Molecular Pathways of Vulnerable Carotid Plaques at Risk of Ischemic Stroke: A Narrative Review

 , , ,
, , , 
Abstract
:1. Introduction
2. Vulnerable Plaques in the Proteomic Era
3. Risk Factors and Pathophysiology of the Vulnerability of Carotid Plaques
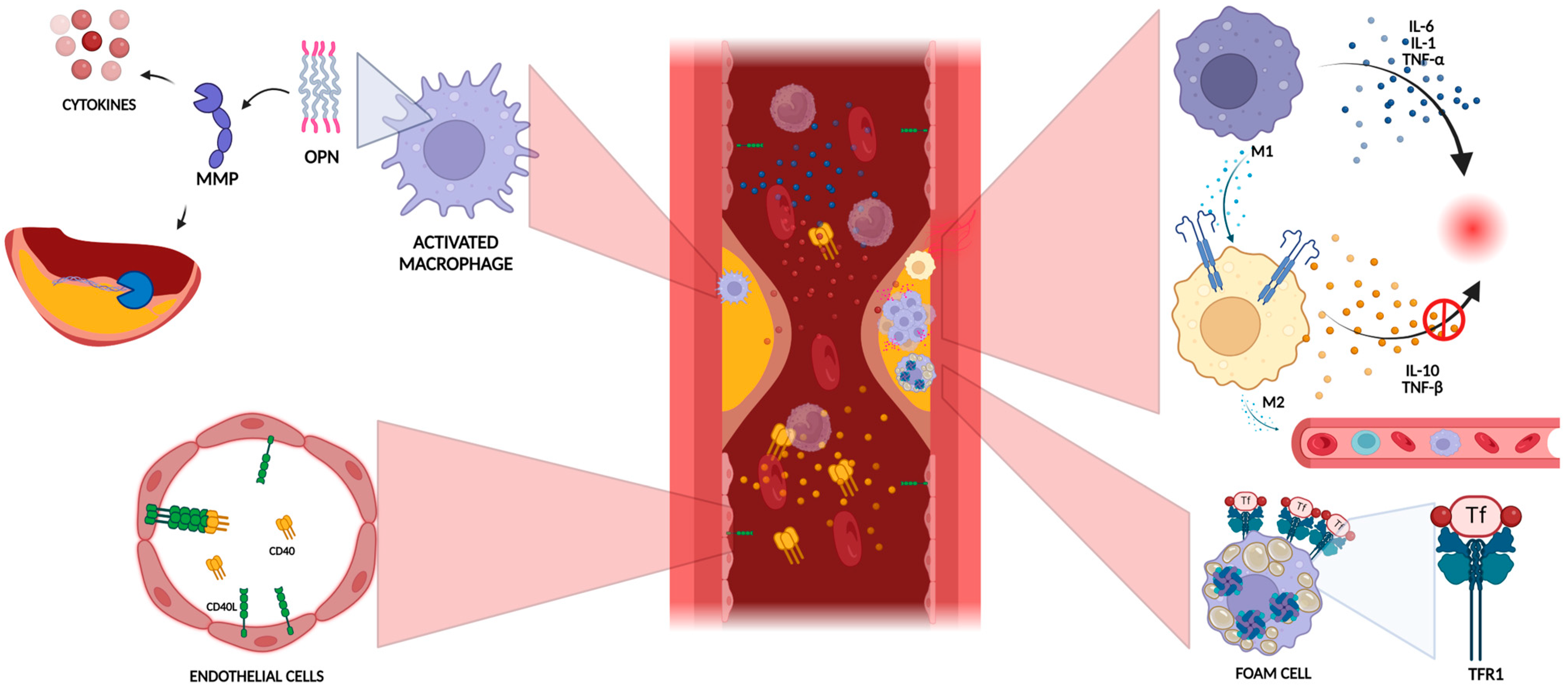
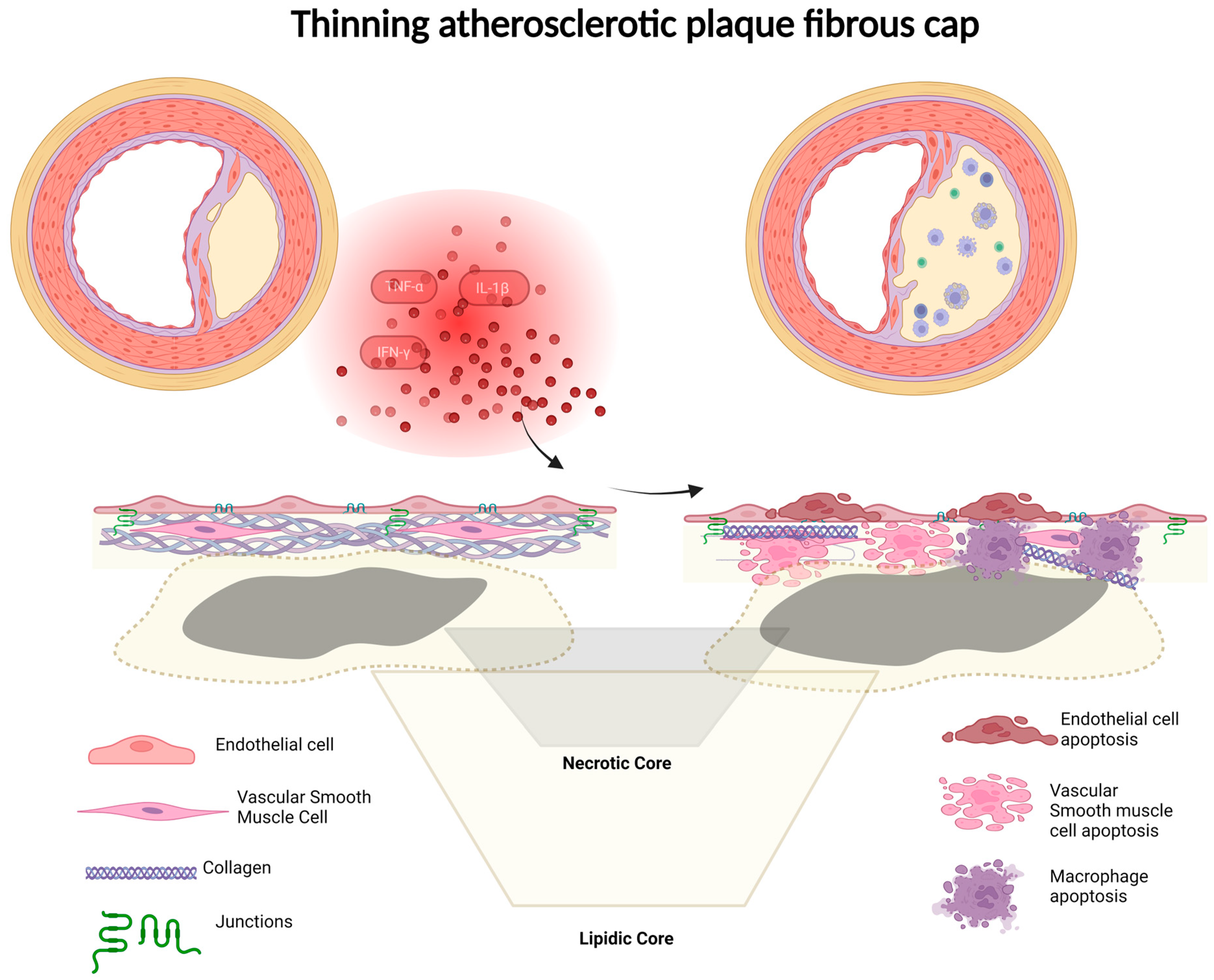
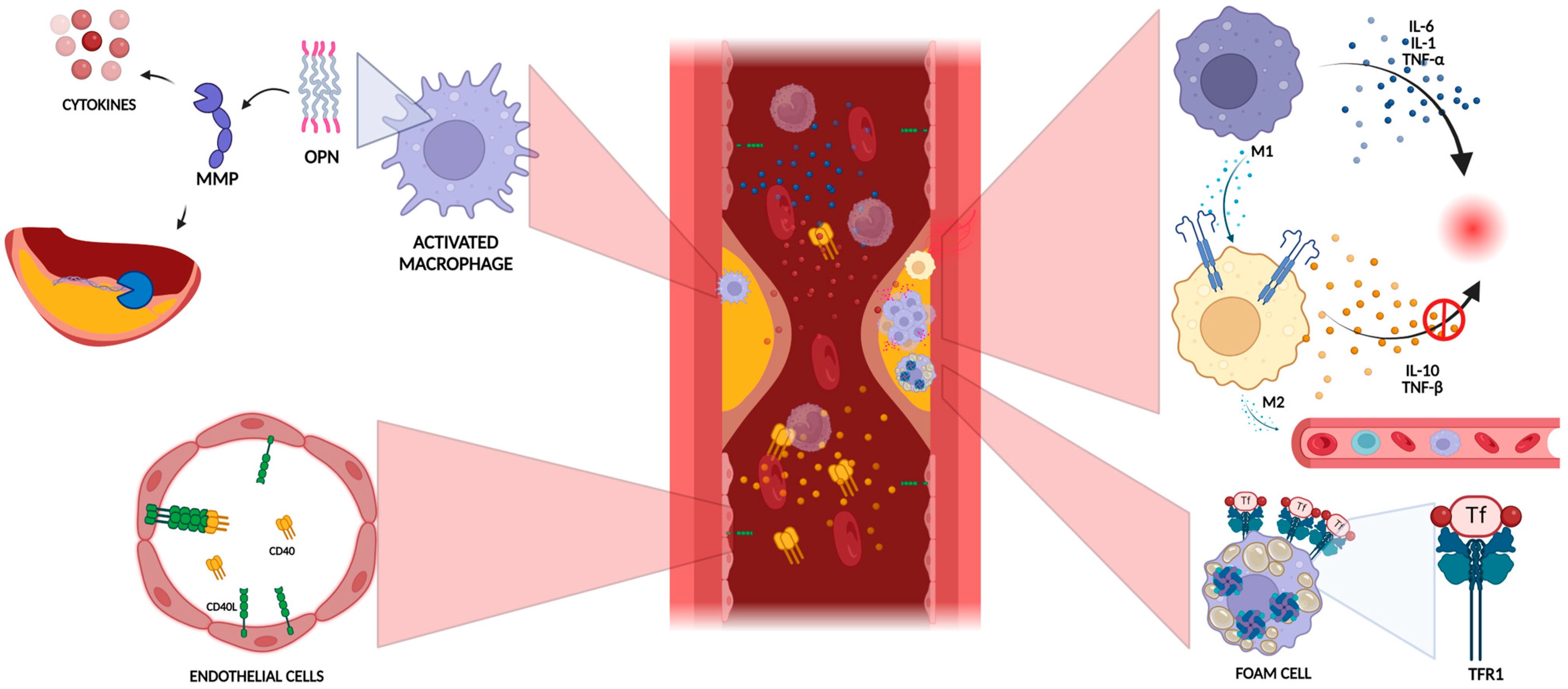
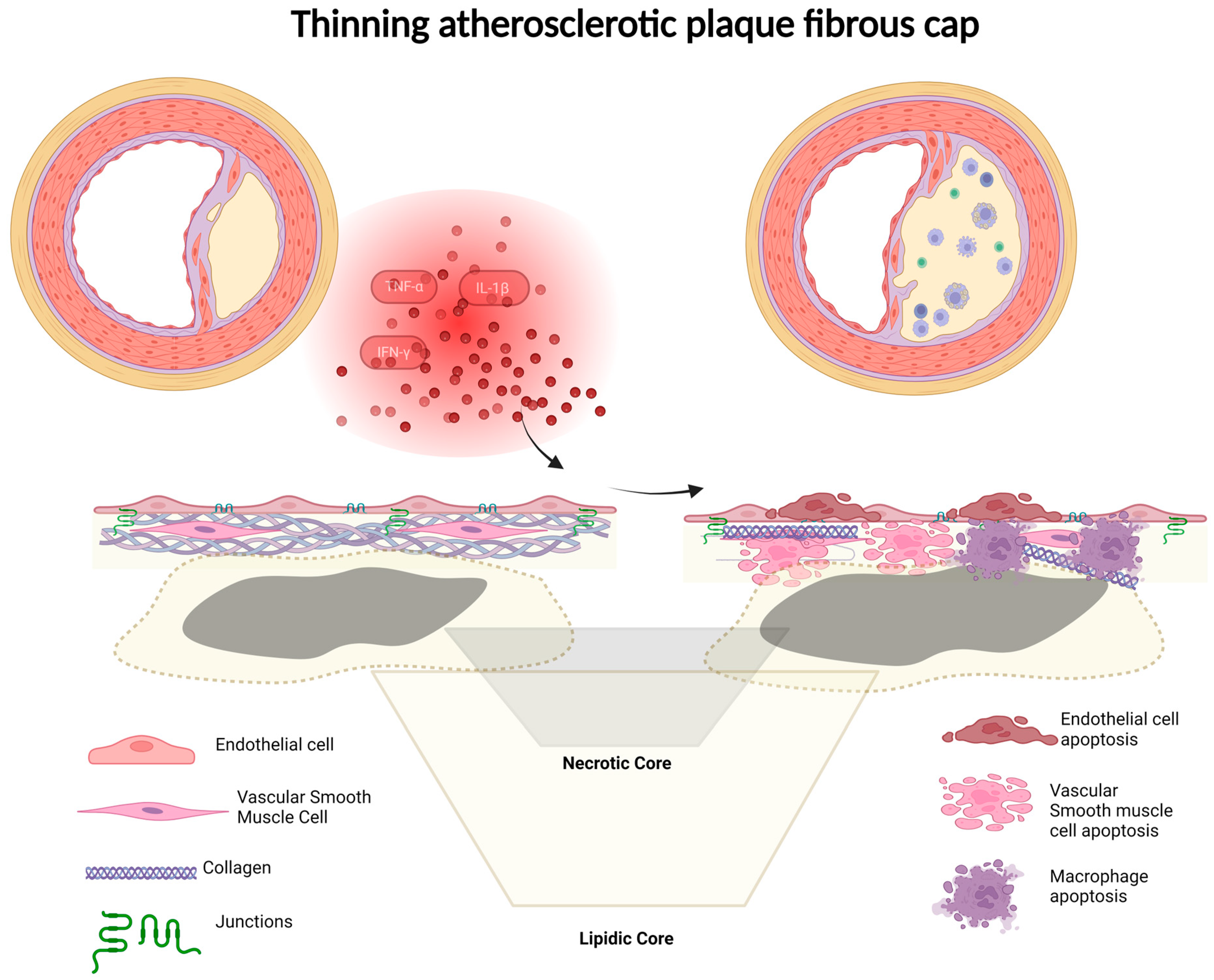
3.1. Role of Inflammation in Plaque Vulnerability
3.2. Extracellular Matrix Degradation
3.3. Lipid Metabolism
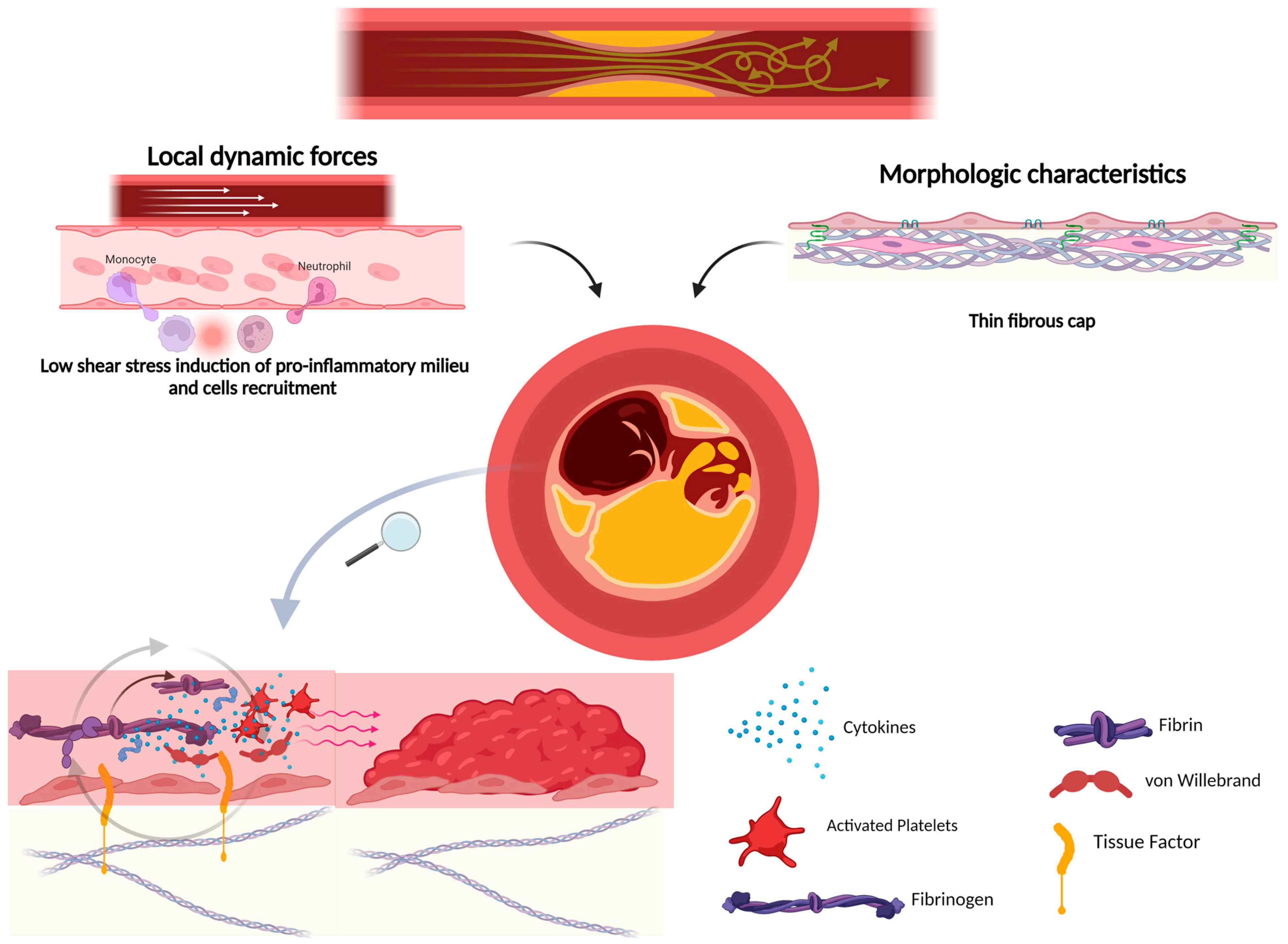
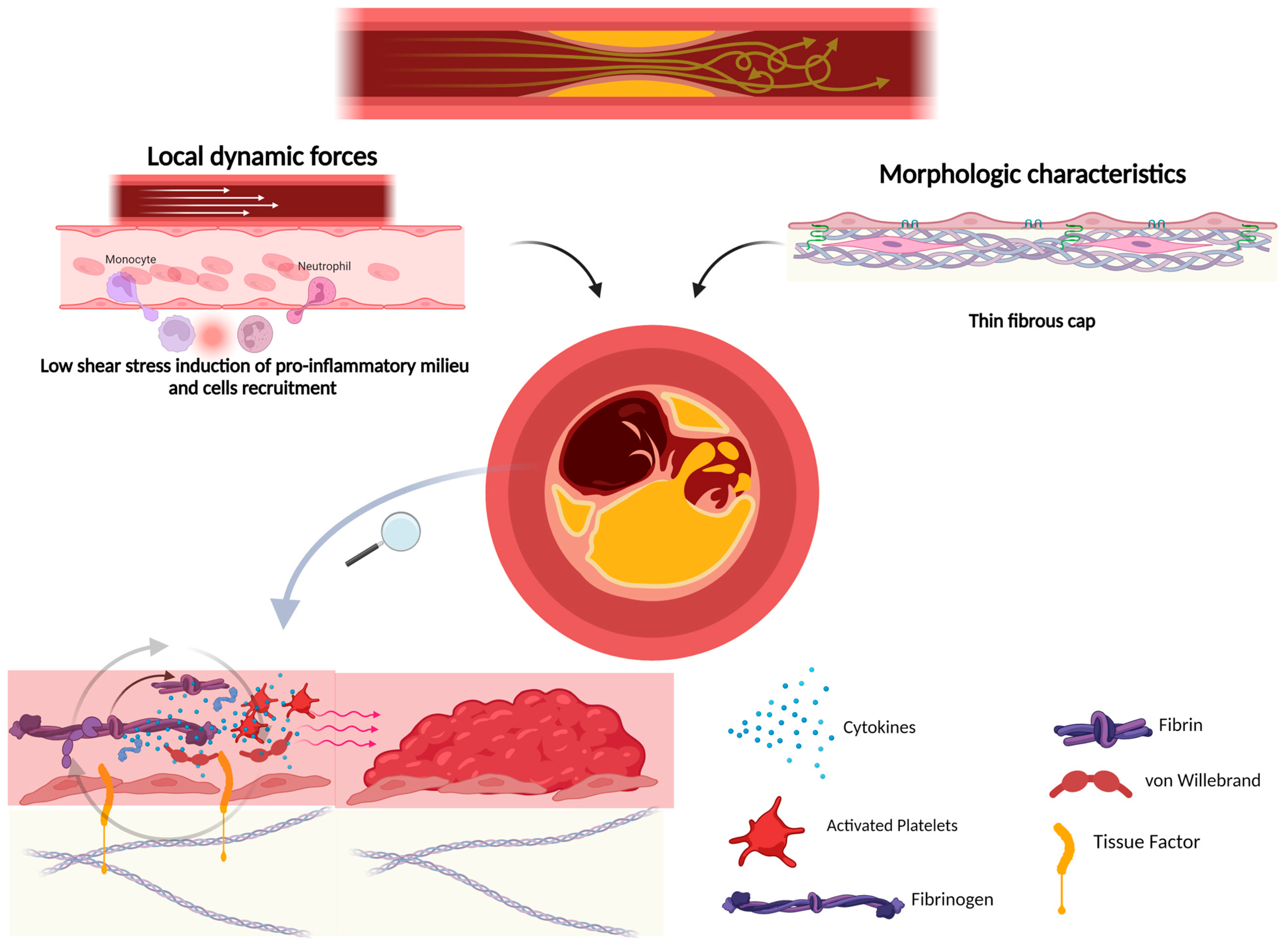
4. Plaque Rupture and Atherothrombosis
5. Plaque Ulceration
6. Microembolism
7. Fibrous Cap Thickness
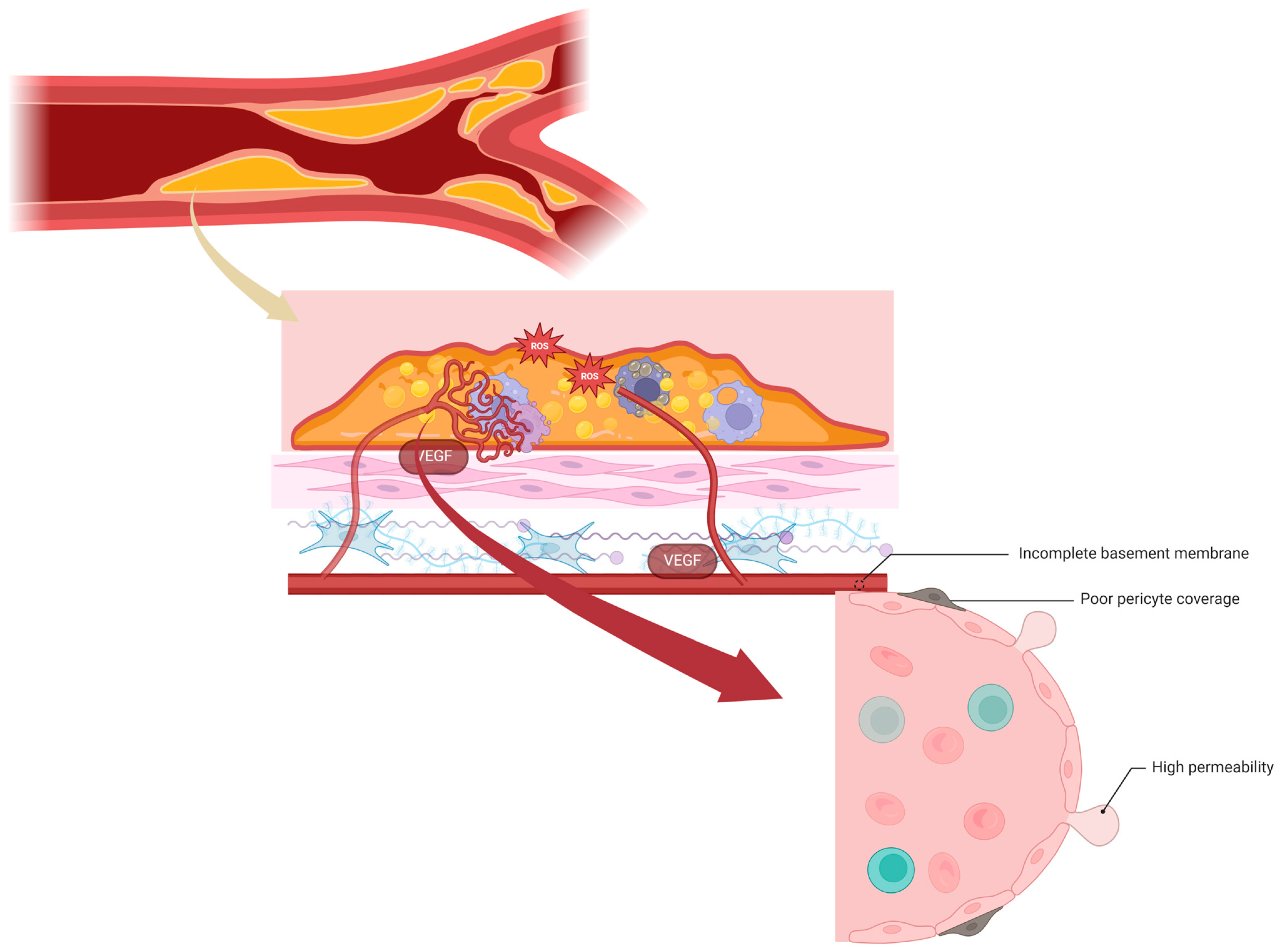
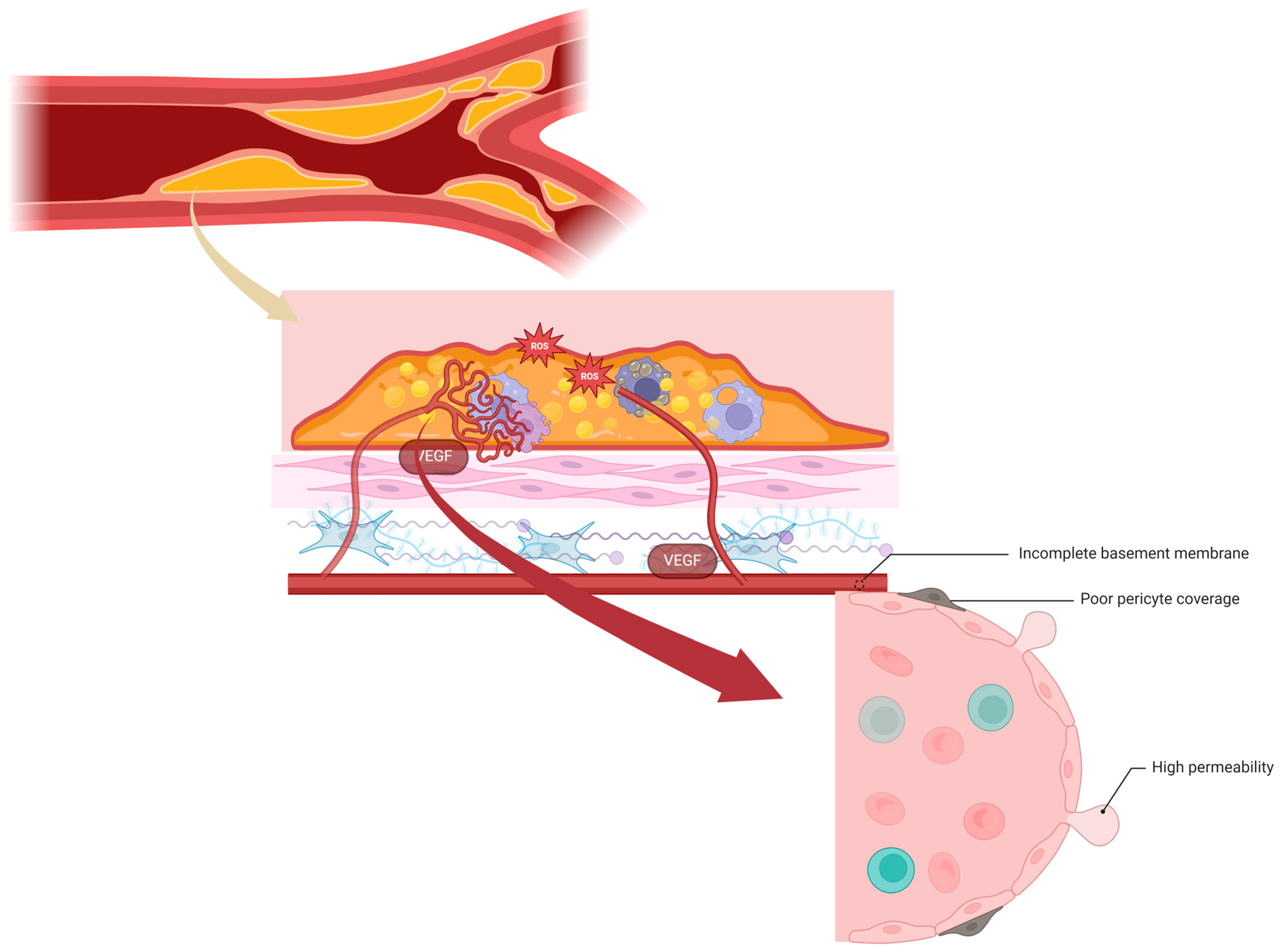
8. Neoangiogenesis and Intraplaque Hemorrhage
9. Lipidic Core
10. Microcalcification
11. Carotid Stenosis
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Katan, M.; Luft, A. Global burden of stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Burke, A.P.; Kolodgie, F.D.; Farb, A. Vulnerable plaque: The pathology of unstable coronary lesions. J. Interv. Cardiol. 2002, 15, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Willey, J.Z.; Pasterkamp, G. The Role of the Vulnerable Carotid Plaque in Embolic Stroke of Unknown Source. J. Am. Coll. Cardiol. 2022, 79, 2200–2202. [Google Scholar] [CrossRef] [PubMed]
- Kamtchum-Tatuene, J.; Noubiap, J.J.; Wilman, A.H.; Saqqur, M.; Shuaib, A.; Jickling, G.C. Prevalence of High-Risk Plaques and Risk of Stroke in Patients With Asymptomatic Carotid Stenosis: A Meta-analysis. JAMA Neurol. 2020, 77, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Kamtchum-Tatuene, J.; Wilman, A.; Saqqur, M.; Shuaib, A.; Jickling, G.C. Carotid Plaque With High-Risk Features in Embolic Stroke of Undetermined Source: Systematic Review and Meta-Analysis. Stroke 2020, 51, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Farrel, A.; Picton, A.; McCollum, C.N. Relationship between endothelial cell markers and arterial stenosis in peripheral and carotid artery disease. Thromb. Res. 2000, 97, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Saba, L.; Saam, T.; Jäger, H.R.; Yuan, C.; Hatsukami, T.S.; Saloner, D.; Wassrman, B.A.; Bonati, L.H.; Wintermark, M. Imaging biomarkers of vulnerable carotid plaques for stroke risk prediction and their potential clinical implications. Lancet Neurol. 2019, 18, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qiu, J.; Luo, S.; Xie, X.; Zheng, Y.; Zhang, K.; Ye, Z.; Liu, W.; Gregersen, H.; Wang, G. High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis. Regen Biomater. 2016, 3, 257–267. [Google Scholar] [CrossRef]
- Kashiwazaki, D.; Shiraishi, K.; Yamamoto, S.; Kamo, T.; Uchinom, H.; Saito, H.; Akioka, N.; Kuwayama, N.; Noguchi, K.; Kuroda, S. Efficacy of Carotid Endarterectomy for Mild (<50%) Symptomatic Carotid Stenosis with Unstable Plaque. World Neurosurg. 2019, 121, e60–e69. [Google Scholar] [CrossRef]
- Naghavi, M.; Libby, P.; Falk, E.; Casscells, S.W.; Litovsky, S.; Rumberger, J.; Badimon, J.J.; Stafanadis, C.; Moreno, P.; Pasterkamp, G.; et al. From vulnerable plaque to vulnerable patient: A call for new definitions and risk assessment strategies: Part I. Circulation 2003, 108, 1664–1672. [Google Scholar] [CrossRef]
- Miceli, G.; Rizzo, G.; Basso, M.G.; Cocciola, E.; Pennacchio, A.R.; Pintus, C.; Tuttolomondo, A. Artificial Intelligence in Symptomatic Carotid Plaque Detection: A Narrative Review. Appl. Sci. 2023, 13, 4321. [Google Scholar] [CrossRef]
- Lovett, J.K.; Gallagher, P.J.; Hands, L.J.; Walton, J.; Rothwell, P.M. Histological correlates of carotid plaque surface morphology on lumen contrast imaging. Circulation 2004, 110, 2190–2197. [Google Scholar] [CrossRef] [PubMed]
- Miceli, G.; Basso, M.G.; Rizzo, G.; Pintus, C.; Cocciola, E.; Pennacchio, A.R.; Tuttolomondo, A. Artificial Intelligence in Acute Ischemic Stroke Subtypes According to Toast Classification: A Comprehensive Narrative Review. Biomedicines 2023, 11, 1138. [Google Scholar] [CrossRef] [PubMed]
- Jander, S.; Sitzer, M.; Schumann, R.; Schroeter, M.; Siebler, M.; Steinmetz, H.; Stoll, G. Inflammation in high-grade carotid stenosis. A possible role for macrophages and T cells in plaque de-stabilization. Stroke 1998, 29, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, M.; Scimeca, M.; Toschi, N.; Bonanno, E.; Giacobbi, E.; Servadei, F.; Ippoliti, A.; Santeusanio, G.; Mauriello, A.; Anemona, L. Effects of Risk Factors on In Situ Expression of Proinflammatory Markers Correlated to Carotid Plaque Instability. Appl. Immunohistochem. Mol. Morphol. 2021, 29, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Tomas, L.; Edsfeldt, A.; Mollet, I.G.; Matic, L.P.; Prehn, C.; Adamski, J.; Paulsson-Berne, G.; Hedin, U.; Nilsson, J.; Bengtsson, E.; et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur. Heart J. 2018, 39, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Tengryd, C.; Nielsen, S.H.; Cavalera, M.; Bengtsson, E.; Genovese, F.; Karsdal, M.; Dunér, P.; Orho-Melander, M.; Nilsson, J.; Esfeldt, A.; et al. The proteoglycan mimecan is associated with carotid plaque vulnerability and increased risk of future cardiovascular death. Atherosclerosis 2020, 313, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernanez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Shirai, T.; Hilhorst, M.; Harrison, D.G.; Goronzy, J.J.; Wayand, C.M. Macrophages in vascular inflammation–from atherosclerosis to vasculitis. Autoimmunity 2015, 48, 139–151. [Google Scholar] [CrossRef]
- Arabpour, M.; Saghazadeh, A.; Rezaei, N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cell-derived exosomes. Int. Immunopharmacol. 2021, 97, 107823. [Google Scholar] [CrossRef] [PubMed]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.Q.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J.; Johns, M.; Kampfer, T.; Nguyen, T.; Game, L.; Schaer, D.J.; Mason, J.C.; Haskard, D.O. Activating transcription factor 1 directs mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 2012, 110, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Tyka, M.; Helmes, C.M.; Akhavanpoor, M.; Rupp, G.; Domschke, G.; Linden, F.; Wolf, A.; Doesch, A.; Lasitschka, F.; et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. 2015, 21, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ling, L.; Zhu, W.; Ying, T.; Yu, T.; Sun, M.; Zhu, X.; Du, Y.; Zhang, L. M1/M2 re-polarization of kaempferol biomimetic NPs in anti-inflammatory therapy of atherosclerosis. J. Control. Release 2023, 353, 1068–1083. [Google Scholar] [CrossRef]
- Bisgaard, L.S.; Mogensen, C.K.; Rosendahl, A.; Cucak, H.; Nielsen, L.B.; Rasmussen, S.E.; Pedersen, T.X. Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression-implications for atherosclerosis research. Sci. Rep. 2016, 6, 35234. [Google Scholar] [CrossRef] [PubMed]
- Stöger, J.L.; Gijbels, M.J.; VanderVelden, S.; Manca, M.; VanderLoos, C.M.; Biessen, E.A.L.; Daemen, M.J.A.P.; Lutgens, E.; DeWinther, M.P.J. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Dubois, S.G.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Qin, Y.; Zhang, Z.; Zhang, H.; Liu, H.; Li, C. Early identification of carotid vulnerable plaque in asymptomatic patients. BMC Cardiovasc. Disord. 2020, 20, 429. [Google Scholar] [CrossRef] [PubMed]
- Butler, W.T. The nature and significance of osteopontin. Connect. Tissue Res. 1989, 23, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Strobescu-Ciobanu, C.; Giusca, S.E.; Caruntu, I.D.; Amalinei, C.; Rusu, A.; Cojocaru, E.; Popa, R.F.; Lupasacu, C.D. Osteopontin and osteoprotegerin in atherosclerotic plaque—Are they significant markers of plaque vulnerability? Rom. J. Morphol. Embryol. 2020, 61, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001, 58, 4–43. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, U.; Libby, P. CD40 signaling and plaque instability. Circ. Res. 2001, 89, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Zhang, Y.W.; Zhang, P.; Deng, B.Q.; Ding, S.; Wang, Z.K.; Wu, T.; Wang, J. CD40 ligand as a potential biomarker for atherosclerotic instability. Neurol. Res. 2013, 35, 693–700. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Z.H.; Ren, Z.; Qu, S.L.; Liu, M.H.; Liu, L.S.; Jiang, Z.S. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol. Cell. Biol. 2013, 33, 1104–1113. [Google Scholar] [CrossRef]
- Ding, Z.; Wang, X.; Schnackenberg, L.; Khaidakov, M.; Liu, S.; Singla, S.; Dai, Y.; Metha, J.L. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7 g. Int. J. Cardiol. 2013, 168, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Ruiz, C.; Sabin, J.A.; Matas, M. High-sensitivity C-reactive protein in high-grade carotid stenosis: Risk marker for unstable carotid plaque. J. Vasc. Surg. 2003, 38, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Liu, Y.H.; Hosseini, H.; Kanellakis, P.; Cao, A.; Peter, K.; Tipping, P.; Bobik, A.; Toh, B.H.; Kyaw, T. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc. Res. 2016, 111, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.D.; Xiong, W.D.; Xiong, S.S.; Chen, G.H. Research Progress on the Risk Factors and Outcomes of Human Carotid Atherosclerotic Plaques. Chin. Med. J. 2017, 130, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Dengler, T.J.; Wangler, S.; Lasitschka, F.; Bea, F.; Wambsganss, N.; Hakimi, M.; Bockler, D.; Katus, H.A.; Gleissner, C.A. Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res. Cardiol. 2011, 106, 125–134. [Google Scholar] [CrossRef]
- Edsfeldt, A.; Nitulescu, M.; Grufman, H.; Gronberg, C.; Persson, A.; Nilsson, M.; Persson, M.; Bjorkbacka, H.; Goncalves, I. Soluble urokinase plasminogen activator receptor is associated with inflammation in the vulnerable human atherosclerotic plaque. Stroke 2012, 43, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Aukrust, P.; Dahl, T.B.; Bjerkeli, V.; Sagen, E.B.L.; Michelsen, A.; Russel, D.; Krohg-Sorensen, K.; Holm, S.; Skjelland, M.; et al. High levels of S100A12 are associated with recent plaque symptomatology in patients with carotid atherosclerosis. Stroke 2012, 43, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Shen, G.; Hu, Q.; Xiao, H.; Qin, X. The Circulating Biomarker Fractalkine and Platelet-Derived Growth Factor BB are Correlated with Carotid Plaque Vulnerability Assessed by Computed Tomography Angiography. J. Stroke Cerebrovasc. Dis. 2022, 31, 1052–3057. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Guo, L.; Chen, L.; Li, J.; Li, Q.; Pi, Y.; Zhu, J.; Zhang, L. A novel role of FKN/ CX3CRI in promoting osteogenic transformation of VSMCs and atherosclerotic calcification. Cell Calcium 2020, 91, 102265. [Google Scholar] [CrossRef]
- Ledard, N.; Lbioz, A.; Blondeau, B.; Babiak, M.; Moulin, C.; Vallin, B.; Guillas, I.; Mateo, V.; Jumeau, C.; Blirando, K.; et al. Slug, a cancer-related transcription factor, is involved in vascular smooth muscle cell transdifferentiation induced by platelet-derived growth factor-BB during atherosclerosis. J. Am. Heart Assoc. 2020, 9, 014276. [Google Scholar] [CrossRef]
- Müller, A.; Krämer, S.D.; Meletta, R.; Beck, K.; Selinova, S.V.; Rancic, Z.; Kaufmann, P.A.; Vos, B.; Meding, J.; Stellfeld, T.; et al. Gene expression levels of matrix metalloproteinases in human atherosclerotic plaques and evaluation of radiolabeled inhibitors as imaging agents for plaque vulnerability. Nucl. Med. Biol. 2014, 41, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, X.; Ma, Z.; Niu, J.; Ma, S.; Wenjie, W.; Chen, J. Combination of tanshinone IIA and astragaloside IV attenuate atherosclerotic plaque vulnerability in ApoE(−/−) mice by activating PI3K/AKT signaling and suppressing TRL4/NF-κB signaling. Biomed. Pharmacother. 2020, 123, 109729. [Google Scholar] [CrossRef] [PubMed]
- Loftus, I.M.; Naylor, A.R.; Goodall, S.; Jones, L.; Bell, P.R.; Thompson, M.M. Increased matrix metalloproteinase-9 activity in unstable carotid plaques. A potential role in acute plaque disruption. Stroke 2000, 31, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Devel, L.; Czarny, B.; George, S.J.; Jackson, C.L.; Rogakos, V.; Beau, F.; Yiotakis, A.; Newby, A.C.; Dive, V. A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Pelisek, J.; Deutsch, L.; Ansel, A.; Pongratz, J.; Stadlbauer, T.; Gebhard, H.; Matevossian, E.; Eckstein, H.H. Expression of a metalloproteinase family of ADAMTS in human vulnerable carotid lesions. J. Cardiovasc. Med. 2017, 18, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ashlin, T.G.; Kwan, A.P.; Ramji, D.P. Regulation of ADAMTS-1, -4 and -5 expression in human macrophages: Differential regulation by key cytokines implicated in atherosclerosis and novel synergism between TL1A and IL-17. Cytokine 2013, 64, 234–242. [Google Scholar] [CrossRef]
- Dong, H.; Du, T.; Premaratne, S.; Zhao, C.X.; Tian, Q.; Li, Y.; Yan, S.; Zhang, W.W. Relationship between ADAMTS4 and carotid atherosclerotic plaque vulnerability in humans. J. Vasc. Surg. 2018, 67, 1120–1126. [Google Scholar] [CrossRef]
- Demircan, K.; Topcu, V.; Takigawa, T.; Akyol, S.; Yonezawa, T.; Ozturk, G.; Ugurcu, V.; Hasgul, R.; Yigitoglu, M.R.; Akyol, O.; et al. ADAMTS4 and ADAMTS5 knockout mice are protected from versican but not aggrecan or brevican proteolysis during spinal cord injury. BioMed Res. Int. 2014, 2014, 693746. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Karathanasis, S.K.; Ding, Z.; Arulandu, A.; Varughese, K.I.; Metha, J.L. LOX-1 in Atherosclerosis and Myocardial Ischemia: Biology, Genetics, and Modulation. J. Am. Coll. Cardiol. 2017, 69, 2759–2768. [Google Scholar] [CrossRef]
- Jin, P.; Cong, S. LOX-1 and atherosclerotic-related diseases. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 491, 24–29. [Google Scholar] [CrossRef]
- Norata, G.D.; Raselli, S.; Grigore, L.; Garlaschelli, K.; Vianello, D.; Bertocco, S.; Zambon, A.; Catapano, A.L. Small dense LDL and VLDL predict common carotid artery IMT and elicit an inflammatory response in peripheral blood mononuclear and endothelial cells. Atherosclerosis 2009, 206, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.; Puato, M.; Faggin, E.; Faggin, E.; Grego, F.; Rattazzi, M.; Pauletto, P. Lipoprotein remnants and dense LDL are associated with features of unstable carotid plaque: A flag for non-HDL-C. Atherosclerosis 2013, 230, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Cai, H.; Wang, F.; Liu, R.; Xu, X.; Li, M.; Han, Y.; Yin, Q.; Ye, R.; Liu, X. Cholesterol Crystals are Associated with Carotid Plaque Vulnerability: An Optical Coherence Tomography Study. J. Stroke Cerebrovasc. Dis. 2020, 29, 104579. [Google Scholar] [CrossRef] [PubMed]
- Janoudi, A.; Shamoun, F.E.; Kalavakunta, J.K.; Abela, G.S. Cholesterol crystal-induced arterial inflammation and destabilization of atherosclerotic plaque. Eur. Heart J. 2016, 37, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Q.; Xu, J.; Zhu, W.; Jiang, J.; Tang, L.; Chen, M. Contrast-enhanced ultrasound perfusion patterns and serum lipid signatures of vulnerable carotid artery plaque in predicting stroke: A cohort study of carotid stenosis in Chinese patients. Clin. Hemorheol. Microcirc. 2020, 75, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, A.; Hulsmans, M.; Holvoet, P.; Monaco, C. Biomechanical factors and macrophages in plaque stability. Cardiovasc. Res. 2013, 99, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Kamm, R.D. Vascular mechanics for the cardiologist. J. Am. Coll. Cardiol. 1994, 23, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Slager, C.J.; Wentzel, J.J.; Gijsen, F.J.; Schuurbiers, J.C.; van der Wal, A.C.; van der Steen, A.F.; Serruys, P.W. The role of shear stress in the generation of rupture-prone vulnerable plaques. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Slager, C.J.; Wentzel, J.J.; Gijsen, F.J.; Thury, A.; van der Wal, A.C.; Schaar, J.A.; Serruys, P.W. The role of shear stress in the destabilization of vulnerable plaques and related therapeutic implications. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 456–464. [Google Scholar] [CrossRef]
- Otsuka, F.; Finn, A.V.; Yazdani, S.K.; Nakano, M.; Kolodgie, F.D.; Virmani, R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat. Rev. Cardiol. 2012, 9, 439–453. [Google Scholar] [CrossRef]
- Freynhofer, M.K.; Bruno, V.; Wojta, J.; Huber, K. The role of platelets in atherothrombotic events. Curr. Pharm. Des. 2012, 18, 5197–5214. [Google Scholar] [CrossRef] [PubMed]
- Miceli, G.; Basso, M.G.; Rizzo, G.; Pintus, C.; Tuttolomondo, A. The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies. Int. J. Mol. Sci. 2022, 23, 14914. [Google Scholar] [CrossRef] [PubMed]
- Penz, S.; Reininger, A.J.; Brandl, R.; Goyal, P.; Rabie, T.; Bernlochner, I.; Rother, E.; Goetz, C.; Engelmann, B.; Smethurst, P.A.; et al. Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. FASEB J. 2005, 19, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Badimon, J.J.; Lettino, M.; Toschi, V.; Fuster, V.; Berrozpe, M.; Chesebro, J.H.; Badimon, L. Local inhibition of tissue factor reduces the thrombogenicity of disrupted human atherosclerotic plaques: Effects of tissue factor pathway inhibitor on plaque thrombogenicity under flow conditions. Circulation 1999, 99, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Camino-Lopez, S.; Llorente-Cortes, V.; Sendra, J.; Badimon, L. Tissue factor induction by aggregated LDL depends on LDL receptor-related protein expression (LRP1) and Rho A trans-location in human vascular smooth muscle cells. Cardiovasc. Res. 2007, 73, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Hemker, H.C.; van Rijn, J.L.; Rosing, J.; van Dieijen, G.; Bevers, E.M.; Zwaal, R.F. Platelet membrane involvement in blood coagulation. Blood Cells 1983, 9, 303–317. [Google Scholar] [PubMed]
- Grad, E.; Danenberg, H.D. C-reactive protein and atherothrombosis: Cause or effect? Blood Rev. 2013, 27, 23–29. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Van Avondt, K.; Hartl, D. Mechanisms and disease relevance of neutrophil extracellular trap formation. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), 12919. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Shimonaga, K.; Matsushige, T.; Takahashi, H.; Hashimoto, Y.; Yoshiyama, M.; Ono, C.; Sakamoto, S. Peptidylarginine Deiminase 4 as a Possible Biomarker of Plaque Instability in Carotid Artery Stenosis. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2021, 30, 1052–3057. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldan, A.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 Has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, M.; Fotakis, P.; Liu, W.; Endo-Umeda, K.; Dou, H.; Abramowicz, S.; Xiao, T.; Libby, P.; Wang, N.; Tall, A.R.; et al. Cholesterol accumulation in macrophages drives NETosis in atherosclerotic plaques via IL-1β secretion. Cardiovasc. Res. 2023, 119, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Usman, A.; Das, T.; Patterson, A.J.; Gillard, J.H.; Graves, M.J. Imaging Carotid Atherosclerosis Plaque Ulceration: Comparison of Advanced Imaging Modalities and Recent Developments. AJNR Am. J. Neuroradiol. 2017, 38, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Rafailidis, V.; Chryssogonidis, I.; Tegos, T.; Kouskouras, K.; Charitanti-Kouridou, A. Imaging of the ulcerated carotid atherosclerotic plaque: A literature review. Insights Imaging 2017, 8, 213–225. [Google Scholar] [CrossRef]
- Fisher, M.; Paganini-Hill, A.; Martin, A.; Cosgrove, M.; Toole, J.F.; Barnett, H.J.; Norris, J. Carotid plaque pathology: Thrombosis, ulceration, and stroke pathogenesis. Stroke 2005, 36, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Nieddu, G.; Formato, M.; Lepedda, A.J. Searching for Atherosclerosis Biomarkers by Proteomics: A Focus on Lesion Pathogenesis and Vulnerability. Int. J. Mol. Sci. 2023, 24, 15175. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Duran, M.C.; Blanco-Colio, L.M.; Meilhac, O.; Leclercq, A.; Michel, J.B.; Jensen, O.N.; Hernandez-Merida, S.; Tuñón, J.; Vivanco, F.; et al. Identification by a differential proteomic approach of heat shock protein 27 as a potential marker of atherosclerosis. Circulation 2004, 110, 2216–2219. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Simionescu, M.; Sima, A.V. Morphology of atherosclerotic lesions. In Inflammation and Atherosclerosis; Springer: Vienna, Austria, 2012; pp. 19–37. [Google Scholar]
- Tabas, I.; Tall, A.; Accili, D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ. Res. 2010, 106, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Masuda, J.; Raines, E.W.; Gown, A.M.; Katsuda, S.; Sasahara, M.; Malden, L.T.; Masuko, H.; Sato, H. Localization of PDGF-B protein in macrophages in all phases of atherogenesis. Science 1990, 248, 1009–1012. [Google Scholar] [CrossRef]
- Gilbertson-Beadling, S.K.; Fisher, C. A potential role for N-cadherin in mediating endothelial cell-smooth muscle cell interactions in the rat vasculature. Lab. Investig. 1993, 69, 203–209. [Google Scholar]
- Mann, J.M.; Davies, M.J. Vulnerable plaque. Relation of characteristics to degree of stenosis in human coronary arteries. Circulation 1996, 94, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, G.; Robenek, H.; Beuck, M.; Krause, R.; Schurek, A.; Niemann, R. Ca++ antagonists and ACAT inhibitors promote cholesterol efflux from macrophages by different mechanisms. I. Characterization of cellular lipid metabolism. Arteriosclerosis 1988, 8, 46–56. [Google Scholar] [CrossRef]
- Laitinen, M.; Zachary, I.; Breier, G.; Pakkanen, T.; Häkkinen, T.; Luoma, J.; Abedi, H.; Risau, W.; Soma, M.; Laakso, M.; et al. VEGF gene transfer reduces intimal thickening via increased production of nitric oxide in carotid arteries. Hum. Gene Ther. 1997, 8, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Bazan, H.A.; Brooks, A.J.; Vongbunyong, K.; Tee, C.; Douglas, H.F.; Klingenber, N.C.; Woods, T.C. A pro-inflammatory and fibrous cap thinning transcriptome profile accompanies carotid plaque rupture leading to stroke. Sci. Rep. 2022, 12, 13499. [Google Scholar] [CrossRef]
- Xu, J.; Lu, X.; Shi, G.P. Vasa vasorum in atherosclerosis and clinical significance. Int. J. Mol. Sci. 2015, 16, 11574–11608. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harb. Perspect. Med. 2012, 2, a006544. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. BioMed Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Narula, J.; Farb, A. When neoangiogenesis ricochets. Am. Heart J. 1998, 136, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Thomas, A.C. Plaque fissuring–the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br. Heart J. 1985, 53, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Mofidi, R.; Crotty, T.B.; McCarthy, P.; Sheehan, S.J.; Mehigan, D.; Keaveny, T.V. Association between plaque instability, angiogenesis, and symptomatic carotid occlusive disease. Br. J. Surg. 2001, 88, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Mattavelli, E.; Grigore, L.; Pellegatta, F.; Magni, P.; Catapano, A.L. Targeted Plasma Proteomics to Predict the Development of Carotid Plaques. Stroke 2022, 53, e411–e414. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Libby, P.; Falk, E.; Casscells, S.W.; Litovsky, S.; Rumberger, J.; Badimon, J.J.; Stefanadis, C.; Moreno, P.; Pasterkamp, G.; et al. From vulnerable plaque to vulnerable patient: A call for new definitions and risk assessment strategies: Part II. Circulation 2003, 108, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Witztum, J.L. The oxidation hypothesis of atherosclerosis. Lancet 1994, 344, 793–795. [Google Scholar] [CrossRef]
- Berliner, J.A.; Navab, M.; Fogelman, A.M.; Frank, J.S.; Demer, L.L.; Edwards, P.A.; Watson, A.D.; Lusis, A.J. Atherosclerosis: Basic mechanisms. Oxidation, inflammation, and genetics. Circulation 1995, 91, 2488–2496. [Google Scholar] [CrossRef]
- Isoviita, P.M.; Nuotio, K.; Saksi, J.; Turunen, R.; Ijäs, P.; Pitkäniemi, J.; Soinne, L.; Kaste, M.; Kovanen, P.T.; Lindsberg, P.J. An imbalance between CD36 and ABCA1 protein expression favors lipid accumulation in stroke-prone ulcerated carotid plaques. Stroke 2010, 41, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Lammers, B.; Zhao, Y.; Meurs, I.; VanBerker, T.J.C.; VanEck, M. ATP-binding cassette transporters A1 and G1, HDL metabolism, cholesterol efflux, and inflammation: Important targets for the treatment of atherosclerosis. Curr. Drug Targets 2011, 12, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.H.; Tallis, G.; Yalamoori, V.; Anantharamaiah, G.M.; Segrest, J.P. Liposome-like particles isolated from human atherosclerotic plaques are structurally and compositionally similar to surface remnants of triglyceride-rich lipoproteins. Arterioscler. Thromb. 1994, 14, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Chisolm, G.M.; Ma, G.; Irwin, K.C.; Martin, L.L.; Gunderson, K.G.; Linberg, L.F.; Morel, D.W.; DiCorleto, P.E. 7 beta-hydroperoxycholest-5-en-3 beta-ol, a component of human atherosclerotic lesions, is the primary cytotoxin of oxidized human low-density lipoprotein. Proc. Natl. Acad. Sci. USA 1994, 91, 11452–11456. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Davies, T.S. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015, 26, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Torii, S.; Kutyna, M.; Sakamoto, A.; Finn, A.V.; Virmani, R. Coronary Artery Calcification and its Progression: What Does it Really Mean? JACC Cardiovasc. Imaging 2018, 11, 127–142. [Google Scholar] [CrossRef]
- Nadra, I.; Boccaccini, A.R.; Philippidis, P.; Whelan, L.C.; McCarthy, G.M.; Haskard, D.O.; Landis, R.C. Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis 2008, 196, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Pazár, B.; Ea, H.K.; Narayan, S.; Kolly, L.; Bagnoud, N.; Chobaz, V.; Roger, T.; Lioté, F.; So, A.; Busso, N. Basic calcium phosphate crystals induce monocyte/macrophage IL-1β secretion through the NLRP3 inflammasome in vitro. J. Immunol. 2011, 186, 2495–2502. [Google Scholar] [CrossRef]
- Viegas, C.S.B.; Costa, R.M.; Santos, L.; Videira, P.A.; Silva, Z.; Araújo, N.; Macedo, A.L.; Matos, A.P.; Vermeer, C.; Simes, D.C. Gla-rich protein function as an anti-inflammatory agent in monocytes/macrophages: Implications for calcification-related chronic inflammatory diseases. PLoS ONE 2018, 13, e0192875. [Google Scholar] [CrossRef]
- Ding, J.; Ghali, O.; Lencel, P.; Broux, O.; Chauveau, C.; Devedjian, J.C.; Hardouin, P.; Magne, D. TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci. 2009, 84, 499–504. [Google Scholar] [CrossRef]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Nadra, I.; Mason, J.C.; Philippidis, P.; Florey, O.; Smythe, C.D.; McCarthy, G.M.; Landis, R.C.; Haskard, D.O. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: A vicious cycle of inflammation and arterial calcification? Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Ewence, A.E.; Bootman, M.; Roderick, H.L.; Skepper, J.N.; McCarthy, G.; Epple, M.; Neumann, M.; Shanahan, C.M.; Proudfoot, D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: A potential mechanism in atherosclerotic plaque destabilization. Circ. Res. 2008, 103, e28–e34. [Google Scholar] [CrossRef] [PubMed]
- Reid, P.; Holen, I. Pathophysiological roles of osteoprotegerin (OPG). Eur. J. Cell Biol. 2009, 88, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Hong, B.K.; Kang, T.S.; Kwon, K.; Kim, H.K.; Jang, Y.; Choi, D.; Park, H.Y.; Kang, S.M.; Cho, S.Y.; et al. Expression of osteopontin in calcified coronary atherosclerotic plaques. J. Korean Med. Sci. 2000, 15, 485–493. [Google Scholar] [CrossRef]
- Momiyama, Y.; Ohmori, R.; Fayad, Z.A.; Kihara, T.; Tanaka, N.; Kato, R.; Taniguchi, H.; Nagata, M.; Nakamura, H.; Ohsuzu, F. Associations between plasma osteopontin levels and the severities of coronary and aortic atherosclerosis. Atherosclerosis 2010, 210, 668–670. [Google Scholar] [CrossRef]
- Maruyama, M.; Rhee, C.; Utsunomiya, T.; Zhang, N.; Ueno, M.; Yao, Z.; Goodman, S.B. Modulation of the Inflammatory Response and Bone Healing. Front. Endocrinol. 2020, 11, 386. [Google Scholar] [CrossRef]
- Gerstenfeld, L.C.; Cho, T.J.; Kon, T.; Aizawa, T.; Cruceta, J.; Graves, B.D.; Einhorn, T.A. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs 2001, 169, 285–294. [Google Scholar] [CrossRef]
- Li, X.; Guo, D.; Hu, Y.; Chen, Y. Oxidative Stress and Inflammation Are Associated with Coexistent Severe Multivessel Coronary Artery Stenosis and Right Carotid Artery Severe Stenosis in Elderly Patients. Oxidative Med. Cell. Longev. 2021, 2021, 2976447. [Google Scholar] [CrossRef]
- Handberg, A.; Skjelland, M.; Michelsen, A.E.; Sagen, E.L.; Krohg-Sørensen, K.; Russell, D.; Dahl, A.; Ueland, T.; Oie, E.; Aukrust, P.; et al. Soluble CD36 in plasma is increased in patients with symptomatic atherosclerotic carotid plaques and is related to plaque instability. Stroke 2008, 39, 3092–3095. [Google Scholar] [CrossRef]
- Jin, G. The relationship between serum CXCL16 level and carotid vulnerable plaque in patients with ischemic stroke. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3911–3915. [Google Scholar] [PubMed]
- Profumo, E.; Buttari, B.; Tosti, M.E.; Salvati, B.; Capoano, R.; Riganò, R. Increased circulating levels of MIP-1α and CD14 are associated with the presence of severe stenosis and hypoechoic plaques in patients with carotid atherosclerosis. Int. J. Immunopathol. Pharmacol. 2023, 37, 3946320231160411. [Google Scholar] [CrossRef] [PubMed]
- Nuotio, K.; Lindsberg, P.J.; Carpén, O.; Soinne, L.; Lehtonen-Smeds, E.M.; Saimanen, E.; Lassila, R.; Sairanen, T.; Sarna, S.; Salonen, O.; et al. Adhesion molecule expression in symptomatic and asymptomatic carotid stenosis. Neurology 2003, 60, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Sayed, S.; Cockerill, G.W.; Torsney, E.; Poston, R.; Thompson, M.M.; Loftus, I.M. Elevated tissue expression of thrombomodulatory factors correlates with acute symptomatic carotid plaque phenotype. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Rocchiccioli, S.; Pelosi, G.; Rosini, S.; Marconi, M.; Viglione, F.; Citti, L.; Ferrari, M.; Trivella, M.G.; Cecchettini, A. Secreted proteins from carotid endarterectomy: An untargeted approach to disclose molecular clues of plaque progression. J. Transl. Med. 2013, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Dellas, C.; Schremmer, C.; Hasenfuss, G.; Konstantinides, S.V.; Schafer, K. Lack of urokinase plasminogen activator promotes progression and instability of atherosclerotic lesions in apolipoprotein E-knockout mice. Thromb Haemost. 2007, 98, 220–227. [Google Scholar] [CrossRef]
- van Dijk, A.C.; Donkel, S.J.; Zadi, T.; Sonneveld, M.A.H.; Schreuder, F.H.B.M.; Chohan, M.F.; Koudstaal, P.J.; Leebeek, F.W.G.; Saxena, R.; Hendrikse, J.; et al. Association between fibrinogen and fibrinogen γ’ and atherosclerotic plaque morphology and composition in symptomatic carotid artery stenosis: Plaque-At-RISK study. Thromb. Res. 2019, 177, 130–135. [Google Scholar] [CrossRef]
- Spencer, M.P.; Thomas, G.I.; Nicholls, S.C.; Sauvage, L.R. Detection of middle cerebral artery emboli during carotid endarterectomy using transcranial Doppler ultrasonography. Stroke 1990, 21, 415–423. [Google Scholar] [CrossRef]
- Puz, P.; Lasek-Bal, A.; Urbanek, T.; Kazibutowska, Z. Assessment of cerebral embolism and vascular reserve parameters in patients with carotid artery stenosis. Neurol. Neurochir. Pol. 2016, 50, 356–362. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, S.; Feng, S.; Sun, D.; Zhuang, A.; Zeng, Q.; Zhang, Y.; Huang, H.; Nie, H.; Zhou, F. Microembolic signal monitoring of TOAST classified cerebral infarction patients. Mol. Med. Rep. 2013, 8, 1135–1142. [Google Scholar] [CrossRef]
- Markus, H.S.; Droste, D.W.; Kaps, M.; Larrue, V.; Lees, K.R.; Siebler, M.; Ringelstein, E.B. Dual antiplatelet therapy with clopidogrel and aspirin in symptomatic carotid stenosis evaluated using doppler embolic signal detection: The clopidogrel and aspirin for reduction of emboli in symptomatic carotid stenosis (CARESS) trial. Circulation 2005, 111, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.S.; Brown, M.M. Differentiation between different pathological cerebral embolic materials using transcranial Doppler in an in vitro model. Stroke 1993, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Droste, D.W.; Ritter, M.; Kemeny, V.; Schulte-Altedorneburg, G.; Ringelstein, E.B. Microembolus detections at follow-up in 19 patients with acute stroke: Correlation with stroke aetiology and antithrombotic treatment. Cerebrovasc. Dis. 2000, 10, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.A.; Jurk, K.; Schriek, C.; Nabavi, D.; Droste, D.W.; Kherel, B.E.; Ringelstein, E.B. Microembolic signals on transcranial Doppler ultrasound are correlated with platelet activation markers, but not with platelet- leukocyte associates: A study in patients with acute stroke and patients with asymptomatic carotid stenosis. Neurol. Res. 2009, 31, 11–16. [Google Scholar] [CrossRef]
- Kinsella, J.A.; Tobin, W.O.; Tierney, S.; Feeley, T.M.; Egan, B.; Collins, D.R.; Couglan, T.; O’Neill, D.; Harbison, J.; Madhavan, P.; et al. Increased platelet activation in early symptomatic vs. asymptomatic carotid stenosis and relationship with microembolic status: Results from the Platelets and Carotid Stenosis Study. J. Thromb. Haemost. 2013, 11, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.X.; Lim, S.T.; Kinsella, J.A.; Murphy, D.; Enright, H.M.; McCabe, D.J.H.; HEIST study group. Increased plate- let count and reticulated platelets in recently symptomatic versus asymptomatic carotid artery stenosis and in cerebral microembolic signal-negative patient subgroups: Results from the HaEmostasis In carotid STenosis (HEIST) study. J. Neurol. 2018, 265, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.X.; Lim, S.T.; Kinsella, J.A.; Tierney, S.; Egan, B.; Feeley, T.M.; Murphy, S.M.; Walsh, R.A.; Collins, D.R.; Coughlan, T.; et al. Increased leucocyte-platelet complex formation in recently symptomatic versus asymptomatic carotid stenosis patients and in micro-emboli negative subgroups. Thromb. Haemost. 2019, 119, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Padro, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, W.H.; Zhao, Y.D.; Chen, X.Y.; Leung, T.W.; Chen, C.; Fu, J.; Markus, H.; Hao, Q.; Wong, K.A.S.; et al. The effectiveness of dual antiplatelet treatment in acute ischemic stroke patients with intracranial arterial stenosis: A subgroup analysis of CLAIR study. Int. J. Stroke 2013, 8, 663–668. [Google Scholar] [CrossRef]
- Goertler, M.; Baeumer, M.; Kross, R.; Blaser, T.; Lutze, G.; Jost, S.; Wallesch, C.W. Rapid decline of cerebral microemboli of arterial origin after intravenous acetylsalicylic acid. Stroke 1999, 30, 66–69. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Lukasik, M.; Rozalski, M.; Luzak, B.; Michalak, M.; Ambrosius, W.; Watala, C.; Kosubski, W. Enhanced platelet-derived microparticle formation is associated with carotid atherosclerosis in convalescent stroke patients. Platelets 2013, 24, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Schiro, A.; Wilkinson, F.L.; Weston, R.; Smyth, J.V.; Serracino-Inglott, F.; Alexander, M.Y. Elevated levels of endothelial-derived microparticles, and serum CXCL9 and SCGF-β are associated with unstable asymptomatic carotid plaques. Sci. Rep. 2015, 5, 16658. [Google Scholar] [CrossRef]
- Kandiyil, N.; MacSweeney, S.T.; Heptinstall, S.; May, J.; Foz, S.C.; Auer, D.P. Circulating Microparticles in Patients with Symptomatic Carotid Disease Are Related to Embolic Plaque Activity and Recent Cerebral Ischaemia. Cerebrovasc. Dis. Extra 2019, 9, 9–18. [Google Scholar] [CrossRef]
- Huang, Z.S.; Jeng, J.S.; Wang, C.H.; Yip, P.K.; Wu, T.H.; Lee, T.K. Correlations between peripheral differential leukocyte counts and carotid atherosclerosis in non-smokers. Atherosclerosis 2001, 158, 431–436. [Google Scholar] [CrossRef]
- Aronow, H.D.; Shishehbor, M.; Davis, D.A.; Katzan, I.L.; Bhatt, D.L.; Abou-Chebl, A.; Derk, K.W.; Whitlow, P.L.; Yadav, H.S. Leukocyte count predicts microembolic Doppler signals during carotid stenting: A link between inflammation and embolization. Stroke 2005, 36, 1910–1914. [Google Scholar] [CrossRef]
- Nasr, N.; Ruidavets, J.B.; Arnal, J.F.; Sie, P.; Larrue, V. Association of neutrophil count with microembolization in patients with symptomatic carotid artery stenosis. Atherosclerosis 2009, 207, 519–523. [Google Scholar] [CrossRef]
- Wuttge, D.M.; Zhou, X.; Sheikine, Y.; Wagsater, D.; Stemme, V.; Hedin, U.; Stemme, S.; Hansson, G.K.; Sirsjo, A. CXCL16/SR-PSOXisaninterferon-gamma- regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 750–755. [Google Scholar] [CrossRef]
- Minami, M.; Kume, N.; Shimaoka, T.; Kataoka, H.; Hayashida, K.; Akiyama, Y.; Nagata, I.; Ando, K.; Nobuyoshi, M.; Hanyuu, M.; et al. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Pan, X.; Xing, X.; Wu, M.; Wang, Y.; Ma, C. Elevation of serum CXCL16 level correlates well with atherosclerotic ischemic stroke. Arch. Med. Sci. 2014, 10, 47–52. [Google Scholar] [CrossRef]
- Ma, A.; Yang, S.; Wang, Y.; Wang, X.; Pan, X. Increase of serum CXCL16 level correlates well to microembolic signals in acute stroke patients with carotid artery stenosis. Clin. Chim. Acta. 2016, 460, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Ma, A.; Zhao, H.; Li, H.; Song, S.; Pan, X. Correlation between microembolic signal and immune inflammation in acute ischemic stroke. Int. J. Cerebrovasc. Dis. 2015, 23, 677–681. [Google Scholar]
- Pini, R.; Faggioli, G.L.; Fittipaldi, S.; Pasquinelli, G.; Tonon, C.; Beltrandi, E.; Mauro, R.; Stella, A. Inflammatory Mediators and Cerebral Embolism in Carotid Stenting: New Markers of Risk. J. Endovasc. Ther. 2013, 20, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Gerosa, C.; Cerrone, G.; Suri, J.S.; Aimola, V.; Cau, F.; Coni, P.; Piras, M.; Cau, R.; Balestrieri, A.; Scano, A.; et al. The human carotid atherosclerotic plaque: An observational review of histological scoring systems. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Burke, A.P.; Kolodgie, F.D.; Farb, A. Pathology of the thin-cap fibroatheroma: A type of vulnerable plaque. J. Interv. Cardiol. 2003, 16, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Puig, N.; Jiménez-Xarrié, E.; Camps-Renom, P.; Benitez, S. Search for Reliable Circulating Biomarkers to Predict Carotid Plaque Vulnerability. Int. J. Mol. Sci. 2020, 21, 8236. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Watson, M.G.; Byrne, H.M.; Macaskill, C.; Myerscough, M.R. A two- phase model of early fibrous cap formation in atherosclerosis. J. Theor. Biol. 2018, 456, 123–136. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Sanahan, C.M. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Ulrich, V.; Rotllan, N.; Araldi, E.; Luciano, A.; Skroblin, P.; Abonnenc, M.; Perrotta, P.; Yin, X.; Bauer, A.; Leslie, K.L.; et al. Chronic miR-29 antagonism promotes favorable plaque remodeling in atherosclerotic mice. EMBO Mol. Med. 2016, 8, 643–653. [Google Scholar] [CrossRef]
- Rekhter, M.D. Collagen synthesis in atherosclerosis: Too much and not enough. Cardiovasc. Res. 1999, 41, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Shimokado, K.; Raines, E.W.; Madtes, D.K.; Barrett, T.B.; Benditt, E.P.; Ross, R. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell 1985, 43, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Yap, A.S.; Brieher, W.M.; Gumbiner, B.M. Molecular and functional analysis of cadherin-based adherens junctions. Annu. Rev. Cell Dev. Biol. 1997, 13, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Sukhova, G.; Lee, R.T.; Galis, Z.S. Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J. Cardiovasc. Pharmacol. 1995, 25, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Guyton, J.R.; Klemp, K.F. Development of the atherosclerotic core region. Chemical and ultrastructural analysis of microdissected atherosclerotic lesions from human aorta. Arterioscler. Thromb. 1994, 14, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hellstrand, M.; Rymo, L.; Rubbia, L.; Gabbiani, G. Interferon gamma inhibits both proliferation and expression of differentiation-specific alpha-smooth muscle actin in arterial smooth muscle cells. J. Exp. Med. 1989, 170, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Negative regulation of cell growth by TGF beta. Biochim. Biophys. Acta 1996, 242, 185–199. [Google Scholar] [CrossRef]
- Bennett, M.R.; Evan, G.I.; Newby, A.C. Deregulated expression of the c-myc oncogene abolishes inhibition of proliferation of rat vascular smooth muscle cells by serum reduction, interferon-gamma, heparin, and cyclic nucleotide analogues and induces apoptosis. Circ. Res. 1994, 74, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Morioka, M.; Hamada, J.; Hashiguchi, A.; Hasegawa, Y.; Todaka, T.; Yano, S.; Kai, Y.; Miura, M.; Fujioka, S.; Ushio, Y. Contribution of angiotensin-converting enzyme and angiotensin II to ischemic stroke: Their role in the formation of stable and unstable carotid atherosclerotic plaques. Surg. Neurol. 2004, 62, 292–301. [Google Scholar] [CrossRef]
- Hao, H.; Iihara, K.; Ishibashi-Ueda, H.; Saito, F.; Hirota, S. Correlation of thin fibrous cap possessing adipophilin-positive macrophages and intraplaque hemorrhage with high clinical risk for carotid endarterectomy. J. Neurosurg. 2011, 114, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Badacz, R.; Przewłocki, T.; Legutko, J.; Żmudka, K.; Kabłak-Ziembicka, A. microRNAs Associated with Carotid Plaque Development and Vulnerability: The Clinician’s Perspective. Int. J. Mol. Sci. 2022, 23, 15645. [Google Scholar] [CrossRef] [PubMed]
- Faber, B.C.; Cleutjens, K.B.; Niessen, R.L.; Aarts, P.L.; Boon, W.; Greenberg, A.S.; Kitslaar, P.J.; TorDOIr, J.H.; Daemen, M.J. Identification of genes potentially involved in rupture of human atherosclerotic plaques. Circ. Res. 2001, 89, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Killingsworth, M.C.; Lord, R.S.A.; Grabs, A.J. Matrix vesicles in the fibrous cap of atherosclerotic plaque: Possible contribution to plaque rupture. J. Cell. Mol. Med. 2008, 12, 2073–2082. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Daemen, M.J. Novel concepts in atherogenesis: Angiogenesis and hypoxia in atherosclerosis. J. Pathol. 2009, 218, 7–29. [Google Scholar] [CrossRef] [PubMed]
- Roiniotis, J.; Dinh, H.; Masendycz, P.; Turner, A.; Elsegood, C.L.; Scholz, G.M.; Hamilton, J.A. Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J. Immunol. 2009, 182, 7974–7981. [Google Scholar] [CrossRef] [PubMed]
- Nakano, D.; Hayashi, T.; Tazawa, N.; Yamashita, C.; Inamoto, S.; Okuda, N.; Mori, T.; Sohmiya, K.; Kitaura, Y.; Okada, Y.; et al. Chronic hypoxia accelerates the progression of atherosclerosis in apolipoprotein E-knockout mice. Hypertens. Res. 2005, 28, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Hellings, W.E.; Peeters, W.; Moll, F.L.; Piers, S.R.; van Setten, J.; Van der Spek, P.J.; de Vries, J.P.; Seldenrijk, K.A.; De Bruin, P.C.; Vink, A.; et al. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: A prognostic study. Circulation 2010, 121, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W.; Eringa, E.C.; Daemen, M.J. Neovascularization of the atherosclerotic plaque: Interplay between atherosclerotic lesion, adventitia-derived microvessels and perivascular fat. Curr. Opin. Lipidol. 2015, 26, 405–411. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Kolodgie, F.D.; Bijnens, A.P.; Maxfield, K.; Pacheco, E.; Kutys, B.; Duimel, H.; Frederik, P.M.; van Hinsbergh, V.W.; Virmani, R.; et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J. Am. Coll. Cardiol. 2009, 53, 1517–1527. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [PubMed]
- Melder, R.J.; Yuan, J.; Munn, L.L.; Jain, R.K. Erythrocytes enhance lymphocyte rolling and arrest in vivo. Microvasc. Res. 2000, 59, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noé, B. Pathology of human plaque vulnerability: Mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, A.; Houard, X.; Philippe, M.; Ollivier, V.; Sebbag, U.; Meilhac, O.; Michel, J.B. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J. Leukoc. Biol. 2007, 82, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, D.M.; De Meyer, G.R.; Kockx, M.M.; Herman, A.G.; Martinet, W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Liu, X.; Song, Y.; Zhai, C.; Zhang, L. VEGF-A/VEGFR-2 and FGF-2/FGFR-1 but not PDGF-BB/PDGFR-beta play important roles in promoting immature and inflammatory intraplaque angiogenesis. PLoS ONE 2018, 13, e0201395. [Google Scholar] [CrossRef] [PubMed]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaquehemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.L.; Reidy, M.A. Basic fibroblast growth factor: Its role in the control of smooth muscle cell migration. Am. J. Pathol. 1993, 143, 1024–1031. [Google Scholar] [PubMed]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef]
- Parma, L.; Peters, H.A.B.; Sluiter, T.J.; Simons, K.H.; Lazzari, P.; de Vries, M.R.; Quax, P.H.A. bFGF blockade reduces intraplaque angiogenesis and macrophage infiltration in atherosclerotic vein graft lesions in ApoE3*Leiden mice. Sci. Rep. 2020, 10, 15968. [Google Scholar] [CrossRef]
- Jeziorska, M.; Woolley, D.E. Local neovascularization and cellular composition within vulnerable regions of atherosclerotic plaques of human carotid arteries. J. Pathol. 1999, 188, 189–196. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic plaque progression and vulnerability to rupture: Angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Corada, M.; Caveda, L.; Breviario, F.; Ayalon, O.; Geiger, B.; Dejana, E. The molecular organization of endothelial cell-to-cell junctions: Differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin). J. Cell Biol. 1995, 129, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Cherian, S.M.; Inder, S.J.; Lord, R.S. Neovascular expression of VE-cadherin in human atherosclerotic arteries and its relation to intimal inflammation. Cardiovasc. Res. 1999, 43, 1003–1017. [Google Scholar] [CrossRef]
- McCarthy, M.J.; Loftus, I.M.; Thompson, M.M.; Jones, L.; London, N.J.; Bell, P.R.; Naylor, A.R.; Brindle, N.P. Angiogenesis and the atherosclerotic carotid plaque: An association between symptomatology and plaque morphology. J. Vasc. Surg. 1999, 30, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Matic, L.P.; Iglesias, J.M.; Vesterlund, M.; Lengquist, M.; Hong, M.G.; Saieed, S.; Sanchez-Rivera, L.; Berg, M.; Razuvaev, A.; Kronqvist, M.; et al. Novel Multiomics Profiling of Human Carotid Atherosclerotic Plaques and Plasma Reveals Biliverdin Reductase B as a Marker of Intraplaque Hemorrhage. JACC Basic Transl. Sci. 2018, 3, 464–480. [Google Scholar] [CrossRef]
- Grosskreutz, C.L.; Anand-Apte, B.; Dupláa, C.; Quinn, T.P.; Terman, B.I.; Zetter, B.; D’Amore, P.A. Vascular endothelial growth factor-induced migration of vascular smooth muscle cells in vitro. Microvasc. Res. 1999, 58, 128–136. [Google Scholar] [CrossRef]
- Carmeliet, P.; Collen, D. Molecular basis of angiogenesis. Role of VEGF and VE-cadherin. Ann. N. Y. Acad. Sci. 2000, 902, 249–264. [Google Scholar] [CrossRef]
- O’Brien, K.D.; McDonald, T.O.; Chait, A.; Allen, M.D.; Alpers, C.E. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation 1996, 93, 672–682. [Google Scholar] [CrossRef]
- Hu, S.; Liu, Y.; You, T.; Zhu, L. Semaphorin 7A Promotes VEGFA/VEGFR2-Mediated Angiogenesis and Intraplaque Neovascularization in ApoE-/- Mice. Front. Physiol. 2018, 9, 1718. [Google Scholar] [CrossRef]
- Suzuki, E.; Imuta, H.; Fujita, D.; Takahashi, M.; Oba, S.; Kiyosue, A.; Nishimatsu, H. Endogenous Interleukin-1β Is Implicated in Intraplaque Hemorrhage in Apolipoprotein E Gene Null Mice. Circ. J. 2018, 82, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Puré, E. Lipoproteins, macrophage function, and atherosclerosis: Beyond the foam cell? Cell Metab. 2005, 1, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Nuotio, K.; Isoviita, P.M.; Saksi, J.; Ijäs, P.; Pitkäniemi, J.; Sonninen, R.; Soinne, L.; Saimanen, E.; Salonen, O.; Kovanen, P.T.; et al. Adipophilin expression is increased in symptomatic carotid atherosclerosis: Correlation with red blood cells and cholesterol crystals. Stroke 2007, 38, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Heinecke, J.W. ATP-binding cassette transporter A1: A cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 2005, 85, 1343–1372. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, T.; Luoma, J.S.; Hiltunen, M.O.; Macphee, C.H.; Miliner, K.J.; Patel, L.; Rice, S.Q.; Tew, D.G.; Karkola, K.; Yla-Herttuala, S. Lipoprotein-associated phospholipase A2, platelet-activation factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Balboa, M.A.; Varela-Nieto, I.; Lucas, K.K.; Dennis, E.A. Expression and function of phospholipase A2 in brain. FEBS Lett. 2002, 531, 12–17. [Google Scholar] [CrossRef]
- Macphee, C.H.; Suckling, K.E. Lipoprotein-associated phospholipase A2 a target directed at the atherosclerotic plaque. Expert Opin. Ther. Targets 2002, 6, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, X.; Yang, B.; Jiang, D. Relationship between lysophosphatidic acid and matrix metalloproteinase-9 plasma concentrations and carotid atheromatous plaque stability in patients with cerebral infarction. J. Int. Med. Res. 2014, 42, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, B.; Sharrett, A.; Lai, S.; Gomes, A.; Cushman, M.; Folsom, A.; Bild, D.; Kronmal, R.; Sinha, S.; Bluemke, D. Risk factor associations with the presence of a lipid core in carotid plaque of asymptomatic individuals using high-resolution MRI: The multi-ethnic study of atherosclerosis (MESA). Stroke 2008, 39, 329–335. [Google Scholar] [CrossRef]
- Guyton, J.R.; Klemp, K.F. Early extracellular and cellular lipid deposits in aorta of cholesterol-fed rabbits. Am. J. Pathol. 1992, 141, 925–936. [Google Scholar]
- Mitchinson, M.J.; Ball, R.Y.; Carpenter, K.H.; Enright, J.H.; Brabbs, C.E. Ceroid, macrophages and atherosclerosis. Biochem. Soc. Trans. 1990, 18, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Tangirala, R.K.; Mahlberg, F.H.; Glick, J.M.; Jerome, W.G.; Rothblat, G.H. Lysosomal accumulation of unesterified cholesterol in model macrophage foam cells. J. Biol. Chem. 1993, 268, 9653–9660. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.L.; Taylor, S.E.; Ballantine, J.A.; Fussell, B.; Halliwell, B.; Mitchinson, M.J. Lipids and oxidised lipids in human atheroma and normal aorta. Biochim. Biophys. Acta 1993, 1167, 121–130. [Google Scholar] [CrossRef]
- Hollander, W.; Colombo, M.A.; Kirkpatrick, B.; Paddock, J. Soluble proteins in the human atherosclerotic plaque. With spectral reference to immunoglobulins, C3-complement component, alpha 1-antitrypsin and alpha 2-macroglobulin. Atherosclerosis 1979, 34, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Seifert, P.S.; Hugo, F.; Hansson, G.K.; Bhakdi, S. Prelesional complement activation in experimental atherosclerosis. Terminal C5b-9 complement deposition coincides with cholesterol accumulation in the aortic intima of hypercholesterolemic rabbits. Lab. Investig. 1989, 60, 747–754. [Google Scholar] [PubMed]
- Small, D.M. Cellular mechanisms for lipid deposition in atherosclerosis (first of two parts). N. Engl. J. Med. 1977, 297, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, Y.H.; Adams, C.W. The action of human high-density lipoprotein on cholesterol crystals. Part 2. Biochemical observations. Atherosclerosis 1978, 31, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Sato, Y.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Kawakami, R.; Gadhoke, N.V.; Kolodgie, F.D.; Virmani, R.; Finn, A.V. Calcium deposition within coronary atherosclerotic lesion: Implications for plaque stability. Atherosclerosis 2020, 306, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Carlier, S.; Xanthos, S.; Cardoso, L.; Ganatos, P.; Virmani, R.; Einav, S.; Gilchrist, L.; Weinbaum, S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. USA 2006, 103, 14678–14683. [Google Scholar] [CrossRef]
- Maldonado, N.; Kelly-Arnold, A.; Vengrenyuk, Y.; Laudier, D.; Fallon, J.T.; Virmani, R.; Cardoso, L.; Weinbaum, S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: Potential implications for plaque rupture. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H619–H628. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, J.J.; Cunnane, E.M.; McHugh, S.M.; Kavanagh, E.G.; Walsh, S.R.; Walsh, M.T. Mechanical, biological and structural characterization of in vitro ruptured human carotid plaque tissue. Acta Biomater. 2013, 9, 9027–9035. [Google Scholar] [CrossRef] [PubMed]
- Shaalan, W.E.; Cheng, H.; Gewertz, B.; McKinsey, J.F.; Schwartz, L.B.; Katz, D.; Cao, D.; Desai, T.; Glagov, S.; Bassiouny, H.S. Degree of carotid plaque calcification in relation to symptomatic outcome and plaque inflammation. J. Vasc. Surg. 2004, 40, 262–269. [Google Scholar] [CrossRef] [PubMed]
- van den Bouwhuijsen, Q.J.; Bos, D.; Ikram, M.A.; Hofman, A.; Krestin, G.P.; Franco, O.H.; van der Lugt, A.; Vernooij, M.W. Coexistence of Calcification, Intraplaque Hemorrhage and Lipid Core within the Asymptomatic Atherosclerotic Carotid Plaque: The Rotterdam Study. Cerebrovasc. Dis. 2015, 39, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, C.M.; Zheng, W.; Shaalan, W.; Tang, J.; Bassiouny, H.S. Human carotid plaque calcification and vulnerability: Relationship between degree of plaque calcification, fibrous cap inflammatory gene expression and symptomatology. Cerebrovasc. Dis. 2009, 27, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Cardoso, L.; Weinbaum, S. Micro-CT based analysis of a new paradigm for vulnerable plaque rupture: Cellular microcalcifications in fibrous caps. MCB Mol. Cell. Biomech. 2008, 5, 37–47. [Google Scholar]
- Pugliese, G.; Iacobini, C.; Blasetti Fantauzzi, C.; Menini, S. The dark and bright side of atherosclerotic calcification. Atherosclerosis 2015, 238, 220–230. [Google Scholar] [CrossRef]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef]
- Ehara, S.; Kobayashi, Y.; Yoshiyama, M.; Shimada, K.; Shimada, Y.; Fukuda, D.; Nakamura, Y.; Yamashita, H.; Yamagishi, H.; Takeuchi, K.; et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: An intravascular ultrasound study. Circulation 2004, 110, 3424–3429. [Google Scholar] [CrossRef]
- Banach, M.; Serban, C.; Sahebkar, A.; Mikhailidis, D.P.; Ursoniu, S.; Ray, K.K.; Rysz, J.; Toth, P.P.; Muntner, P.; Mosteoru, S.; et al. Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group. Impact of statin therapy on coronary plaque composition: A systematic review and meta-analysis of virtual histology intravascular ultrasound studies. BMC Med. 2015, 13, 229. [Google Scholar] [CrossRef]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of statins on serial coronary calcification during atheroma progression and regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Mujaj, B.; Bos, D.; Selwaness, M.; Leening, M.J.G.; Kavousi, M.; Wentzel, J.J.; van der Lugt, A.; Hofman, A.; Stricker, B.H.; Vernooij, M.W.; et al. Statin use is associated with carotid plaque composition: The Rotterdam Study. Int. J. Cardiol. 2018, 260, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.Z.; Lu, M.; Fong, F.; Qiao, R.; Patel, N.R.; Abeydeera, D.; Iriana, S.; Demer, L.L.; Tintut, Y. Statin Effects on Vascular Calcification: Microarchitectural Changes in Aortic Calcium Deposits in Aged Hyperlipidemic Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e185–e192. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Virmani, R.; Younis, H.; Burke, A.P.; Kamm, R.D.; Lee, R.T. The impact of calcification on the biomechanical stability of atherosclerotic plaques. Circulation 2001, 103, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.V.; Vesey, A.T.; Williams, M.C.; Shah, A.S.; Calvert, P.A.; Craighead, F.H.; Yeoh, S.E.; Wallace, W.; Salter, D.; Fletcher, A.M.; et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: A prospective clinical trial. Lancet 2014, 383, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Reith, S.; Milzi, A.; Dettori, R.; Marx, N.; Burgmaier, M. Predictors for target lesion microcalcifications in patients with stable coronary artery disease: An optical coherence tomography study. Clin. Res. Cardiol. 2018, 107, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Mauriello, A.; Servadei, F.; Zoccai, G.B.; Giacobbi, E.; Anemona, L.; Bonanno, E.; Casella, S. Coronary calcification identifies the vulnerable patient rather than the vulnerable Plaque. Atherosclerosis 2013, 229, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef]
- Kim, K.M. Apoptosis and calcification. Scanning Microsc. 1995, 9, 19. [Google Scholar]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem. Biophys. Res. Commun. 2010, 391, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Yang, X.; Yan, Z.; Zhao, M.; Yue, X.; Cheng, X.; Zheng, Z.; Guan, K.; Dou, J.; Xu, T.; et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int. J. Cardiol. 2013, 168, 2242–2247. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Neureiter, D.; Câmpean, V.; Soder, S.; Amann, K. Expression of cartilage-specific markers in calcified and non-calcified atherosclerotic lesions. Atherosclerosis 2008, 196, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, M.; Roszkowska, M.; Briolay, A.; Bougault, C.; Guignandon, A.; Diaz-Hernandez, J.I.; Diaz-Hernandez, M.; Pikula, S.; Buchet, R.; Hamade, E.; et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef]
- Pettengill, M.; Matute, J.D.; Tresenriter, M.; Hibbert, J.; Burgner, D.; Richmond, P.; Millán, J.L.; Ozonoff, A.; Strunk, T.; Currie, A.; et al. Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PLoS ONE 2018, 13, e0197532. [Google Scholar] [CrossRef] [PubMed]
- Morony, S.; Tintut, Y.; Zhang, Z.; Cattley, R.C.; Van, G.; Dwyer, D.; Stolina, M.; Kostenuik, P.J.; Demer, L.L. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(-/-) mice. Circulation 2008, 117, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Dautova, Y.; Kapustin, A.N.; Pappert, K.; Epple, M.; Okkenhaug, H.; Cook, S.J.; Shanahan, C.M.; Bootman, M.D.; Proudfoot, D. Calcium phosphate particles stimulate interleukin-1β release from human vascular smooth muscle cells: A role for spleen tyrosine kinase and exosome release. J. Mol. Cell Cardiol. 2018, 115, 82–93. [Google Scholar] [CrossRef]
- Clarke, M.C.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef]
- Roijers, R.B.; Debernardi, N.; Cleutjens, J.P.; Schurgers, L.J.; Mutsaers, P.H.; van der Vusse, G.J. Microcalcifications in early intimal lesions of atherosclerotic human coronary arteries. Am. J. Pathol. 2011, 178, 2879–2887. [Google Scholar] [CrossRef]
- Huang, L.H.; Han, J.; Ouyang, J.M.; Gui, B.S. Shape-dependent adhesion and endocytosis of hydroxyapatite nanoparticles on A7R5 aortic smooth muscle cells. J. Cell. Physiol. 2020, 235, 465–479. [Google Scholar] [CrossRef]
- Matsui, Y.; Rittling, S.R.; Okamoto, H.; Inobe, M.; Jia, N.; Shimizu, T.; Akino, M.; Sugawara, T.; Morimoto, J.; Kimura, C.; et al. Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1029–1034. [Google Scholar] [CrossRef]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular calcification: Mechanisms and clinical ramifications. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef]
- Collin-Osdoby, P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ. Res. 2004, 95, 1046–1057. [Google Scholar] [CrossRef]
- Kadoglou, N.P.E.; Gerasimidis, T.; Moumtzouoglou, A.; Kapelouzou, A.; Sailer, N.; Fotiadis, G.; Vitta, I.; Katinios, A.; Kougias, P.; Bandios, S.; et al. Intensive lipid-lowering therapy ameliorates novel calcification markers and GSM score in patients with carotid stenosis. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 661–668. [Google Scholar] [CrossRef]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef]
- Heymann, M.F.; Herisson, F.; Davaine, J.M.; Charrier, C.; Battaglia, S.; Passuti, N.; Lambert, G.; Gouëffic, Y.; Heymann, D. Role of the OPG/RANK/RANKL triad in calcifications of the atheromatous plaques: Comparison between carotid and femoral beds. Cytokine 2012, 58, 300–306. [Google Scholar] [CrossRef]
- Rochette, L.; Meloux, A.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. The role of osteoprotegerin and its ligands in vascular function. Int. J. Mol. Sci. 2019, 20, 705. [Google Scholar] [CrossRef]
- Fukui, N.; Ikeda, Y.; Ohnuki, T.; Hikita, A.; Tanaka, S.; Yamane, S.; Suzuki, R.; Sandell, L.J.; Ochi, T. Pro-inflammatory cytokine tumor necrosis factor-alpha induces bone morphogenetic protein-2 in chondrocytes via mRNA stabilization and transcriptional up-regulation. J. Biol. Chem. 2006, 281, 27229–27241. [Google Scholar] [CrossRef]
- Csiszar, A.; Smith, K.E.; Koller, A.; Kaley, G.; Edwards, J.G.; Ungvari, Z. Regulation of bone morphogenetic protein-2 expression in endothelial cells: Role of nuclear factor-kappaB activation by tumor necrosis factor-alpha, H2O2, and high intravascular pressure. Circulation 2005, 111, 2364–2372. [Google Scholar] [CrossRef]
- Tsuji, K.; Bandyopadhyay, A.; Harfe, B.D.; Cox, K.; Kakar, S.; Gerstenfeld, L.; Einhorn, T.; Tabin, C.J.; Rosen, V. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 2006, 38, 1424–1429. [Google Scholar] [CrossRef]
- Montanaro, M.; Scimeca, M.; Anemona, L.; Servadei, F.; Giacobbi, E.; Bonfiglio, R.; Bonanno, E.; Urbano, N.; Ippoliti, A.; Santeusanio, G.; et al. The Paradox Effect of Calcification in Carotid Atherosclerosis: Microcalcification is Correlated with Plaque Instability. Int. J. Mol. Sci. 2021, 22, 395. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef]
- Okulla, T.; Gass, S.; Böhme, K.; Tiemann, K.; Harbrecht, U.; Klockgether, T.; Pohl, C. Circulating adhesion molecules in patients with internal carotid artery stenosis. Cerebrovasc. Dis. 2002, 14, 9–14. [Google Scholar] [CrossRef]
- Li, X.; Guo, D.; Hu, Y.; Zhou, H.; Chen, Y. Potential Biomarkers and Therapeutic Targets: Inflammation and Oxidative Stress in Left Carotid Artery Stenosis with Coronary Artery Disease. Curr. Pharm. Des. 2023, 29, 966–979. [Google Scholar] [CrossRef]
- Tian, X.; Wang, X.; Shi, Z.; Yu, C.; Li, M.; Chen, L.; Jia, Q.; Liang, G. Tumor necrosis factor-stimulated gene-6-a new serum identification marker to identify severe and symptomatic carotid artery stenosis. Pathol. Res. Pract. 2022, 232, 153838. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, M.; Liu, Q.; Liu, J.; Cui, Y. Neutrophil extracellular traps induce thrombogenicity in severe carotid stenosis. Immun. Inflamm. Dis. 2021, 9, 1025–1036. [Google Scholar] [CrossRef]
- Yayla, Ç.; Açikgöz, S.K.; Yayla, K.G.; Açikgöz, E.; Canpolat, U.; Kirbaş, Ö.; Öksüz, F.; Özcan, F.; Akboğa, M.K.; Topaloğlu, S.; et al. The association between platelet-to-lymphocyte ratio and inflammatory markers with the severity of aortic stenosis. Biomark. Med. 2016, 10, 367–373. [Google Scholar] [CrossRef]
- Papalambros, E.; Georgopoulos, S.; Sigala, F.; Vourliotakis, G.; Chrisostomidis, G.; Venetsanou, K.; Hadjoulis, G.; Felekouras, E.; Bastounis, E. Changes in circulating levels of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 after carotid endarterectomy. Int. J. Mol. Med. 2004, 14, 133–136. [Google Scholar] [CrossRef]
- Puertas-Umbert, L.; Puig, N.; Camacho, M.; Dantas, A.P.; Marín, R.; Martí-Fàbregas, J.; Jiménez-Xarrié, E.; Benitez, S.; Camps-Renom, P.; Jiménez-Altayó, F. Serum from Stroke Patients with High-Grade Carotid Stenosis Promotes Cyclooxygenase-Dependent Endothelial Dysfunction in Non-ischemic Mice Carotid Arteries. Transl. Stroke Res. 2024, 15, 140–152. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features of Plaque Vulnerability | Molecular Patterns Associated with Plaque Instability | References |
|---|---|---|
| Plaque Rupture And Atherothrombosis | GPIba, vWF, TF, thrombin, CRP, IL-1, NETs | [70,72,73,74,75,76,78] |
| Plaque Ulceration | MMP-9, CD40, CD40L, heat shock protein 27, thrombospondin-1, ApoE, vWF, ADAMTS13 | [37,86,87,88,89,90] |
| Fibrous Cap Thickness | TF, IFN-γ, MMPs, reactive oxygen species, IL-18, growth differentiation factor-15, TNF, PDGF B, macrophage-produced heparin-binding epidermal growth-factor-like growth factor, cadherin, prostaglandins, ACE/Ang II, XCR1, CD177 | [91,92,93,94,95,96,97,98,99] |
| Intraplaque Hemorrhage | VEGF, MMPS, bFGF, vWF, VE-cadherin, E-selectin, intercellular adhesion molecule-1, vascular cell adhesion molecule-1, Semaphorin 7A | [100,101,102,103,104,105,106,107,108] |
| Large Lipidic Core | CD36, ABCA, LP-PLA2, LPA, 7β-OH-cholesterol, C3d, C5b-9 neoantigen, ACAT | [109,110,111,112,113,114,115,116,117] |
| Microcalcification | TNF-α, IL-1β, IL-6, BMP2, IL-8, OPN, OPG, TGF-β | [118,119,120,121,122,123,124,125,126,127,128,129,130] |
| Carotid Stenosis | Lipid hydroperoxide, 8-isoprostane, malondialdehyde, monocyte chemotactic protein-4, amyloid A, hs-CRP, TNF-α, prostaglandin E2, IFN-γ, TSG-6, vWF, CXCL16, VEGF, VEGFR2 | [5,131,132,133,134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miceli, G.; Basso, M.G.; Pintus, C.; Pennacchio, A.R.; Cocciola, E.; Cuffaro, M.; Profita, M.; Rizzo, G.; Tuttolomondo, A. Molecular Pathways of Vulnerable Carotid Plaques at Risk of Ischemic Stroke: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 4351. https://doi.org/10.3390/ijms25084351
Miceli G, Basso MG, Pintus C, Pennacchio AR, Cocciola E, Cuffaro M, Profita M, Rizzo G, Tuttolomondo A. Molecular Pathways of Vulnerable Carotid Plaques at Risk of Ischemic Stroke: A Narrative Review. International Journal of Molecular Sciences. 2024; 25(8):4351. https://doi.org/10.3390/ijms25084351
Chicago/Turabian StyleMiceli, Giuseppe, Maria Grazia Basso, Chiara Pintus, Andrea Roberta Pennacchio, Elena Cocciola, Mariagiovanna Cuffaro, Martina Profita, Giuliana Rizzo, and Antonino Tuttolomondo. 2024. "Molecular Pathways of Vulnerable Carotid Plaques at Risk of Ischemic Stroke: A Narrative Review" International Journal of Molecular Sciences 25, no. 8: 4351. https://doi.org/10.3390/ijms25084351