
AAV-Mediated CAG-Targeting Selectively Reduces Polyglutamine-Expanded Protein and Attenuates Disease Phenotypes in a Spinocerebellar Ataxia Mouse Model

 , , ,
, , ,  ,
,  , and
, and 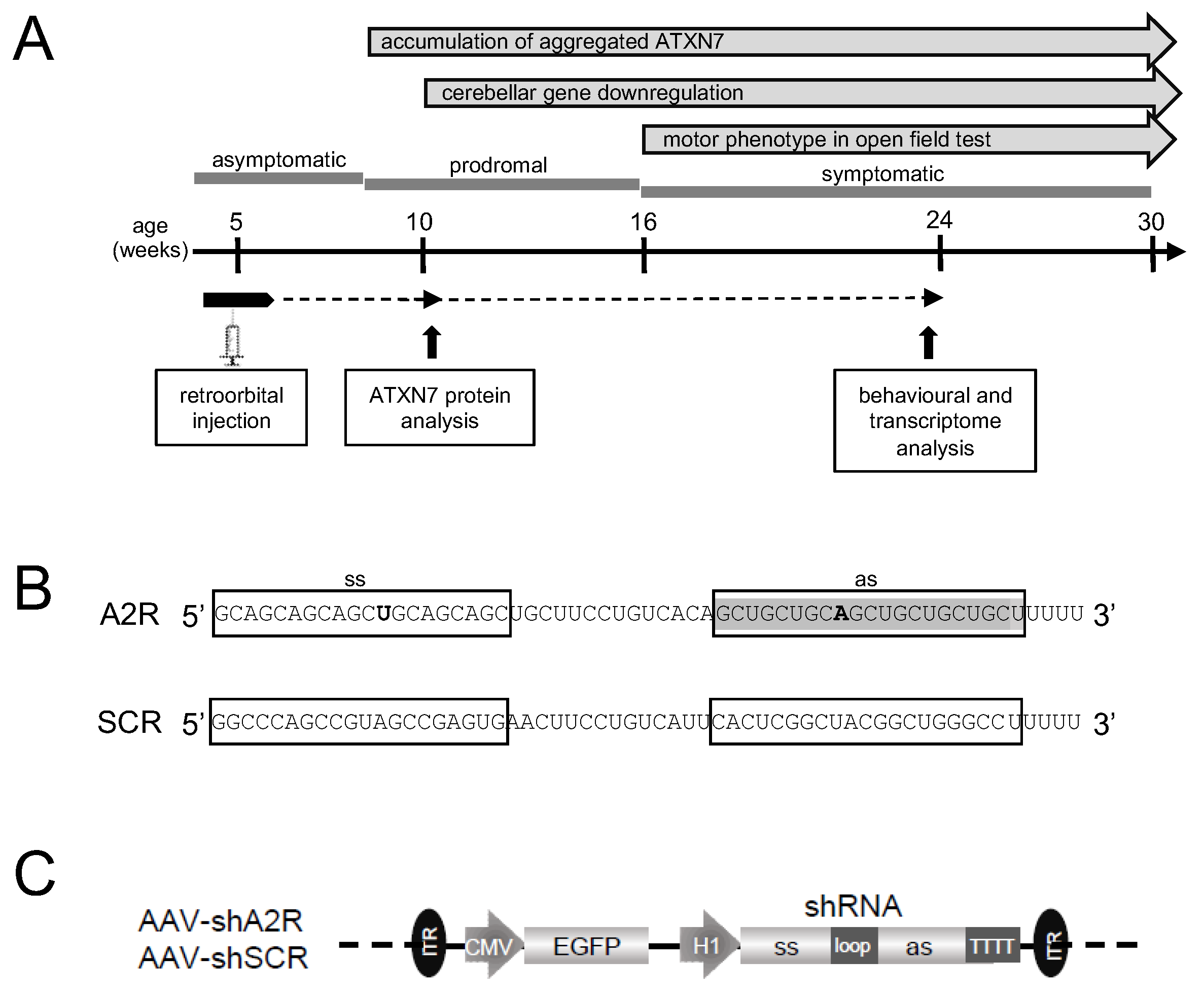{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. A2R shRNA Expressed from AAV PHP.eB Lowers ATXN7 in SCA7 Mouse Primary Cells
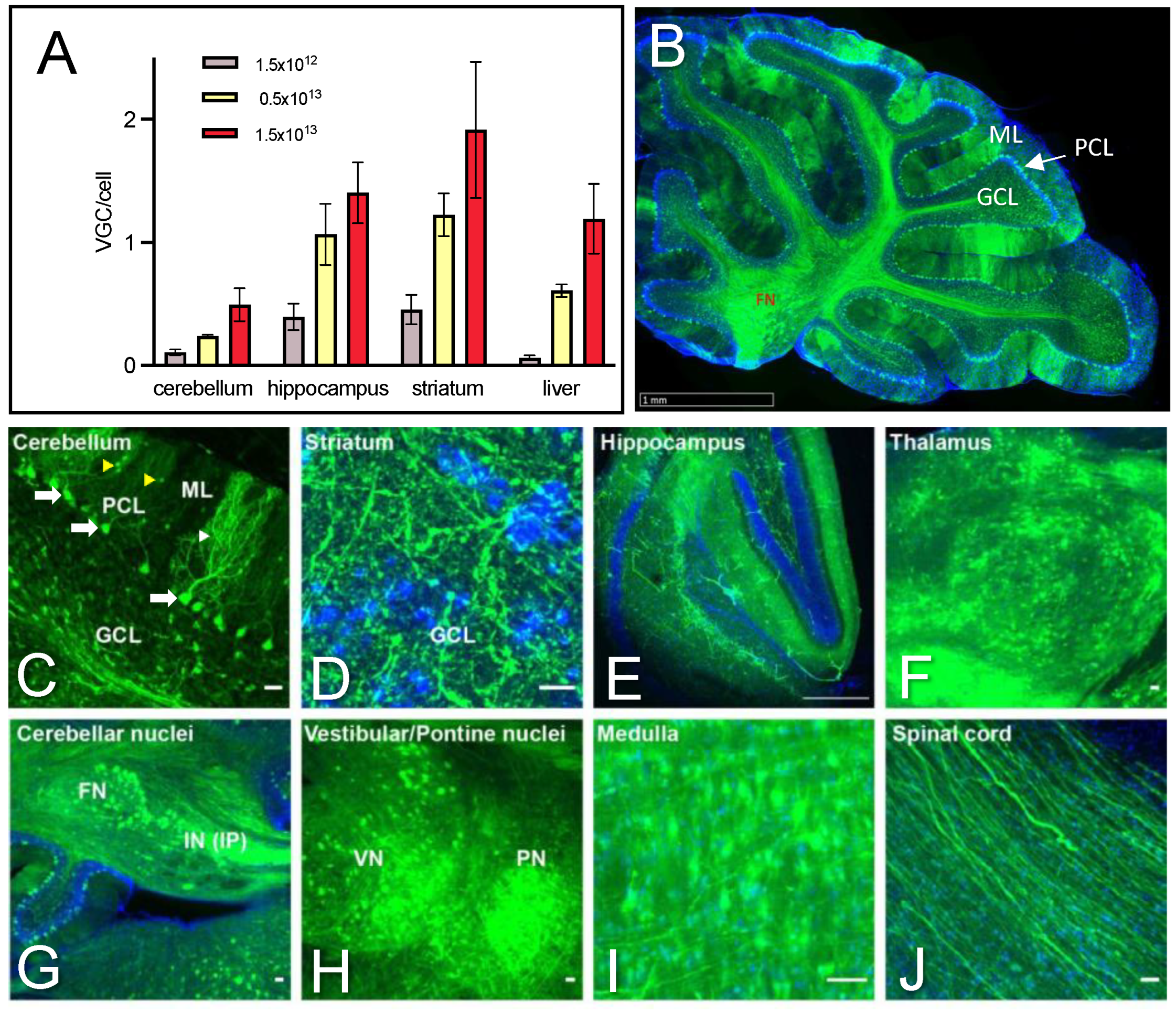
2.2. Dose-Dependent Transduction of the Cerebellum and Safety Profile of AAV PHP.eB Injected Systemically
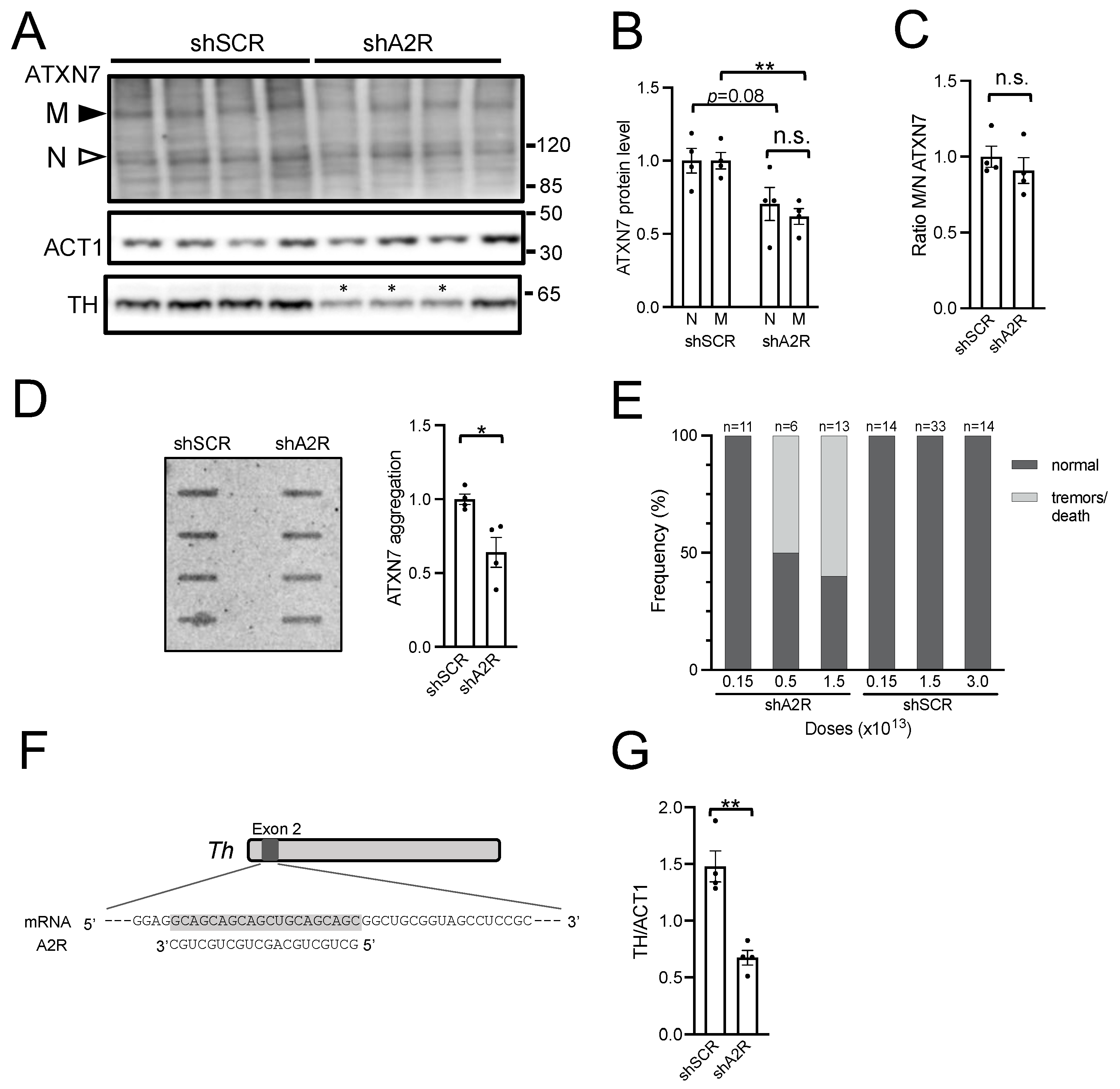
2.3. AAV-shA2R Lowers mATXN7 Cerebellar Protein Level and Induces Severe Adverse Side Effects
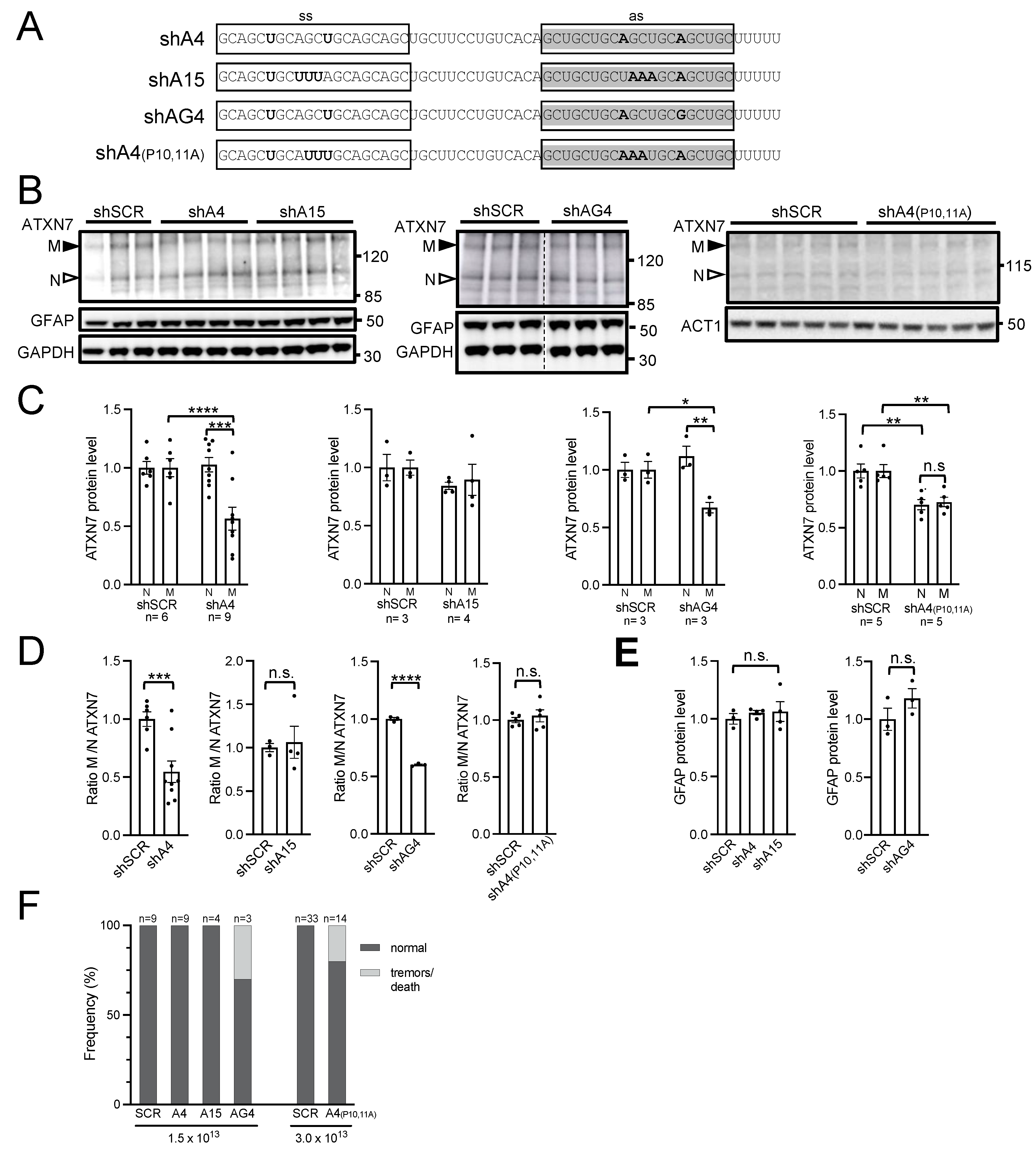
2.4. AAV PHP.eB Expressing A4 shRNA Selectively Lowers mATXN7 Cerebellar Level and Is Well Tolerated
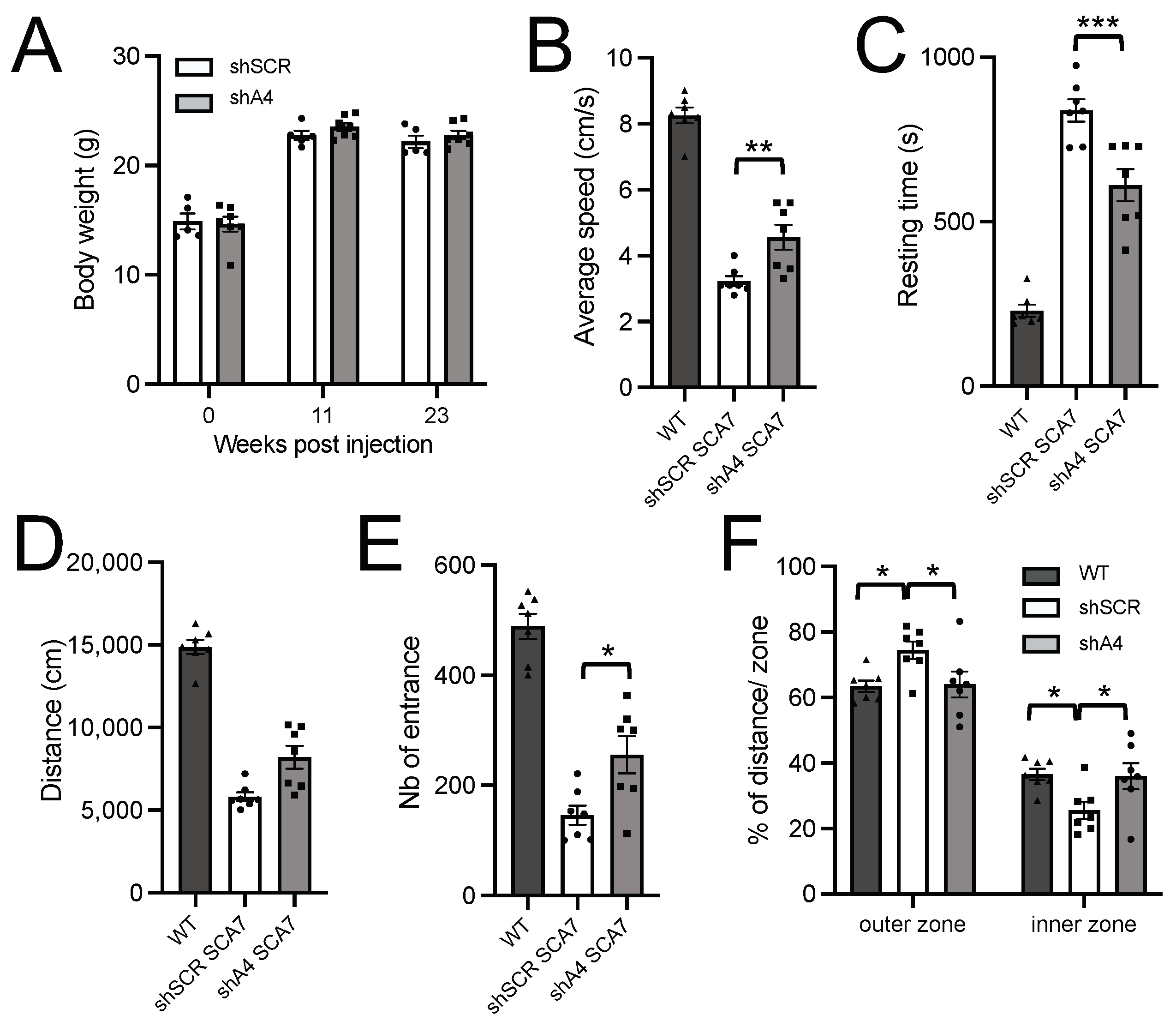
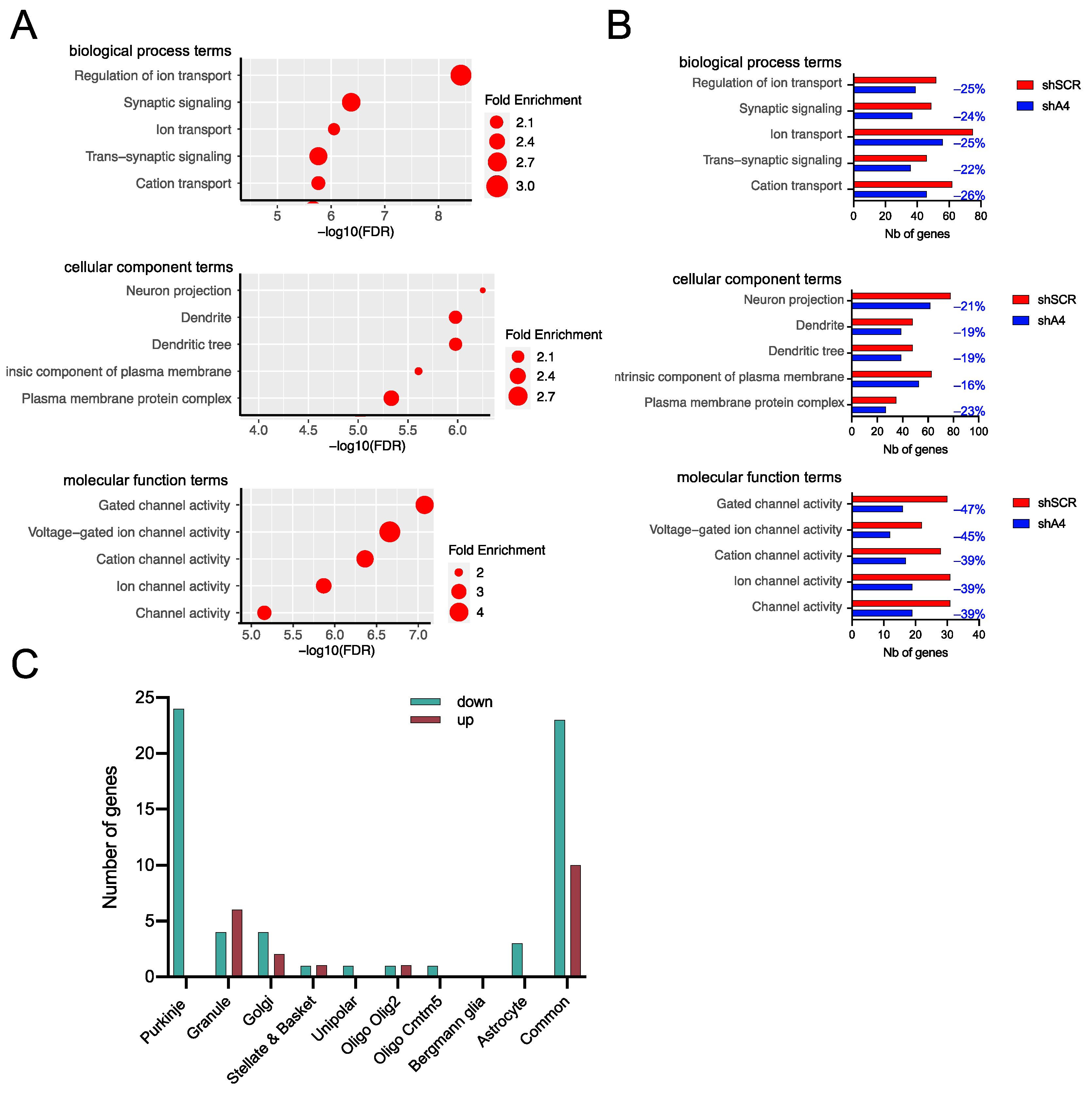
2.5. AAV-shA4 Improves Motor, Behavioral, and Molecular Phenotypes in SCA7 Mice
3. Discussion
3.1. Variability of Different shRNAs to Safely and Effectively Target CAG Expansion In Vivo
3.2. Attenuation of Ataxia and Cerebellar Phenotypes by CAG-Targeting Strategy
3.3. Efficacy and Limitation of AAV PHP.eB Serotype for Preclinical Trial in SCA7 Mouse
3.4. Towards Further Advancement of CAG-Targeting Strategies
4. Materials and Methods
4.1. Mouse Information
4.2. Adeno-Associated Virus Production
4.3. Primary Cell Culture and Transfection/Transduction
4.4. Retro-Orbital Viral Injections in Mice
4.5. Vector Genome Copy Number
4.6. eGFP Fluorescence Acquisition
4.7. Western Blot Analysis
4.8. Filter Trap Assay
4.9. Open Field Test
4.10. RNA-Seq Library Preparation, Sequencing and Data Analysis
4.11. Experimental Design and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Kacher, R.; Lejeune, F.X.; Noel, S.; Cazeneuve, C.; Brice, A.; Humbert, S.; Durr, A. Propensity for somatic expansion increases over the course of life in Huntington disease. eLife 2021, 10, e64674. [Google Scholar] [CrossRef] [PubMed]
- Rub, U.; Schols, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.C.; Nobre, R.J.; de Almeida, L.P. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143, 407–429. [Google Scholar] [CrossRef] [PubMed]
- van der Bent, M.L.; Evers, M.M.; Valles, A. Emerging Therapies for Huntington’s Disease—Focus on N-Terminal Huntingtin and Huntingtin Exon 1. Biologics 2022, 16, 141–160. [Google Scholar] [CrossRef]
- Fiszer, A.; Olejniczak, M.; Galka-Marciniak, P.; Mykowska, A.; Krzyzosiak, W.J. Self-duplexing CUG repeats selectively inhibit mutant huntingtin expression. Nucleic Acids Res. 2013, 41, 10426–10437. [Google Scholar] [CrossRef]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef]
- Ciesiolka, A.; Stroynowska-Czerwinska, A.; Joachimiak, P.; Ciolak, A.; Kozlowska, E.; Michalak, M.; Dabrowska, M.; Olejniczak, M.; Raczynska, K.D.; Zielinska, D.; et al. Artificial miRNAs targeting CAG repeat expansion in ORFs cause rapid deadenylation and translation inhibition of mutant transcripts. Cell Mol. Life Sci. 2021, 78, 1577–1596. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Yu, D.; Chu, Y.; Corey, D.R. Mechanism of allele-selective inhibition of huntingtin expression by duplex RNAs that target CAG repeats: Function through the RNAi pathway. Nucleic Acids Res. 2012, 40, 11270–11280. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Ellison-Klimontowicz, M.E.; Krzyzosiak, W.J. Silencing of genes responsible for polyQ diseases using chemically modified single-stranded siRNAs. Acta Biochim. Pol. 2016, 63, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Wroblewska, J.P.; Nowak, B.M.; Krzyzosiak, W.J. Mutant CAG Repeats Effectively Targeted by RNA Interference in SCA7 Cells. Genes 2016, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Kotowska-Zimmer, A.; Ostrovska, Y.; Olejniczak, M. Universal RNAi Triggers for the Specific Inhibition of Mutant Huntingtin, Atrophin-1, Ataxin-3, and Ataxin-7 Expression. Mol. Ther. Nucleic Acids 2020, 19, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Datson, N.A.; Gonzalez-Barriga, A.; Kourkouta, E.; Weij, R.; van de Giessen, J.; Mulders, S.; Kontkanen, O.; Heikkinen, T.; Lehtimaki, K.; van Deutekom, J.C. The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS ONE 2017, 12, e0171127. [Google Scholar] [CrossRef] [PubMed]
- Kourkouta, E.; Weij, R.; Gonzalez-Barriga, A.; Mulder, M.; Verheul, R.; Bosgra, S.; Groenendaal, B.; Puolivali, J.; Toivanen, J.; van Deutekom, J.C.T.; et al. Suppression of Mutant Protein Expression in SCA3 and SCA1 Mice Using a CAG Repeat-Targeting Antisense Oligonucleotide. Mol. Ther. Nucleic Acids 2019, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Kotowska-Zimmer, A.; Przybyl, L.; Pewinska, M.; Suszynska-Zajczyk, J.; Wronka, D.; Figiel, M.; Olejniczak, M. A CAG repeat-targeting artificial miRNA lowers the mutant huntingtin level in the YAC128 model of Huntington’s disease. Mol. Ther. Nucleic Acids 2022, 28, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sanchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Michalik, A.; Martin, J.J.; Van Broeckhoven, C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur. J. Hum. Genet. 2004, 12, 2–15. [Google Scholar] [CrossRef]
- David, G.; Durr, A.; Stevanin, G.; Cancel, G.; Abbas, N.; Benomar, A.; Belal, S.; Lebre, A.S.; Abada-Bendib, M.; Grid, D.; et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum. Mol. Genet. 1998, 7, 165–170. [Google Scholar] [CrossRef]
- Del-Favero, J.; Krols, L.; Michalik, A.; Theuns, J.; Lofgren, A.; Goossens, D.; Wehnert, A.; Van den Bossche, D.; Van Zand, K.; Backhovens, H.; et al. Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA type II) caused by CAG triplet repeat expansion. Hum. Mol. Genet. 1998, 7, 177–186. [Google Scholar] [CrossRef] [PubMed]
- van de Warrenburg, B.P.; Frenken, C.W.; Ausems, M.G.; Kleefstra, T.; Sinke, R.J.; Knoers, N.V.; Kremer, H.P. Striking anticipation in spinocerebellar ataxia type 7: The infantile phenotype. J. Neurol. 2001, 248, 911–914. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Doussau, F.; Perot, J.B.; Roux, M.J.; Keime, C.; Hache, A.; Piguet, F.; Novati, A.; Weber, C.; Yalcin, B.; et al. SCA7 Mouse Cerebellar Pathology Reveals Preferential Downregulation of Key Purkinje Cell-Identity Genes and Shared Disease Signature with SCA1 and SCA2. J. Neurosci. 2021, 41, 4910–4936. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Farshim, P.P.; Flomen, R.; Jones, S.T.; McAteer, S.J.; Deverman, B.E.; Gradinaru, V.; Bates, G.P. Use of high-content imaging to quantify transduction of AAV-PHP viruses in the brain following systemic delivery. Brain Commun. 2021, 3, fcab105. [Google Scholar] [CrossRef] [PubMed]
- Takazawa, C.; Fujimoto, K.; Homma, D.; Sumi-Ichinose, C.; Nomura, T.; Ichinose, H.; Katoh, S. A brain-specific decrease of the tyrosine hydroxylase protein in sepiapterin reductase-null mice—As a mouse model for Parkinson’s disease. Biochem. Biophys. Res. Commun. 2008, 367, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Nomura, T.; Sumi-Ichinose, C. Metabolism of tetrahydrobiopterin: Its relevance in monoaminergic neurons and neurological disorders. Chem. Rec. 2008, 8, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Ransdell, J.L.; Dranoff, E.; Lau, B.; Lo, W.L.; Donermeyer, D.L.; Allen, P.M.; Nerbonne, J.M. Loss of Navbeta4-Mediated Regulation of Sodium Currents in Adult Purkinje Neurons Disrupts Firing and Impairs Motor Coordination and Balance. Cell Rep. 2017, 19, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Yanicostas, C.; Barbieri, E.; Hibi, M.; Brice, A.; Stevanin, G.; Soussi-Yanicostas, N. Requirement for zebrafish ataxin-7 in differentiation of photoreceptors and cerebellar neurons. PLoS ONE 2012, 7, e50705. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.D.; Dialynas, G.; Weake, V.M.; Liu, J.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Workman, J.L.; Abmayr, S.M. Loss of Drosophila Ataxin-7, a SAGA subunit, reduces H2B ubiquitination and leads to neural and retinal degeneration. Genes Dev. 2014, 28, 259–272. [Google Scholar] [CrossRef]
- Carrillo-Rosas, S.; Weber, C.; Fievet, L.; Messaddeq, N.; Karam, A.; Trottier, Y. Loss of zebrafish Ataxin-7, a SAGA subunit responsible for SCA7 retinopathy, causes ocular coloboma and malformation of photoreceptors. Hum. Mol. Genet. 2019, 28, 912–927. [Google Scholar] [CrossRef]
- van der Heijden, C.D.; Rijpkema, M.; Arias-Vasquez, A.; Hakobjan, M.; Scheffer, H.; Fernandez, G.; Franke, B.; van de Warrenburg, B.P. Genetic variation in ataxia gene ATXN7 influences cerebellar grey matter volume in healthy adults. Cerebellum 2013, 12, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, L.C.; Switonski, P.M.; Adamek, G.; Carvalho, J.; Duvick, L.A.; Jarrah, S.I.; McLoughlin, H.S.; Scoles, D.R.; Pulst, S.M.; Orr, H.T.; et al. Purkinje-Enriched snRNA-seq in SCA7 Cerebellum Reveals Zebrin Identity Loss as a Central Feature of Polyglutamine Ataxias. bioRxiv 2023. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Eilers, J.; Garaschuk, O.; Thoenen, H.; Konnerth, A.; Meyer, M. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc. Natl. Acad. Sci. USA 1997, 94, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Carvalho-de-Souza, J.L.; Wei, C.; Carrasquel-Ursulaez, W.; Lorenzo, Y.; Gonzalez, N.; Kubota, T.; Staisch, J.; Hain, T.; Petrossian, N.; et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc. Natl. Acad. Sci. USA 2020, 117, 6023–6034. [Google Scholar] [CrossRef] [PubMed]
- Kawabata Galbraith, K.; Fujishima, K.; Mizuno, H.; Lee, S.J.; Uemura, T.; Sakimura, K.; Mishina, M.; Watanabe, N.; Kengaku, M. MTSS1 Regulation of Actin-Nucleating Formin DAAM1 in Dendritic Filopodia Determines Final Dendritic Configuration of Purkinje Cells. Cell Rep. 2018, 24, 95–106.e9. [Google Scholar] [CrossRef] [PubMed]
- Bushart, D.D.; Chopra, R.; Singh, V.; Murphy, G.G.; Wulff, H.; Shakkottai, V.G. Targeting potassium channels to treat cerebellar ataxia. Ann. Clin. Transl. Neurol. 2018, 5, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Meera, P.; Altindag, B.; Chopra, R.; Perkins, E.M.; Paul, S.; Scoles, D.R.; Tarapore, E.; Magri, J.; Huang, H.; et al. MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias. Proc. Natl. Acad. Sci. USA 2018, 115, E12407–E12416. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Xiong, H.; Shin, Y.; Wang, D.; Deerinck, T.; Takahashi, H.; Ellisman, M.H.; Lipton, S.A.; Tong, G.; Descalzi, G.; et al. Roles of KChIP1 in the regulation of GABA-mediated transmission and behavioral anxiety. Mol. Brain 2010, 3, 23. [Google Scholar] [CrossRef][Green Version]
- Hirono, M.; Karube, F.; Yanagawa, Y. Modulatory Effects of Monoamines and Perineuronal Nets on Output of Cerebellar Purkinje Cells. Front. Neural Circuits 2021, 15, 661899. [Google Scholar] [CrossRef]
- Mulherkar, S.; Uddin, M.D.; Couvillon, A.D.; Sillitoe, R.V.; Tolias, K.F. The small GTPases RhoA and Rac1 regulate cerebellar development by controlling cell morphogenesis, migration and foliation. Dev. Biol. 2014, 394, 39–53. [Google Scholar] [CrossRef]
- Surdyka, M.; Jesion, E.; Niewiadomska-Cimicka, A.; Trottier, Y.; Kalinowska-Poska, Z.; Figiel, M. Selective transduction of cerebellar Purkinje and granule neurons using delivery of AAV-PHP.eB and AAVrh10 vectors at axonal terminal locations. Front. Mol. Neurosci. 2022, 15, 947490. [Google Scholar] [CrossRef] [PubMed]
- Perot, J.B.; Niewiadomska-Cimicka, A.; Brouillet, E.; Trottier, Y.; Flament, J. Longitudinal MRI and 1H-MRS study of SCA7 mouse forebrain reveals progressive multiregional atrophy and early brain metabolite changes indicating early neuronal and glial dysfunction. PLoS ONE 2024, 19, e0296790. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Pennesi, M.E.; Weeber, E.J.; Xu, B.; Atkinson, R.; Chen, S.; Armstrong, D.L.; Wu, S.M.; Sweatt, J.D.; Zoghbi, H.Y. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 2003, 37, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.P.; Bolch, S.N.; Zhu, P.; Weidert, F.; Dinculescu, A.; Lobanova, E.S. Systemic Delivery of Genes to Retina Using Adeno-Associated Viruses. Adv. Exp. Med. Biol. 2019, 1185, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Monckton, D.G.; Cayuela, M.L.; Gould, F.K.; Brock, G.J.; Silva, R.; Ashizawa, T. Very large (CAG)(n) DNA repeat expansions in the sperm of two spinocerebellar ataxia type 7 males. Hum. Mol. Genet. 1999, 8, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015, e52434. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMB-Net. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niewiadomska-Cimicka, A.; Fievet, L.; Surdyka, M.; Jesion, E.; Keime, C.; Singer, E.; Eisenmann, A.; Kalinowska-Poska, Z.; Nguyen, H.H.P.; Fiszer, A.; et al. AAV-Mediated CAG-Targeting Selectively Reduces Polyglutamine-Expanded Protein and Attenuates Disease Phenotypes in a Spinocerebellar Ataxia Mouse Model. Int. J. Mol. Sci. 2024, 25, 4354. https://doi.org/10.3390/ijms25084354
Niewiadomska-Cimicka A, Fievet L, Surdyka M, Jesion E, Keime C, Singer E, Eisenmann A, Kalinowska-Poska Z, Nguyen HHP, Fiszer A, et al. AAV-Mediated CAG-Targeting Selectively Reduces Polyglutamine-Expanded Protein and Attenuates Disease Phenotypes in a Spinocerebellar Ataxia Mouse Model. International Journal of Molecular Sciences. 2024; 25(8):4354. https://doi.org/10.3390/ijms25084354
Chicago/Turabian StyleNiewiadomska-Cimicka, Anna, Lorraine Fievet, Magdalena Surdyka, Ewelina Jesion, Céline Keime, Elisabeth Singer, Aurélie Eisenmann, Zaneta Kalinowska-Poska, Hoa Huu Phuc Nguyen, Agnieszka Fiszer, and et al. 2024. "AAV-Mediated CAG-Targeting Selectively Reduces Polyglutamine-Expanded Protein and Attenuates Disease Phenotypes in a Spinocerebellar Ataxia Mouse Model" International Journal of Molecular Sciences 25, no. 8: 4354. https://doi.org/10.3390/ijms25084354
APA StyleNiewiadomska-Cimicka, A., Fievet, L., Surdyka, M., Jesion, E., Keime, C., Singer, E., Eisenmann, A., Kalinowska-Poska, Z., Nguyen, H. H. P., Fiszer, A., Figiel, M., & Trottier, Y. (2024). AAV-Mediated CAG-Targeting Selectively Reduces Polyglutamine-Expanded Protein and Attenuates Disease Phenotypes in a Spinocerebellar Ataxia Mouse Model. International Journal of Molecular Sciences, 25(8), 4354. https://doi.org/10.3390/ijms25084354