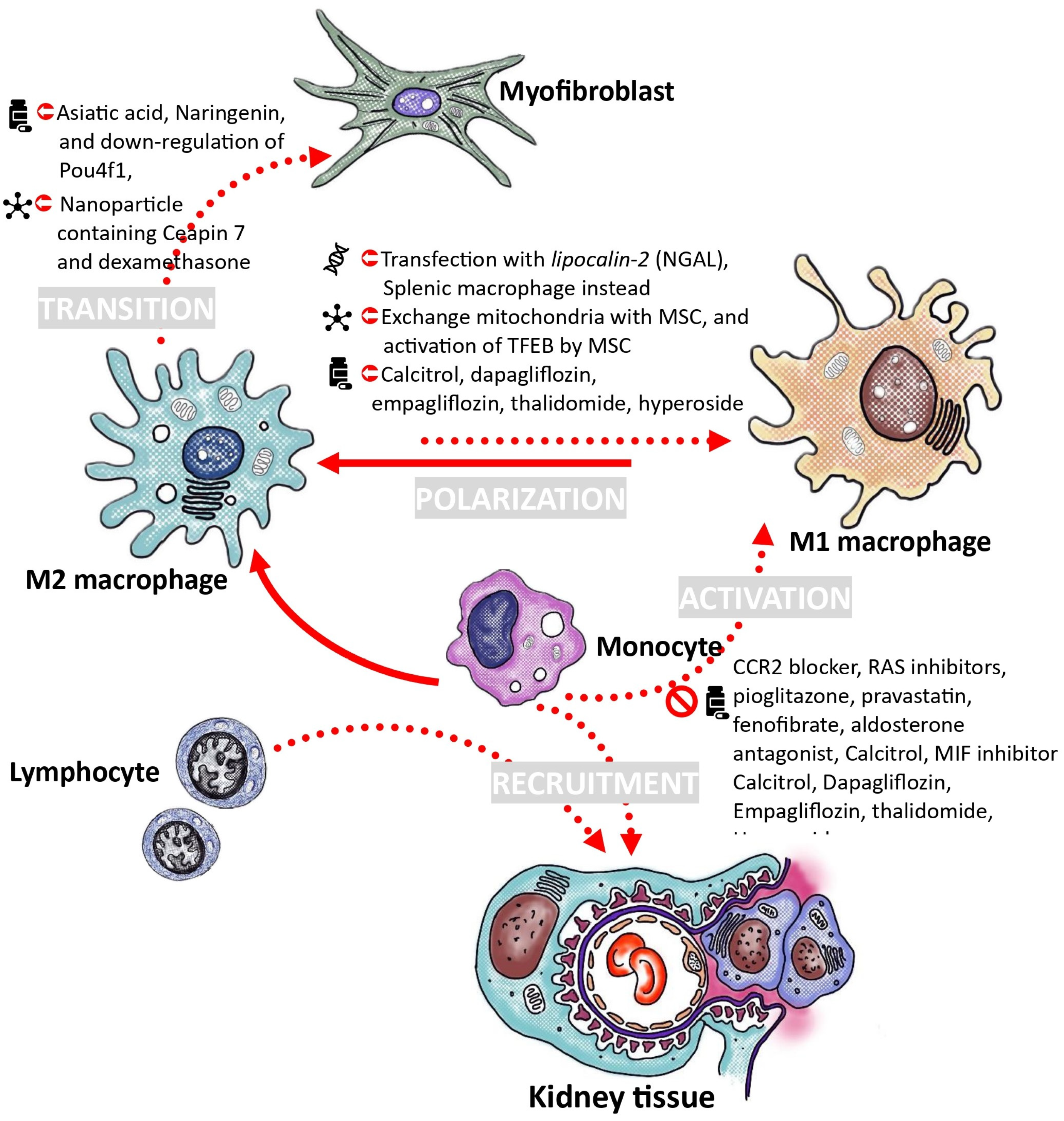
6. Proposals for the Treatment of Diabetic Nephropathy through the Modulation of Macrophages
6.1. Decrease Recruitment and Activation of Monocytes
6.2. Adoption of Macrophage Polarization toward M2 Phenotype
6.3. Utilizing Ex Vivo Macrophage Transfusion for the Treatment of Diabetic Kidney Disease
6.4. Strategies to Inhibit the Transition of Macrophages into Myofibroblasts
6.5. Timing and the Corresponding Microenvironment Play Important Roles
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Woo, K.; Yi, J.A. Epidemiology of end-stage kidney disease. Semin. Vasc. Surg. 2021, 34, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.W.; Yu, X.; Chen, H.C.; Kashihara, N.; Park, H.C.; Liew, A.; Goh, B.L.; Nazareth, M.G.C.; Bunnag, S.; Tan, J.; et al. Dialysis Care and Dialysis Funding in Asia. Am. J. Kidney Dis. 2020, 75, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. (2011) 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Tesch, G.H. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J. Am. Soc. Nephrol. 2005, 16, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; DHT, I.J. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Akat, K.M.; Sun, Z.; Zhang, W.; Schlondorff, D.; Liu, Z.; Tuschl, T.; Lee, K.; He, J.C. Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Sun, Z.; Wang, X.; Zhang, T.; Yuan, W.; Salem, F.; Yu, S.M.; Zhang, W.; Lee, K.; He, J.C. The single-cell landscape of kidney immune cells reveals transcriptional heterogeneity in early diabetic kidney disease. Kidney Int. 2022, 102, 1291–1304. [Google Scholar] [CrossRef]
- Sassy-Prigent, C.; Heudes, D.; Mandet, C.; Belair, M.F.; Michel, O.; Perdereau, B.; Bariety, J.; Bruneval, P. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes 2000, 49, 466–475. [Google Scholar] [CrossRef]
- Awad, A.S.; Kinsey, G.R.; Khutsishvili, K.; Gao, T.; Bolton, W.K.; Okusa, M.D. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am. J. Physiol. Renal Physiol. 2011, 301, F1358–F1366. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.J.; Lee, E.S.; Kim, G.T.; Hyun, M.; Lee, J.H.; Chen, S.; Choi, R.; Kim, H.M.; Lee, E.Y.; Chung, C.H. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol. Dial. Transplant. 2013, 28, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, Y.; Cui, Q.; Xu, J.; Tang, Z.; Wang, Y.; He, C.; Wang, X. Toll-like receptor 4 plays a key role in advanced glycation end products-induced M1 macrophage polarization. Biochem. Biophys. Res. Commun. 2020, 531, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.; Sourris, K.C.; Summers, S.A.; McCarthy, D.; Ward, M.S.; Borg, D.J.; Gallo, L.A.; Fotheringham, A.K.; Pettit, A.R.; Yap, F.Y.; et al. Deletion of bone-marrow-derived receptor for AGEs (RAGE) improves renal function in an experimental mouse model of diabetes. Diabetologia 2014, 57, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Price, D.A.; Porter, L.E.; Gordon, M.; Fisher, N.D.; De’Oliveira, J.M.; Laffel, L.M.; Passan, D.R.; Williams, G.H.; Hollenberg, N.K. The paradox of the low-renin state in diabetic nephropathy. J. Am. Soc. Nephrol. 1999, 10, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Jonas, U.; Buhler, F.R.; Resink, T.J. Activation of human peripheral monocytes by angiotensin II. FEBS Lett. 1994, 347, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Gomez, R.A.; Norling, L.L.; Wilfong, N.; Isakson, P.; Lynch, K.R.; Hock, R.; Quesenberry, P. Leukocytes synthesize angiotensinogen. Hypertension 1993, 21, 470–475. [Google Scholar] [CrossRef]
- Iwai, N.; Inagami, T.; Ohmichi, N.; Kinoshita, M. Renin is expressed in rat macrophage/monocyte cells. Hypertension 1996, 27 Pt 1, 399–403. [Google Scholar] [CrossRef]
- Dezso, B.; Jacobsen, J.; Poulsen, K. Evidence for the presence of angiotensins in normal, unstimulated alveolar macrophages and monocytes. J. Hypertens. 1989, 7, 5–11. [Google Scholar] [CrossRef]
- Alvarez, A.; Cerda-Nicolas, M.; Naim Abu Nabah, Y.; Mata, M.; Issekutz, A.C.; Panes, J.; Lobb, R.R.; Sanz, M.J. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood 2004, 104, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pueyo, M.E.; Gonzalez, W.; Nicoletti, A.; Savoie, F.; Arnal, J.F.; Michel, J.B. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Shikata, K.; Matsuda, M.; Akiyama, K.; Sugimoto, H.; Kushiro, M.; Makino, H. Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia 1998, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Luyckx, V.A.; Ots, M.; Lee, K.W.; Ziai, F.; Troy, J.L.; Brenner, B.M.; MacKenzie, H.S. Renin-angiotensin blockade lowers MCP-1 expression in diabetic rats. Kidney Int. 1999, 56, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.J.; Wilkinson-Berka, J.L.; Ricardo, S.D.; Cox, A.J.; Gilbert, R.E. Progression of tubulointerstitial injury by osteopontin-induced macrophage recruitment in advanced diabetic nephropathy of transgenic (mRen-2)27 rats. Nephrol. Dial. Transplant. 2002, 17, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, K.; Xiao, J.; Xin, J.; Zhang, L.; Li, X.; Li, L.; Si, J.; Wang, L.; Ma, K. Angiotensin II induces RAW264.7 macrophage polarization to the M1-type through the connexin 43/NF-kappaB pathway. Mol. Med. Rep. 2020, 21, 2103–2112. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Zuo, Y.; Ma, L.J.; Kaseda, R.; Fogo, A.B.; Ichikawa, I.; Linton, M.F.; Fazio, S.; Kon, V. Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Lu, H.; Chen, R.; Tian, Y.; Jiang, Y.; Zhang, S.; Ni, D.; Su, Z.; Shao, X. Angiotensin II enhances the acetylation and release of HMGB1 in RAW264.7 macrophage. Cell Biol. Int. 2018, 42, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Wang, H.; Huang, Y.; Huang, J.H.; Ren, H.L.; Xu, Q.; Su, X.M.; Wang, A.M.; Ren, F.; Zhou, M.S. Myeloid Angiotensin II Type 1 Receptor Mediates Macrophage Polarization and Promotes Vascular Injury in DOCA/Salt Hypertensive Mice. Front. Pharmacol. 2022, 13, 879693. [Google Scholar] [CrossRef]
- Bakris, G.; Burgess, E.; Weir, M.; Davidai, G.; Koval, S.; Investigators, A.S. Telmisartan is more effective than losartan in reducing proteinuria in patients with diabetic nephropathy. Kidney Int. 2008, 74, 364–369. [Google Scholar] [CrossRef]
- Cucak, H.; Nielsen Fink, L.; Hojgaard Pedersen, M.; Rosendahl, A. Enalapril treatment increases T cell number and promotes polarization towards M1-like macrophages locally in diabetic nephropathy. Int. Immunopharmacol. 2015, 25, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Saruta, T. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy. J. Int. Med. Res. 2001, 29, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Johansson, U.; Peng, X.R.; Bamberg, K.; Huang, Y. An additive effect of eplerenone to ACE inhibitor on slowing the progression of diabetic nephropathy in the db/db mice. Am. J. Transl. Res. 2016, 8, 1339–1354. [Google Scholar]
- Liu, S.; Li, L.; Lou, P.; Zhao, M.; Wang, Y.; Tang, M.; Gong, M.; Liao, G.; Yuan, Y.; Li, L.; et al. Elevated branched-chain alpha-keto acids exacerbate macrophage oxidative stress and chronic inflammatory damage in type 2 diabetes mellitus. Free Radic. Biol. Med. 2021, 175, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Song, Z.; Zhou, M.; Yang, Y.; Zhao, Y.; Liu, B.; Zhang, X. Infiltrating macrophages in diabetic nephropathy promote podocytes apoptosis via TNF-alpha-ROS-p38MAPK pathway. Oncotarget 2017, 8, 53276–53287. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, R.; Hayashi, K.; Itoh, H. Effects of High Glucose and Lipotoxicity on Diabetic Podocytes. Nutrients 2021, 13, 241. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Brosius, F.C., 3rd; Menon, R.K. The glomerular podocyte as a target of growth hormone action: Implications for the pathogenesis of diabetic nephropathy. Curr. Diabetes Rev. 2011, 7, 50–55. [Google Scholar] [CrossRef]
- Nishad, R.; Mukhi, D.; Kethavath, S.; Raviraj, S.; Paturi, A.S.V.; Motrapu, M.; Kurukuti, S.; Pasupulati, A.K. Podocyte derived TNF-alpha mediates monocyte differentiation and contributes to glomerular injury. FASEB J. 2022, 36, e22622. [Google Scholar] [CrossRef]
- Ruster, C.; Bondeva, T.; Franke, S.; Tanaka, N.; Yamamoto, H.; Wolf, G. Angiotensin II upregulates RAGE expression on podocytes: Role of AT2 receptors. Am. J. Nephrol. 2009, 29, 538–550. [Google Scholar] [CrossRef]
- Wei, M.; Li, Z.; Xiao, L.; Yang, Z. Effects of ROS-relative NF-kappaB signaling on high glucose-induced TLR4 and MCP-1 expression in podocyte injury. Mol. Immunol. 2015, 68 Pt A, 261–271. [Google Scholar] [CrossRef]
- Gu, L.; Hagiwara, S.; Fan, Q.; Tanimoto, M.; Kobata, M.; Yamashita, M.; Nishitani, T.; Gohda, T.; Ni, Z.; Qian, J.; et al. Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrol. Dial. Transplant. 2006, 21, 299–313. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Gao, T.; Cooper, T.K.; Brian Reeves, W.; Awad, A.S. Macrophages directly mediate diabetic renal injury. Am. J. Physiol. Renal Physiol. 2013, 305, F1719–F1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, T.; Li, D.; Du, X.; Wang, T.; Li, C.; Song, X.; Xu, L.; Yi, F.; Liang, X.; et al. Tim-3 aggravates podocyte injury in diabetic nephropathy by promoting macrophage activation via the NF-kappaB/TNF-alpha pathway. Mol. Metab. 2019, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, M.J.; Keyel, P.A.; Mishra, S.K.; Watkins, S.C.; Heuser, J.E.; Traub, L.M. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic 2006, 7, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Pagie, S.; Gerard, N.; Charreau, B. Notch signaling triggered via the ligand DLL4 impedes M2 macrophage differentiation and promotes their apoptosis. Cell Commun. Signal 2018, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Zhang, L.; Huang, Y.; Li, X.H.; Liu, Y.F.; Zhang, S.M.; Zhao, Y.E.; Chen, X.J.; Liu, Y.; He, L.Y.; et al. Epsin1-mediated exosomal sorting of Dll4 modulates the tubular-macrophage crosstalk in diabetic nephropathy. Mol. Ther. 2023, 31, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.J.; Xu, C.T.; Du, C.L.; Dong, J.H.; Xu, S.B.; Hu, B.F.; Feng, R.; Zang, D.D.; Meng, X.M.; Huang, C.; et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics 2022, 12, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Egido, J. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 2003, 35, 881–900. [Google Scholar] [CrossRef] [PubMed]
- Tanifuji, C.; Suzuki, Y.; Geot, W.M.; Horikoshi, S.; Sugaya, T.; Ruiz-Ortega, M.; Egido, J.; Tomino, Y. Reactive oxygen species-mediated signaling pathways in angiotensin II-induced MCP-1 expression of proximal tubular cells. Antioxid. Redox Signal. 2005, 7, 1261–1268. [Google Scholar] [CrossRef]
- Haruhara, K.; Suzuki, T.; Wakui, H.; Azushima, K.; Kurotaki, D.; Kawase, W.; Uneda, K.; Kobayashi, R.; Ohki, K.; Kinguchi, S.; et al. Deficiency of the kidney tubular angiotensin II type1 receptor-associated protein ATRAP exacerbates streptozotocin-induced diabetic glomerular injury via reducing protective macrophage polarization. Kidney Int. 2022, 101, 912–928. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Zhao, M.; Wu, Y.; Xu, Y.; Li, X.; Fu, L.; Han, L.; Zhou, W.; Hu, Q.; et al. Macrophage-derived exosomes promote activation of NLRP3 inflammasome and autophagy deficiency of mesangial cells in diabetic nephropathy. Life Sci. 2023, 330, 121991. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.J.; Zhu, M.; Xu, X.X.; Meng, X.M.; Wu, Y.G. Exosomes from high glucose-treated macrophages activate glomerular mesangial cells via TGF-beta1/Smad3 pathway in vivo and in vitro. FASEB J. 2019, 33, 9279–9290. [Google Scholar] [CrossRef] [PubMed]
- Mauer, S.M. Structural-functional correlations of diabetic nephropathy. Kidney Int. 1994, 45, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Young, B.; Johnson, R.; Alpers, C.; Eng, E.; Floege, J.; Couser, W. Mesangial cell (MC) proliferation precedes development of glomerulosclerosis (GS) in experimental diabetic nephropathy (DN). J. Am. Soc. Nephrol. 1992, 3, 770. [Google Scholar]
- Wang, A.J.; Ren, J.; Abbadi, A.; Wang, A.; Hascall, V.C. Heparin affects cytosolic glucose responses of hyperglycemic dividing mesangial cells. J. Biol. Chem. 2019, 294, 6591–6597. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hascall, V.C. Hyaluronan structures synthesized by rat mesangial cells in response to hyperglycemia induce monocyte adhesion. J. Biol. Chem. 2004, 279, 10279–10285. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Mori, K.; Mukoyama, M.; Kasahara, M.; Yokoi, H.; Nakao, K. Macrophage-mediated glucolipotoxicity via myeloid-related protein 8/toll-like receptor 4 signaling in diabetic nephropathy. Clin. Exp. Nephrol. 2014, 18, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.L.; Tang, P.M.; Li, C.J.; You, Y.K.; Li, J.; Huang, X.R.; Ni, J.; Feng, M.; Liu, B.C.; Lan, H.Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017, 91, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Hata, Y.; Kuwabara, T.; Mori, K.; Kan, Y.; Sato, Y.; Umemoto, S.; Fujimoto, D.; Kanki, T.; Nishiguchi, Y.; Yokoi, H.; et al. Ablation of Myeloid Cell MRP8 Ameliorates Nephrotoxic Serum-induced Glomerulonephritis by Affecting Macrophage Characterization through Intraglomerular Crosstalk. Sci. Rep. 2020, 10, 3056. [Google Scholar] [CrossRef]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef]
- Satriano, J.A.; Hora, K.; Shan, Z.; Stanley, E.R.; Mori, T.; Schlondorff, D. Regulation of monocyte chemoattractant protein-1 and macrophage colony-stimulating factor-1 by IFN-gamma, tumor necrosis factor-alpha, IgG aggregates, and cAMP in mouse mesangial cells. J. Immunol. 1993, 150, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H. Macrophages and diabetic nephropathy. Semin. Nephrol. 2010, 30, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Harris, D.C. Macrophages in renal disease. J. Am. Soc. Nephrol. 2011, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Imani, F.; Horii, Y.; Suthanthiran, M.; Skolnik, E.Y.; Makita, Z.; Sharma, V.; Sehajpal, P.; Vlassara, H. Advanced glycosylation endproduct-specific receptors on human and rat T-lymphocytes mediate synthesis of interferon gamma: Role in tissue remodeling. J. Exp. Med. 1993, 178, 2165–2172. [Google Scholar] [CrossRef]
- Navarro-Gonzalez, J.F.; Mora-Fernandez, C.; Muros de Fuentes, M.; Garcia-Perez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; Navarro, J.F. Inflammation and pathogenesis of diabetic nephropathy. Metabolism 2004, 53, 265–266, author reply 266–267. [Google Scholar] [CrossRef] [PubMed]
- Suchanek, O.; Ferdinand, J.R.; Tuong, Z.K.; Wijeyesinghe, S.; Chandra, A.; Clauder, A.K.; Almeida, L.N.; Clare, S.; Harcourt, K.; Ward, C.J.; et al. Tissue-resident B cells orchestrate macrophage polarisation and function. Nat. Commun. 2023, 14, 7081. [Google Scholar] [CrossRef]
- Peng, B.; Ming, Y.; Yang, C. Regulatory B cells: The cutting edge of immune tolerance in kidney transplantation. Cell Death Dis. 2018, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol. 2012, 23, 194–203. [Google Scholar] [CrossRef]
- Kurts, C.; Ginhoux, F.; Panzer, U. Kidney dendritic cells: Fundamental biology and functional roles in health and disease. Nat. Rev. Nephrol. 2020, 16, 391–407. [Google Scholar] [CrossRef]
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.D.; Primiano, M.J.; Hall, J.P.; Franklin, B.S.; Knolle, P.A.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef]
- Khan, J.; Sharma, P.K.; Mukhopadhaya, A. Vibrio cholerae porin OmpU mediates M1-polarization of macrophages/monocytes via TLR1/TLR2 activation. Immunobiology 2015, 220, 1199–1209. [Google Scholar] [CrossRef]
- Tsuji, Y.; Kuramochi, M.; Golbar, H.M.; Izawa, T.; Kuwamura, M.; Yamate, J. Acetaminophen-Induced Rat Hepatotoxicity Based on M1/M2-Macrophage Polarization, in Possible Relation to Damage-Associated Molecular Patterns and Autophagy. Int. J. Mol. Sci. 2020, 21, 8998. [Google Scholar] [CrossRef]
- Mahon, O.R.; Kelly, D.J.; McCarthy, G.M.; Dunne, A. Osteoarthritis-associated basic calcium phosphate crystals alter immune cell metabolism and promote M1 macrophage polarization. Osteoarthr. Cartil. 2020, 28, 603–612. [Google Scholar] [CrossRef]
- Abuduhalike, R.; Abudouwayiti, A.; Juan, S.; MaheMuti, A. Study on the Mechanism of NLRP3/IL-1/NF-kappaB Signaling Pathway and Macrophage Polarization in the Occurrence and Development of VTE. Ann. Vasc. Surg. 2023, 89, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, W.; Wang, S.; Zhang, P.; Wang, Q.; Xiao, J.; Zhang, C.; Zheng, X.; Xu, X.; Xue, S.; et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019, 27, 1176–1189.e5. [Google Scholar] [CrossRef] [PubMed]
- Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J.F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front. Immunol. 2019, 10, 3. [Google Scholar] [CrossRef]
- Qu, X.; Xu, G.; Hou, X.; Chen, G.; Fan, T.; Yang, X.; Chen, Z. M1 Macrophage-Derived Interleukin-6 Promotes the Osteogenic Differentiation of Ligamentum Flavum Cells. Spine 2022, 47, E527–E535. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Holdbrooks, A.T.; Liu, Y.; Reynolds, S.L.; Yanagisawa, L.L.; Benveniste, E.N. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012, 189, 3439–3448. [Google Scholar] [CrossRef]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, Secreted by Pro-Inflammatory Macrophages, Targets Endoglin in Human Macrophages and Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef]
- Mily, A.; Kalsum, S.; Loreti, M.G.; Rekha, R.S.; Muvva, J.R.; Lourda, M.; Brighenti, S. Polarization of M1 and M2 Human Monocyte-Derived Cells and Analysis with Flow Cytometry upon Mycobacterium tuberculosis Infection. J. Vis. Exp. 2020, 163, e61807. [Google Scholar] [CrossRef]
- Kim, H.; Wang, S.Y.; Kwak, G.; Yang, Y.; Kwon, I.C.; Kim, S.H. Exosome-Guided Phenotypic Switch of M1 to M2 Macrophages for Cutaneous Wound Healing. Adv. Sci. 2019, 6, 1900513. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, E.; Hotter, G.; Jung, M.; Herrero-Fresneda, I.; Torras, J.; Sola, A. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J. Pathol. 2008, 214, 104–113. [Google Scholar] [CrossRef]
- Sapudom, J.; Karaman, S.; Mohamed, W.K.E.; Garcia-Sabate, A.; Quartey, B.C.; Teo, J.C.M. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. NPJ Regen. Med. 2021, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Wang, X.; Wang, Y.; Niu, A.; Wang, S.; Zou, C.; Harris, R.C. IL-4/IL-13-mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int. 2017, 91, 375–386. [Google Scholar] [CrossRef]
- Yue, Y.; Yang, X.; Feng, K.; Wang, L.; Hou, J.; Mei, B.; Qin, H.; Liang, M.; Chen, G.; Wu, Z. M2b macrophages reduce early reperfusion injury after myocardial ischemia in mice: A predominant role of inhibiting apoptosis via A20. Int. J. Cardiol. 2017, 245, 228–235. [Google Scholar] [CrossRef]
- Ambarus, C.A.; Santegoets, K.C.; van Bon, L.; Wenink, M.H.; Tak, P.P.; Radstake, T.R.; Baeten, D.L. Soluble immune complexes shift the TLR-induced cytokine production of distinct polarized human macrophage subsets towards IL-10. PLoS ONE 2012, 7, e35994. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Cao, Q.; Zheng, D.; Sun, Y.; Wang, C.; Yu, X.; Wang, Y.; Lee, V.W.; Zheng, G.; Tan, T.K.; et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int. 2013, 84, 745–755. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, H.; Wang, C.; Li, H.; Zhang, Q.; Bai, J. M2A and M2C Macrophage Subsets Ameliorate Inflammation and Fibroproliferation in Acute Lung Injury Through Interleukin 10 Pathway. Shock. 2017, 48, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed]
- She, Y.; Yu, M.; Wang, L.; Wang, Y.; Fang, P.; Zhang, Z. Emerging Protective Actions of PGC-1alpha in Diabetic Nephropathy. Oxid. Med. Cell Longev. 2022, 2022, 6580195. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Ko, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1alpha axis in db/db mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chen, Y.; Peng, T.; Li, L.; Zhu, W.; Liu, F.; Liu, S.; An, X.; Luo, R.; Cheng, J.; et al. Mitochondrial ROS-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin. Sci. 2019, 133, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, Y.; Jiang, Y.; Zhu, X.; Liu, Y.; Zhang, X. Mitophagy regulates macrophage phenotype in diabetic nephropathy rats. Biochem. Biophys. Res. Commun. 2017, 494, 42–50. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, L.; Zhu, L.; Liu, F.; Tang, X.; Liao, G.; Liu, J.; Cheng, J.; Chen, Y.; Lu, Y. Mesenchymal stem cells elicit macrophages into M2 phenotype via improving transcription factor EB-mediated autophagy to alleviate diabetic nephropathy. Stem Cells 2020, 38, 639–652. [Google Scholar] [CrossRef]
- Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Cheng, J.; Lu, Y. Mitochondrial transfer from mesenchymal stem cells to macrophages restricts inflammation and alleviates kidney injury in diabetic nephropathy mice via PGC-1alpha activation. Stem Cells 2021, 39, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Monks, J.; Rosner, D.; Geske, F.J.; Lehman, L.; Hanson, L.; Neville, M.C.; Fadok, V.A. Epithelial cells as phagocytes: Apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ. 2005, 12, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A., Jr.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 2018, 49, 666–677.e6. [Google Scholar] [CrossRef]
- Ngai, D.; Schilperoort, M.; Tabas, I. Efferocytosis-induced lactate enables the proliferation of pro-resolving macrophages to mediate tissue repair. Nat. Metab. 2023, 5, 2206–2219. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, H.; Tang, Y.; Yao, P. Potential Mechanisms and Effects of Efferocytosis in Atherosclerosis. Front. Endocrinol. 2020, 11, 585285. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Apoptosis and efferocytosis in mouse models of atherosclerosis. Curr. Drug Targets 2007, 8, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, Y.; Guo, F.; Zhao, L.; Qin, G. Single-Cell RNA Sequencing Reveals RAC1 Involvement in Macrophages Efferocytosis in Diabetic Kidney Disease. Inflammation 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Friggeri, A.; Banerjee, S.; Biswas, S.; de Freitas, A.; Liu, G.; Bierhaus, A.; Abraham, E. Participation of the receptor for advanced glycation end products in efferocytosis. J. Immunol. 2011, 186, 6191–6198. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.Y.; He, S.Y.; Hu, Q.Y.; Lu, Y.; Niu, Y.X.; Li, X.Y.; Zhang, H.M.; Qin, L.; Su, Q. Advanced Glycation End Products (AGEs) Inhibit Macrophage Efferocytosis of Apoptotic beta Cells through Binding to the Receptor for AGEs. J. Immunol. 2022, 208, 1204–1213. [Google Scholar] [CrossRef]
- Chen, Y.; Qiao, F.; Zhao, Y.; Wang, Y.; Liu, G. HMGB1 is activated in type 2 diabetes mellitus patients and in mesangial cells in response to high glucose. Int. J. Clin. Exp. Pathol. 2015, 8, 6683–6691. [Google Scholar]
- Liu, B.; Gan, X.; Zhao, Y.; Gao, J.; Yu, H. Inhibition of HMGB1 reduced high glucose-induced BMSCs apoptosis via activation of AMPK and regulation of mitochondrial functions. J. Physiol. Biochem. 2021, 77, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, G.; Chen, R.; Zhou, J.; Chen, T.; Cheng, Y.; Lou, Q.; Wang, H. PARP1 Is Upregulated by Hyperglycemia Via N6-methyladenosine Modification and Promotes Diabetic Retinopathy. Discov. Med. 2022, 34, 115–129. [Google Scholar] [PubMed]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp. Cell Res. 2018, 373, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.; Banerjee, S.; Friggeri, A.; Bell, C.; Abraham, E.; Zerfaoui, M. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol. Med. 2012, 18, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. 2018, 29, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Catano Canizales, Y.G.; Uresti Rivera, E.E.; Garcia Jacobo, R.E.; Portales Perez, D.P.; Yadira, B.; Rodriguez Rivera, J.G.; Amaro, R.G.; Enciso Moreno, J.A.; Garcia Hernandez, M.H. Increased Levels of AIM2 and Circulating Mitochondrial DNA in Type 2 Diabetes. Iran. J. Immunol. 2018, 15, 142–155. [Google Scholar]
- Bae, J.H.; Jo, S.I.; Kim, S.J.; Lee, J.M.; Jeong, J.H.; Kang, J.S.; Cho, N.J.; Kim, S.S.; Lee, E.Y.; Moon, J.S. Circulating Cell-Free mtDNA Contributes to AIM2 Inflammasome-Mediated Chronic Inflammation in Patients with Type 2 Diabetes. Cells 2019, 8, 328. [Google Scholar] [CrossRef]
- El-Deeb, O.S.; Hafez, Y.M.; Eltokhy, A.K.; Awad, M.M.; El-Shaer, R.A.A.; Abdel Ghafar, M.T.; Atef, M.M. Stimulator of interferon genes/Interferon regulatory factor 3 (STING-IRF3) and inflammasome-activation mediated pyroptosis biomarkers: A network of integrated pathways in diabetic nephropathy. J. Diabetes Metab. Disord. 2023, 22, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zheng, Z.; Xue, M.; Zhang, S.; Hu, F.; Li, Y.; Yang, Y.; Zou, M.; Li, S.; Wang, L.; et al. Extracellular Vesicles from Albumin-Induced Tubular Epithelial Cells Promote the M1 Macrophage Phenotype by Targeting Klotho. Mol. Ther. 2019, 27, 1452–1466. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, J.; Zheng, Z.; Tao, Y.; Zhang, S.; Zou, M.; Yang, Y.; Xue, M.; Hu, F.; Li, Y.; et al. Tubular epithelial cell-derived extracellular vesicles induce macrophage glycolysis by stabilizing HIF-1alpha in diabetic kidney disease. Mol. Med. 2022, 28, 95. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.S.; Recamonde-Mendoza, M.; De Souza, B.M.; Crispim, D. MicroRNA expression profiles and type 1 diabetes mellitus: Systematic review and bioinformatic analysis. Endocr. Connect. 2017, 6, 773–790. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Leung, S.W. Identification of microRNA biomarkers in type 2 diabetes: A meta-analysis of controlled profiling studies. Diabetologia 2015, 58, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock. 2016, 46, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Zhong, M.; Suo, Q.; Lv, K. Expression profiles of miRNAs in polarized macrophages. Int. J. Mol. Med. 2013, 31, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, W.; Liao, J.; Tang, F.; Gao, G.; Peng, J.; Fu, X.; Zhan, Y.; Chen, Z.; Xu, W.; et al. MicroRNA-21: A Critical Pathogenic Factor of Diabetic Nephropathy. Front. Endocrinol. 2022, 13, 895010. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jing, N.; Shen, A.; Guo, F.; Song, Y.; Pan, M.; Ma, X.; Zhao, L.; Zhang, H.; Wu, L.; et al. MiR-21-5p in macrophage-derived extracellular vesicles affects podocyte pyroptosis in diabetic nephropathy by regulating A20. J. Endocrinol. Investig. 2021, 44, 1175–1184. [Google Scholar] [CrossRef]
- Liechty, C.; Hu, J.; Zhang, L.; Liechty, K.W.; Xu, J. Role of microRNA-21 and Its Underlying Mechanisms in Inflammatory Responses in Diabetic Wounds. Int. J. Mol. Sci. 2020, 21, 3328. [Google Scholar] [CrossRef]
- Lu, J.; Xie, L.; Sun, S. The inhibitor miR-21 regulates macrophage polarization in an experimental model of chronic obstructive pulmonary disease. Tob. Induc. Dis. 2021, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Lanting, L.L.; Jia, Y.; Yadav, S.; Reddy, M.A.; Magilnick, N.; Boldin, M.; Natarajan, R. Anti-Inflammatory Role of MicroRNA-146a in the Pathogenesis of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Meng, L.F.; Lu, X.H.; Liu, L.H.; Ci, X.; Zhuo, Z. Protective effect of miR-146 against kidney injury in diabetic nephropathy rats through mediating the NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3215–3222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Le, X.; Zheng, S.; Zhang, K.; He, J.; Liu, M.; Tu, C.; Rao, W.; Du, H.; Ouyang, Y.; et al. MicroRNA-146a-5p-modified human umbilical cord mesenchymal stem cells enhance protection against diabetic nephropathy in rats through facilitating M2 macrophage polarization. Stem Cell Res. Ther. 2022, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Mahtal, N.; Lenoir, O.; Tinel, C.; Anglicheau, D.; Tharaux, P.L. MicroRNAs in kidney injury and disease. Nat. Rev. Nephrol. 2022, 18, 643–662. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, S.W.; Kim, H.Y.; Lee, S.Y.; Lee, W.S.; Hong, K.W.; Kim, C.D. SIRT1 inhibits differentiation of monocytes to macrophages: Amelioration of synovial inflammation in rheumatoid arthritis. J. Mol. Med. 2016, 94, 921–931. [Google Scholar] [CrossRef]
- Ka, S.O.; Song, M.Y.; Bae, E.J.; Park, B.H. Myeloid SIRT1 regulates macrophage infiltration and insulin sensitivity in mice fed a high-fat diet. J. Endocrinol. 2015, 224, 109–118. [Google Scholar] [CrossRef]
- Lee, A.S.; Jung, Y.J.; Kim, D.; Nguyen-Thanh, T.; Kang, K.P.; Lee, S.; Park, S.K.; Kim, W. SIRT2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem. Biophys. Res. Commun. 2014, 450, 1363–1369. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Gandhirajan, A.; Kibler, C.; Wang, X.; Vachharajani, V. Sirtuin 2 Dysregulates Autophagy in High-Fat-Exposed Immune-Tolerant Macrophages. Cells 2021, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Huang, G.; Wei, T.; Gao, J.; Huang, C.; Sun, M.; Zhu, L.; Shen, W. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Hu, G.; He, J.; Wang, T.; Zuo, Y.; Cao, Y.; Zheng, Q.; Tu, J.; Ma, J.; Cai, R.; et al. SENP1-Sirt3 signaling promotes alpha-ketoglutarate production during M2 macrophage polarization. Cell Rep. 2022, 39, 110660. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Colby, J.K.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The Role of PPAR-delta in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int. J. Mol. Sci. 2018, 19, 3339. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1beta Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Zhou, X.; Ou, M.; Xiao, G.; Li, F.; Wang, Z.; Wang, Z.; Liu, L.; Zhang, G. Sirtuin 7 Regulates Nitric Oxide Production and Apoptosis to Promote Mycobacterial Clearance in Macrophages. Front. Immunol. 2021, 12, 779235. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, Y.; Wang, H.; Zhang, W.; He, L.; Wu, J.; Liu, Y. Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int. J. Oncol. 2019, 55, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yang, Q.; Yang, Y.; Gao, Z.; Ma, Y.; Zhang, L.; Liang, W.; Ding, G. Sirt6 Suppresses High Glucose-Induced Mitochondrial Dysfunction and Apoptosis in Podocytes through AMPK Activation. Int. J. Biol. Sci. 2019, 15, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, Y.; Yang, P. High glucose-induced oxidative stress represses sirtuin deacetylase expression and increases histone acetylation leading to neural tube defects. J. Neurochem. 2016, 137, 371–383. [Google Scholar] [CrossRef]
- Chen, C.; Cao, J.; Gao, Y.; Xie, X.; Zhao, Z.; Yuan, Q.; He, Y.; Zu, X.; Liu, J. High glucose promotes macrophage M1 polarization through miR-32/Mef2d/cAMP signaling pathway. Genes Dis. 2024, 11, 539–541. [Google Scholar] [CrossRef]
- Geng, K.; Ma, X.; Jiang, Z.; Huang, W.; Gu, J.; Wang, P.; Luo, L.; Xu, Y.; Xu, Y. High glucose-induced STING activation inhibits diabetic wound healing through promoting M1 polarization of macrophages. Cell Death Discov. 2023, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Braga, T.T.; Correa-Costa, M.; Guise, Y.F.; Castoldi, A.; de Oliveira, C.D.; Hyane, M.I.; Cenedeze, M.A.; Teixeira, S.A.; Muscara, M.N.; Perez, K.R.; et al. MyD88 signaling pathway is involved in renal fibrosis by favoring a TH2 immune response and activating alternative M2 macrophages. Mol. Med. 2012, 18, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ren, J.; Gui, Y.; Wei, W.; Shu, B.; Lu, Q.; Xue, X.; Sun, X.; He, W.; Yang, J.; et al. Wnt/beta-Catenin-Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Yuan, S.; Wang, C.; Chang, J.; Tong, Y.; Liu, R.; Sang, T.; Li, L.; Li, J.; et al. TGF-beta1 peptide-based inhibitor P144 ameliorates renal fibrosis after ischemia-reperfusion injury by modulating alternatively activated macrophages. Cell Prolif. 2022, 55, e13299. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Cheng, S.; Lovett, D.H. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am. J. Pathol. 2003, 162, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Luo, N.S.; Ying, R.; Xie, Y.; Chen, J.Y.; Wang, X.Q.; Gu, Z.J.; Mai, J.T.; Liu, W.H.; Wu, M.X.; et al. Macrophage-derived foam cells impair endothelial barrier function by inducing endothelial-mesenchymal transition via CCL-4. Int. J. Mol. Med. 2017, 40, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Wynes, M.W.; Frankel, S.K.; Riches, D.W. IL-4-induced macrophage-derived IGF-I protects myofibroblasts from apoptosis following growth factor withdrawal. J. Leukoc. Biol. 2004, 76, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Eitner, F.; Alpers, C.E. A new look at platelet-derived growth factor in renal disease. J. Am. Soc. Nephrol. 2008, 19, 12–23. [Google Scholar] [CrossRef]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.T.; Hughes, J.; Sethi, T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef]
- Meng, X.M.; Mak, T.S.; Lan, H.Y. Macrophages in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 285–303. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. (2011) 2014, 4, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Wang, S.; Huang, X.R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.T.; Hanjani, N.A.; Movahed, E.; Doroudian, M. The role of macrophage subtypes and exosomes in immunomodulation. Cell Mol. Biol. Lett. 2022, 27, 83. [Google Scholar] [CrossRef]
- Luo, L.; Wang, S.; Hu, Y.; Wang, L.; Jiang, X.; Zhang, J.; Liu, X.; Guo, X.; Luo, Z.; Zhu, C.; et al. Precisely Regulating M2 Subtype Macrophages for Renal Fibrosis Resolution. ACS Nano 2023, 17, 22508–22526. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Dong, F.; Lu, J.; Wei, L.; Tian, L.; Ge, H.; Zou, Y.; Ma, X.; Yang, Y.; Zhou, L.; et al. Polarized M2c macrophages have a promoting effect on the epithelial-to-mesenchymal transition of human renal tubular epithelial cells. Immunobiology 2018, 223, 826–833. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef]
- Lu, Y.P.; Wu, H.W.; Zhu, T.; Li, X.T.; Zuo, J.; Hasan, A.A.; Reichetzeder, C.; Delic, D.; Yard, B.; Klein, T.; et al. Empagliflozin reduces kidney fibrosis and improves kidney function by alternative macrophage activation in rats with 5/6-nephrectomy. Biomed. Pharmacother. 2022, 156, 113947. [Google Scholar] [CrossRef] [PubMed]
- Ninichuk, V.; Clauss, S.; Kulkarni, O.; Schmid, H.; Segerer, S.; Radomska, E.; Eulberg, D.; Buchner, K.; Selve, N.; Klussmann, S.; et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3′PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am. J. Pathol. 2008, 172, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Fu, Y.X.; Shu, A.M.; Lv, X.; Chen, Y.P.; Gao, Y.Y.; Chen, J.; Wang, W.; Lv, G.H.; Lu, J.F.; et al. Loganin alleviates macrophage infiltration and activation by inhibiting the MCP-1/CCR2 axis in diabetic nephropathy. Life Sci. 2021, 272, 118808. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, S.G.; Ryu, M.; Kulkarni, O.P.; Schmid, H.; Lichtnekert, J.; Gruner, S.; Green, L.; Mattei, P.; Hartmann, G.; Anders, H.J. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011, 80, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qi, X.; Shao, Y.; Li, Y.; Fu, X.; Feng, S.; Wu, Y. Blockade of TGF-beta-activated kinase 1 prevents advanced glycation end products-induced inflammatory response in macrophages. Cytokine 2016, 78, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Ko, G.J.; Kang, Y.S.; Han, S.Y.; Lee, M.H.; Song, H.K.; Han, K.H.; Kim, H.K.; Han, J.Y.; Cha, D.R. Pioglitazone attenuates diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. Nephrol. Dial. Transplant. 2008, 23, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Ni, Z.; Qian, J.; Tomino, Y. Pravastatin inhibits carboxymethyllysine-induced monocyte chemoattractant protein 1 expression in podocytes via prevention of signalling events. Nephron Exp. Nephrol. 2007, 106, e1–e10. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Gao, X.; Wang, S.; Huang, M.; Sun, Z.; Dong, H.; Yu, H.; Wang, G. PPAR-alpha Agonist Fenofibrate Prevented Diabetic Nephropathy by Inhibiting M1 Macrophages via Improving Endothelial Cell Function in db/db Mice. Front. Med. 2021, 8, 652558. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.R.; Kang, Y.S.; Han, S.Y.; Jee, Y.H.; Han, K.H.; Kim, H.K.; Han, J.Y.; Kim, Y.S. Role of aldosterone in diabetic nephropathy. Nephrology 2005, 10 (Suppl. S2), S37–S39. [Google Scholar] [CrossRef]
- Kang, M.K.; Li, J.; Kim, J.L.; Gong, J.H.; Kwak, S.N.; Park, J.H.; Lee, J.Y.; Lim, S.S.; Kang, Y.H. Purple corn anthocyanins inhibit diabetes-associated glomerular monocyte activation and macrophage infiltration. Am. J. Physiol. Renal Physiol. 2012, 303, F1060–F1069. [Google Scholar] [CrossRef]
- Riek, A.E.; Oh, J.; Darwech, I.; Moynihan, C.E.; Bruchas, R.R.; Bernal-Mizrachi, C. 25(OH) vitamin D suppresses macrophage adhesion and migration by downregulation of ER stress and scavenger receptor A1 in type 2 diabetes. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt A, 172–179. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, M.; Wang, M.; Chen, L.; Liu, H.; Ren, Y.; Shi, K.; Jiang, H. Inhibition of macrophage migration inhibitory factor reduces diabetic nephropathy in type II diabetes mice. Inflammation 2014, 37, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, J.; Li, Y.Y.; Xia, L.L.; Wu, Y.G. Bruton’s tyrosine kinase regulates macrophage-induced inflammation in the diabetic kidney via NLRP3 inflammasome activation. Int. J. Mol. Med. 2021, 48, 177. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Shikata, K.; Miyasaka, K.; Okada, S.; Sasaki, M.; Kodera, R.; Hirota, D.; Kajitani, N.; Takatsuka, T.; Kataoka, H.U.; et al. Cholecystokinin plays a novel protective role in diabetic kidney through anti-inflammatory actions on macrophage: Anti-inflammatory effect of cholecystokinin. Diabetes 2012, 61, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Yu, M.R.; Kim, H.J.; Lee, J.H.; Park, B.W.; Wu, I.H.; Matsumoto, M.; King, G.L. Beta 2-adrenergic receptor agonists are novel regulators of macrophage activation in diabetic renal and cardiovascular complications. Kidney Int. 2017, 92, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.Z.; Chen, Q.; Lan, H.Y. Macrophage Migration Inhibitory Factor (MIF) as a Stress Molecule in Renal Inflammation. Int. J. Mol. Sci. 2022, 23, 4908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Guo, Y.F.; Song, Z.X.; Zhou, M. Vitamin D prevents podocyte injury via regulation of macrophage M1/M2 phenotype in diabetic nephropathy rats. Endocrinology 2014, 155, 4939–4950. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Y.; Zhu, X.; Guo, Y.; Yang, Y.; Jiang, Y.; Liu, B. Active vitamin D regulates macrophage M1/M2 phenotypes via the STAT-1-TREM-1 pathway in diabetic nephropathy. J. Cell Physiol. 2019, 234, 6917–6926. [Google Scholar] [CrossRef]
- Yuan, Y.; Sun, M.; Jin, Z.; Zheng, C.; Ye, H.; Weng, H. Dapagliflozin ameliorates diabetic renal injury through suppressing the self-perpetuating cycle of inflammation mediated by HMGB1 feedback signaling in the kidney. Eur. J. Pharmacol. 2023, 943, 175560. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jin, J.; Kim, D.H.; Kim, D.; Lee, Y.M.; Byun, J.K.; Choi, Y.K.; Park, K.G. SGLT2 inhibitors prevent LPS-induced M1 macrophage polarization and alleviate inflammatory bowel disease by downregulating NHE1 expression. Inflamm. Res. 2023, 72, 1981–1997. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. eBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Sheng, H.; Liang, C.; Liu, H.; Moran Guerrero, J.A.; Lu, Z.; Mao, W.; Dai, Z.; Liu, X.; et al. Hyperoside Suppresses Renal Inflammation by Regulating Macrophage Polarization in Mice with Type 2 Diabetes Mellitus. Front. Immunol. 2021, 12, 733808. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lei, X.Q.; Yang, J.K.; Jia, J.; Zhong, X.; Tan, R.Z.; Wang, L. Astragalus mongholicus Bunge and Panax notoginseng formula (A&P) improves renal mesangial cell damage in diabetic nephropathy by inhibiting the inflammatory response of infiltrated macrophages. BMC Complement. Med. Ther. 2022, 22, 17. [Google Scholar] [CrossRef]
- Liao, H.; Li, Y.; Zhang, X.; Zhao, X.; Zheng, D.; Shen, D.; Li, R. Protective Effects of Thalidomide on High-Glucose-Induced Podocyte Injury through In Vitro Modulation of Macrophage M1/M2 Differentiation. J. Immunol. Res. 2020, 2020, 8263598. [Google Scholar] [CrossRef]
- Jiandong, L.; Yang, Y.; Peng, J.; Xiang, M.; Wang, D.; Xiong, G.; Li, S. Trichosanthes kirilowii lectin ameliorates streptozocin-induced kidney injury via modulation of the balance between M1/M2 phenotype macrophage. Biomed. Pharmacother. 2019, 109, 93–102. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Liu, M.; Shao, Y.; Dong, X. Schisandrin C attenuates renal damage in diabetic nephropathy by regulating macrophage polarization. Am. J. Transl. Res. 2021, 13, 210–222. [Google Scholar] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed]
- Metzinger-Le Meuth, V.; Burtey, S.; Maitrias, P.; Massy, Z.A.; Metzinger, L. microRNAs in the pathophysiology of CKD-MBD: Biomarkers and innovative drugs. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 337–345. [Google Scholar] [CrossRef]
- M’Baya-Moutoula, E.; Louvet, L.; Molinie, R.; Guerrera, I.C.; Cerutti, C.; Fourdinier, O.; Nourry, V.; Gutierrez, L.; Morliere, P.; Mesnard, F.; et al. A multi-omics analysis of the regulatory changes induced by miR-223 in a monocyte/macrophage cell line. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2664–2678. [Google Scholar] [CrossRef]
- Zheng, D.; Wang, Y.; Cao, Q.; Lee, V.W.; Zheng, G.; Sun, Y.; Tan, T.K.; Wang, Y.; Alexander, S.I.; Harris, D.C. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp. Nephrol. 2011, 118, e87–e99. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.P.; Zheng, G.; Lee, V.W.; Ouyang, L.; Chang, D.H.; Mahajan, D.; Coombs, J.; Wang, Y.M.; Alexander, S.I.; et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007, 72, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, C.; Wang, X.M.; Lee, V.W.; Wang, Y.; Zheng, G.; Tan, T.K.; et al. Failed renoprotection by alternatively activated bone marrow macrophages is due to a proliferation-dependent phenotype switch in vivo. Kidney Int. 2014, 85, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Guiteras, R.; Sola, A.; Flaquer, M.; Hotter, G.; Torras, J.; Grinyo, J.M.; Cruzado, J.M. Macrophage Overexpressing NGAL Ameliorated Kidney Fibrosis in the UUO Mice Model. Cell Physiol. Biochem. 2017, 42, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Guiteras, R.; Sola, A.; Flaquer, M.; Manonelles, A.; Hotter, G.; Cruzado, J.M. Exploring macrophage cell therapy on Diabetic Kidney Disease. J. Cell Mol. Med. 2019, 23, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xia, Y.; Lin, X.; Feng, X.H.; Wang, Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab. Investig. 2014, 94, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Jiang, H.; Pan, J.; Huang, X.R.; Wang, Y.C.; Huang, H.F.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y.; Chen, J.H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.F.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, L.; Zhou, X.; Ponnusamy, M.; Tang, J.; Zhuang, M.A.; Tolbert, E.; Bayliss, G.; Bai, J.; Zhuang, S. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 2016, 89, 68–81. [Google Scholar] [CrossRef]
- Chung, A.C.; Huang, X.R.; Zhou, L.; Heuchel, R.; Lai, K.N.; Lan, H.Y. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol. Dial. Transplant. 2009, 24, 1443–1454. [Google Scholar] [CrossRef]
- Li, J.H.; Huang, X.R.; Zhu, H.-J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-β-dependent and -independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chung, A.C.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Zhang, Y.; Huang, X.R.; Ren, G.L.; Li, J.; Lan, H.Y. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget 2015, 6, 36984–36997. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999, 18, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Z.; Yang, J.; Mitch, W.E.; Wang, Y. JAK3/STAT6 Stimulates Bone Marrow-Derived Fibroblast Activation in Renal Fibrosis. J. Am. Soc. Nephrol. 2015, 26, 3060–3071. [Google Scholar] [CrossRef]
- Liang, H.; Liu, B.; Gao, Y.; Nie, J.; Feng, S.; Yu, W.; Wen, S.; Su, X. Jmjd3/IRF4 axis aggravates myeloid fibroblast activation and m2 macrophage to myofibroblast transition in renal fibrosis. Front. Immunol. 2022, 13, 978262. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, J.; Liang, H.; Xiao, P.; Lai, X.; Nie, J.; Yu, W.; Gao, Y.; Wen, S. Natural killer T cell/IL-4 signaling promotes bone marrow-derived fibroblast activation and M2 macrophage-to-myofibroblast transition in renal fibrosis. Int. Immunopharmacol. 2021, 98, 107907. [Google Scholar] [CrossRef]
- Zuloff-Shani, A.; Adunsky, A.; Even-Zahav, A.; Semo, H.; Orenstein, A.; Tamir, J.; Regev, E.; Shinar, E.; Danon, D. Hard to heal pressure ulcers (stage III–IV): Efficacy of injected activated macrophage suspension (AMS) as compared with standard of care (SOC) treatment controlled trial. Arch. Gerontol. Geriatr. 2010, 51, 268–272. [Google Scholar] [CrossRef]
- Leor, J.; Rozen, L.; Zuloff-Shani, A.; Feinberg, M.S.; Amsalem, Y.; Barbash, I.M.; Kachel, E.; Holbova, R.; Mardor, Y.; Daniels, D.; et al. Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation 2006, 114 (Suppl. S1), I94–I100. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Wang, B.; Xie, Y.; Cao, X.; Wang, M. The Role of M1 and M2 Myocardial Macrophages in Promoting Proliferation and Healing via Activating Epithelial-to-Mesenchymal Transition. Biomedicines 2023, 11, 2666. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Y.; Shen, S.E.; Huang, C.F.; Liu, Y.N.; Chen, Y.C.; Luo, L.; Zeng, Y.; Wang, A.P. Effect of activated autologous monocytes/macrophages on wound healing in a rodent model of experimental diabetes. Diabetes Res. Clin. Pract. 2013, 102, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Jetten, N.; Roumans, N.; Gijbels, M.J.; Romano, A.; Post, M.J.; de Winther, M.P.; van der Hulst, R.R.; Xanthoulea, S. Wound administration of M2-polarized macrophages does not improve murine cutaneous healing responses. PLoS ONE 2014, 9, e102994. [Google Scholar] [CrossRef] [PubMed]
- Agren, M.S.; Mirastschijski, U.; Karlsmark, T.; Saarialho-Kere, U.K. Topical synthetic inhibitor of matrix metalloproteinases delays epidermal regeneration of human wounds. Exp. Dermatol. 2001, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Denans, N.; Tran, N.T.T.; Swall, M.E.; Diaz, D.C.; Blanck, J.; Piotrowski, T. An anti-inflammatory activation sequence governs macrophage transcriptional dynamics during tissue injury in zebrafish. Nat. Commun. 2022, 13, 5356. [Google Scholar] [CrossRef] [PubMed]
- Lurier, E.B.; Dalton, D.; Dampier, W.; Raman, P.; Nassiri, S.; Ferraro, N.M.; Rajagopalan, R.; Sarmady, M.; Spiller, K.L. Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology 2017, 222, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Z.; Zhou, H.; Pan, H.; Han, W.; Deng, Y.; Li, Q.; Xue, J.; Ge, X.; Wang, S.; et al. First-in-human study of GFH018, a small molecule inhibitor of transforming growth factor-β receptor I inhibitor, in patients with advanced solid tumors. BMC Cancer 2024, 24, 444. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Kim, S.W.; Seok, S.H. A new era of macrophage-based cell therapy. Exp. Mol. Med. 2023, 55, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Callegari, I.O.M.; Rocha, G.Z.; Oliveira, A.G. Physical exercise, health, and disease treatment: The role of macrophages. Front. Physiol. 2023, 14, 1061353. [Google Scholar] [CrossRef]
- da Costa Fernandes, C.J.; da Cruz Rodrigues, K.C.; de Melo, D.G.; de Campos, T.D.P.; Dos Santos Canciglieri, R.; Simabuco, F.M.; da Silva, A.S.R.; Cintra, D.E.; Ropelle, E.R.; Pauli, J.R.; et al. Short-term strength exercise reduces the macrophage M1/M2 ratio in white adipose tissue of obese animals. Life Sci. 2023, 329, 121916. [Google Scholar] [CrossRef]
- Silveira, L.S.; Antunes Bde, M.; Minari, A.L.; Dos Santos, R.V.; Neto, J.C.; Lira, F.S. Macrophage Polarization: Implications on Metabolic Diseases and the Role of Exercise. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 115–132. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.-W.; Yang, T.-M.; Ho, C.; Shih, Y.-H.; Lin, C.-L.; Hsu, Y.-C. Targeting Macrophages: Therapeutic Approaches in Diabetic Kidney Disease. Int. J. Mol. Sci. 2024, 25, 4350. https://doi.org/10.3390/ijms25084350
Lin D-W, Yang T-M, Ho C, Shih Y-H, Lin C-L, Hsu Y-C. Targeting Macrophages: Therapeutic Approaches in Diabetic Kidney Disease. International Journal of Molecular Sciences. 2024; 25(8):4350. https://doi.org/10.3390/ijms25084350
Chicago/Turabian StyleLin, Da-Wei, Tsung-Ming Yang, Cheng Ho, Ya-Hsueh Shih, Chun-Liang Lin, and Yung-Chien Hsu. 2024. "Targeting Macrophages: Therapeutic Approaches in Diabetic Kidney Disease" International Journal of Molecular Sciences 25, no. 8: 4350. https://doi.org/10.3390/ijms25084350
APA StyleLin, D.-W., Yang, T.-M., Ho, C., Shih, Y.-H., Lin, C.-L., & Hsu, Y.-C. (2024). Targeting Macrophages: Therapeutic Approaches in Diabetic Kidney Disease. International Journal of Molecular Sciences, 25(8), 4350. https://doi.org/10.3390/ijms25084350