
The Contribution of Type 2 Diabetes to Parkinson’s Disease Aetiology
Abstract
:1. Introduction
1.1. Physiology and Pathophysiology of Alfa Synuclein
Formation of α-syn Oligomers, Fibrillar Conglomerates, and LBs
1.2. Brain Glucose Metabolism
1.2.1. Glycolysis
1.2.2. The Tricarboxylic Acid Cycle in the MT Matrix
1.2.3. Oxidative Phosphorylation
1.2.4. The Pentose Phosphate Pathway
1.3. Insulin Resistance-Associated PD Pathology in Patients Comorbid with T2D
2. Diabetes Risk Factors for PD
3. Clinical Signs and Symptoms of PD and Diabetes Are Either Similar or Distinct
3.1. The Order of Appearance of Gastrointestinal, Cognitive, and Motor Symptoms in PD
3.2. Symptoms of PD and Diabetes
3.3. Brain Pathology
3.4. Selective Loss of High-Metabolism Cells in T2D and PD
4. Overlap of PD and Diabetes Aetiologies
4.1. T2D Does Not Accelerate Lewy Body Formation in PD
4.2. Amylin Neuropathology
4.3. Hyperglycemia
4.4. Increased Protein, Lipid, and Nucleic Acid Glycation
4.5. Insulin Resistance
4.6. Oxidative Stress and Inflammation
4.7. Mitochondrial Dysfunction
4.8. Reduced Efficiency of Autophagy and Proteasome Degradation
4.9. T2D Hyperglycemia Accelerates or Induces the Onset of PD Pathology
5. Pharmacological Interventions for PD
5.1. Conventional Pharmacological Interventions for PD
5.1.1. Levodopa
5.1.2. Dopamine Agonists
5.1.3. Monoamine Oxidase B (MAO-B) Inhibitors
5.1.4. Catechol-O-methyl Transferase (COMT) Inhibitors
5.1.5. Anticholinergics
5.1.6. Amantadine
5.2. Potential Alternative Pharmacological Interventions for PD
5.2.1. Anti-Alfa Synuclein Vaccination and Humanised α-Synuclein Antibodies (Arbo et al. 2022) [245]
5.2.2. Biguanides
5.2.3. Dipeptidyl Peptidase-4 Inhibitors
5.2.4. Flavonoids
5.2.5. Glucagon-like Peptide-1 Receptor Agonists
5.2.6. Hydroxy-3-methyl-glutaryl-coenzyme A Reductase Inhibitors
5.2.7. IL-1β Inhibitors
5.2.8. Insulin
5.2.9. Lenalidomide
5.2.10. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
5.2.11. Sulfonylureas
5.2.12. Thiazolidinediones
6. Conclusions
7. Study Limitations
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| ADTIQ | tetrahydroisoquinoline (ADTIQ) |
| AGE | advanced glycation end product |
| AKT | protein kinase B |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| ATP13A2 | late endosomal/lysosomal P5-type transport ATPase |
| BBB | blood–brain barrier |
| BDNF | brain-derived neurotrophic factor |
| CNS | central nervous system |
| CoA | coenzyme A |
| COMT | catechol-O-methyl transferase |
| CRP | C-reactive protein |
| CSF | cerebrospinal fluid |
| COX | cyclooxygenase |
| DAT | dopamine transporter |
| DJ1 | protein deglycase DJ1 |
| DLB | dementia with Lewy Bodies |
| DNA | deoxyribonucleic acid |
| DPP4s | dipeptidyl peptidase-4 enzyme inhibitors, also known as gliptins |
| ER | endoplasmic reticulum |
| FAD | flavin adenine dinucleotide |
| FADH2 | dihydroflavine-adenine dinucleotide |
| FAT10 | ubiquitin-like modifier HLA-F adjacent transcript 10, or ubiquitin D |
| FOXO1 | forkhead box protein O1 |
| G3P | glycerol-3-phosphate |
| G6PD | glucose-6-phosphate dehydrogenase |
| GA | Golgi apparatus |
| GABA | gamma-aminobutyric acid |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GI | gastrointestinal |
| GLP1 | Glucagon-like Peptide-1 |
| GLUT | glucose transporter |
| GSH | reduced glutathione |
| GSK3β | glycogen synthase kinase 3β |
| GSSG | glutathione disulfide, oxidised glutathione |
| GTP | guanosine-5′-triphosphate |
| HDAC | histone deacetylase |
| HIF1 | hypoxia-inducible factor 1 |
| HMG-CoA | 3-Hydroxy-3-methylglutaryl-coenzyme A |
| IDE | insulin-degrading enzyme |
| IFNγ | interferon gamma |
| IL | interleukin |
| INI | intranasal insulin |
| IR | insulin resistance |
| IRS-1 | insulin receptor substrate 1 |
| KGDHC | alpha-ketoglutarate dehydrogenase complex |
| L-DOPA | levodopa |
| LBs | Lewy Bodies |
| MAPK | mitogen-activated protein kinase |
| MGO | methylglyoxal |
| MIDN | Midnolin |
| MOA-B | monoamine oxidase B |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSA | multiple system atrophy |
| MT | mitochondria, mitochondrial |
| mTORC1 | mTOR Complex 1 |
| MTPTP | mitochondrial permeability transition pore |
| NAD | nicotinamide adenine dinucleotide |
| NADH | reduced nicotinamide adenine dinucleotide |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| NDD | neurodegenerative brain disorder |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR family pyrin domain-containing protein 3 |
| NMDA | N-methyl-D-aspartate |
| Nrf 2 | nuclear factor erythroid 2-related factor |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| PARK2 | encodes cytosolic ubiquitin-E3- ligase, the Parkin protein FAT10 |
| Parkin | 465-amino acid residue E3 ubiquitin ligase |
| PD | Parkinson’s disease |
| PGC1α | peroxisome proliferator-activated receptor-γ coactivator 1-α |
| PGK1 | phosphoglycerate kinase 1 |
| PI3K | phosphatidylinositol 3-kinase |
| PINK1 | protein kinase with a mitochondrial targeting domain |
| PPP | pentose phosphate pathway |
| RAGEs | receptors for advanced glycation end products |
| RBCs | red blood cells |
| RC | respiratory complex of the electron transport chain |
| RNA | ribonucleic acid |
| ROS | reactive oxidative species |
| SNARE | protein containing a characteristic 60–70 residue domain, the SNARE motif |
| SNCA | Synuclein Alpha gene |
| SNpc | pars compacta of substanca nigra |
| T2D | Type 2 diabetes |
| TCA | tricarboxylic acid |
| TNFα | tumour necrosis factor α |
| TXNIP | thioredoxin-binding protein |
| UPR | unfolded protein response |
| VaP | vascular Parkinsonism |
| VMAT2 | vesicular monoamine transporter 2 |
| α-syn | α-synuclein protein monomer |
| α-synO | α-synuclein oligomer |
References
- Yan, Y.; Shimoga, D.; Sharma, A. Parkinson’s Disease and Diabetes Mellitus: Synergistic Effects on Pathophysiology and GI Motility. Curr. Gastroenterol. Rep. 2023, 25, 106–113. [Google Scholar] [CrossRef]
- Cullinane, P.W.; de Pablo Fernandez, E.; Konig, A.; Outeiro, T.F.; Jaunmuktane, Z.; Warner, T.T. Type 2 Diabetes and Parkinson’s Disease: A Focused Review of Current Concepts. Mov. Disord. 2023, 38, 162–177. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s disease and Parkinson’s disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernandez, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Arguero-Sanchez, R.; Schule, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2019, 13, 1399. [Google Scholar] [CrossRef]
- Stevenson, T.J.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Dieriks, B.V.; Curtis, M.A. alpha-synuclein inclusions are abundant in non-neuronal cells in the anterior olfactory nucleus of the Parkinson’s disease olfactory bulb. Sci. Rep. 2020, 10, 6682. [Google Scholar] [CrossRef] [PubMed]
- Ni, A.; Ernst, C. Evidence That Substantia Nigra Pars Compacta Dopaminergic Neurons Are Selectively Vulnerable to Oxidative Stress Because They Are Highly Metabolically Active. Front. Cell. Neurosci. 2022, 16, 826193. [Google Scholar] [CrossRef] [PubMed]
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef]
- Jaumotte, J.D.; Wyrostek, S.L.; Zigmond, M.J. Protection of cultured dopamine neurons from MPP(+) requires a combination of neurotrophic factors. Eur. J. Neurosci. 2016, 44, 1691–1699. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Guzman, J.N.; Sanchez, J.; Schumacker, P.T. Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009290. [Google Scholar] [CrossRef] [PubMed]
- Puopolo, M.; Raviola, E.; Bean, B.P. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J. Neurosci. 2007, 27, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.A.; Guzman, J.N.; Estep, C.M.; Ilijic, E.; Kondapalli, J.; Sanchez-Padilla, J.; Surmeier, D.J. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat. Neurosci. 2012, 15, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; McCarty, V.; Peng, H.; Jefri, M.; Hettige, N.; Antonyan, L.; Crapper, L.; O’Leary, L.A.; Zhang, X.; Zhang, Y.; et al. Lesch-Nyhan disease causes impaired energy metabolism and reduced developmental potential in midbrain dopaminergic cells. Stem Cell Rep. 2021, 16, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Pristera, A.; Lin, W.; Kaufmann, A.K.; Brimblecombe, K.R.; Threlfell, S.; Dodson, P.D.; Magill, P.J.; Fernandes, C.; Cragg, S.J.; Ang, S.L. Transcription factors FOXA1 and FOXA2 maintain dopaminergic neuronal properties and control feeding behavior in adult mice. Proc. Natl. Acad. Sci. USA 2015, 112, E4929–E4938. [Google Scholar] [CrossRef] [PubMed]
- Abdi, I.Y.; Ghanem, S.S.; El-Agnaf, O.M. Immune-related biomarkers for Parkinson’s disease. Neurobiol. Dis. 2022, 170, 105771. [Google Scholar] [CrossRef] [PubMed]
- Roverato, N.D.; Sailer, C.; Catone, N.; Aichem, A.; Stengel, F.; Groettrup, M. Parkin is an E3 ligase for the ubiquitin-like modifier FAT10, which inhibits Parkin activation and mitophagy. Cell Rep. 2021, 34, 108857. [Google Scholar] [CrossRef]
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, M.; Tadayon, R.; Salzano, G.; Shaw, G.S.; Walden, H. A mechanistic review of Parkin activation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129894. [Google Scholar] [CrossRef]
- Sagehashi, N.; Obara, Y.; Maruyama, O.; Nakagawa, T.; Hosoi, T.; Ishii, K. Insulin Enhances Gene Expression of Midnolin, a Novel Genetic Risk Factor for Parkinson’s Disease, via Extracellular Signal-Regulated Kinase, Phosphoinositide 3-Kinase and Multiple Transcription Factors in SH-SY5Y Cells. J. Pharmacol. Exp. Ther. 2022, 381, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Beckett, L.A.; Murray, A.M.; Shannon, K.M.; Goetz, C.G.; Pilgrim, D.M.; Evans, D.A. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N. Engl. J. Med. 1996, 334, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Luthra, N.S.; Corcos, D.M.; Fantuzzi, G. C-reactive protein as the biomarker of choice to monitor the effects of exercise on inflammation in Parkinson’s disease. Front. Immunol. 2023, 14, 1178448. [Google Scholar] [CrossRef]
- Santiago, J.A.; Potashkin, J.A. System-based approaches to decode the molecular links in Parkinson’s disease and diabetes. Neurobiol. Dis. 2014, 72 Pt A, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Khang, R.; Park, C.; Shin, J.H. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience 2015, 294, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Tan, C.; Zhao, L.; Liang, Y.; Liu, G.; Liu, H.; Zhong, Y.; Liu, Z.; Mo, L.; Liu, X.; et al. Glucose metabolism impairment in Parkinson’s disease. Brain Res. Bull. 2023, 199, 110672. [Google Scholar] [CrossRef]
- Sheng, L.; Stewart, T.; Yang, D.; Thorland, E.; Soltys, D.; Aro, P.; Khrisat, T.; Xie, Z.; Li, N.; Liu, Z.; et al. Erythrocytic alpha-synuclein contained in microvesicles regulates astrocytic glutamate homeostasis: A new perspective on Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 102. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of alpha-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: Another mechanism for initiation and progression of Parkinson’s disease? Acta Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef]
- Liu, Z.; Chan, R.B.; Cai, Z.; Liu, X.; Wu, Y.; Yu, Z.; Feng, T.; Yang, Y.; Zhang, J. alpha-Synuclein-containing erythrocytic extracellular vesicles: Essential contributors to hyperactivation of monocytes in Parkinson’s disease. J. Neuroinflamm. 2022, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Di Lazzaro, G.; Marino, G.; Campanelli, F.; Ghiglieri, V. Advances in understanding the function of alpha-synuclein: Implications for Parkinson’s disease. Brain 2023, 146, 3587–3597. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.-I.; et al. α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.H.; Beach, T.G. Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov. Disord. 2016, 31, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef]
- Froula, J.M.; Castellana-Cruz, M.; Anabtawi, N.M.; Camino, J.D.; Chen, S.W.; Thrasher, D.R.; Freire, J.; Yazdi, A.A.; Fleming, S.; Dobson, C.M.; et al. Defining alpha-synuclein species responsible for Parkinson’s disease phenotypes in mice. J. Biol. Chem. 2019, 294, 10392–10406. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Ludtmann, M.H.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [PubMed]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.H.; Fuentes, F.; Vanasco, V.; Alvarez, S.; Alaimo, A.; Cassina, A.; Coluccio Leskow, F.; Velazquez, F. Alpha-synuclein mitochondrial interaction leads to irreversible translocation and complex I impairment. Arch. Biochem. Biophys. 2018, 651, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265. [Google Scholar] [CrossRef]
- Fan, J.; Hu, Z.; Zeng, L.; Lu, W.; Tang, X.; Zhang, J.; Li, T. Golgi apparatus and neurodegenerative diseases. Int. J. Dev. Neurosci. 2008, 26, 523–534. [Google Scholar] [CrossRef]
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23179–23188. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. alpha-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef]
- Liu, M.; Qin, L.; Wang, L.; Tan, J.; Zhang, H.; Tang, J.; Shen, X.; Tan, L.; Wang, C. alpha-synuclein induces apoptosis of astrocytes by causing dysfunction of the endoplasmic reticulum-Golgi compartment. Mol. Med. Rep. 2018, 18, 322–332. [Google Scholar] [PubMed]
- Paiva, I.; Jain, G.; Lazaro, D.F.; Jercic, K.G.; Hentrich, T.; Kerimoglu, C.; Pinho, R.; Szego, E.M.; Burkhardt, S.; Capece, V.; et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol. Dis. 2018, 119, 121–135. [Google Scholar] [CrossRef]
- Lee, Y.J.; Wang, S.; Slone, S.R.; Yacoubian, T.A.; Witt, S.N. Defects in very long chain fatty acid synthesis enhance alpha-synuclein toxicity in a yeast model of Parkinson’s disease. PLoS ONE 2011, 6, e15946. [Google Scholar] [CrossRef]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Cali, T.; Gai, W.P.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19, e44617. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid beta-protein and alpha-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Standaert, D.G. Reaping what you sow: Cross-seeding between aggregation-prone proteins in neurodegeneration. Mov. Disord. 2014, 29, 306. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chorell, E.; Wittung-Stafshede, P. Insulin-degrading enzyme is activated by the C-terminus of alpha-synuclein. Biochem. Biophys. Res. Commun. 2015, 466, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Sharma, S.K.; Chorell, E.; Steneberg, P.; Vernersson-Lindahl, E.; Edlund, H.; Wittung-Stafshede, P. Insulin-degrading enzyme prevents alpha-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 2015, 5, 12531. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Herman, P.; Rothman, D.L.; Agarwal, D.; Hyder, F. Evaluating the gray and white matter energy budgets of human brain function. J. Cereb. Blood Flow. Metab. 2018, 38, 1339–1353. [Google Scholar] [CrossRef]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e3. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow. Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef] [PubMed]
- Mielke, R.; Kessler, J.; Szelies, B.; Herholz, K.; Wienhard, K.; Heiss, W.D. Normal and pathological aging--findings of positron-emission-tomography. J. Neural. Transm. 1998, 105, 821–837. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Marques, A.; Dutheil, F.; Durand, E.; Rieu, I.; Mulliez, A.; Fantini, M.L.; Boirie, Y.; Durif, F. Glucose dysregulation in Parkinson’s disease: Too much glucose or not enough insulin? Parkinsonism Relat. Disord. 2018, 55, 122–127. [Google Scholar] [CrossRef]
- Liu, W.; Tang, J. Association between diabetes mellitus and risk of Parkinson’s disease: A prisma-compliant meta-analysis. Brain Behav. 2021, 11, e02082. [Google Scholar] [CrossRef]
- Sanchez-Gomez, A.; Diaz, Y.; Duarte-Salles, T.; Compta, Y.; Marti, M.J. Prediabetes, type 2 diabetes mellitus and risk of Parkinson’s disease: A population-based cohort study. Parkinsonism Relat. Disord. 2021, 89, 22–27. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Lee, W.Y. Association Between Glycemic Status and the Risk of Parkinson Disease: A Nationwide Population-Based Study. Diabetes Care 2020;43:2169-2175. Diabetes Care 2021, 44, e97. [Google Scholar] [CrossRef] [PubMed]
- Klimek, P.; Kautzky-Willer, A.; Chmiel, A.; Schiller-Fruhwirth, I.; Thurner, S. Quantification of diabetes comorbidity risks across life using nation-wide big claims data. PLoS Comput. Biol. 2015, 11, e1004125. [Google Scholar] [CrossRef]
- Chung, H.S.; Lee, J.S.; Kim, J.A.; Roh, E.; Lee, Y.B.; Hong, S.H.; Yu, J.H.; Kim, N.H.; Yoo, H.J.; Seo, J.A.; et al. Fasting plasma glucose variability in midlife and risk of Parkinson’s disease: A nationwide population-based study. Diabetes Metab. 2021, 47, 101195. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Goldacre, R.; Pakpoor, J.; Noyce, A.J.; Warner, T.T. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018, 91, e139–e142. [Google Scholar] [CrossRef]
- Chohan, H.; Senkevich, K.; Patel, R.K.; Bestwick, J.P.; Jacobs, B.M.; Bandres Ciga, S.; Gan-Or, Z.; Noyce, A.J. Type 2 Diabetes as a Determinant of Parkinson’s Disease Risk and Progression. Mov. Disord. 2021, 36, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Han, K.; Kim, D.; Rhee, S.Y.; Jang, W.; Shin, D.W. Body mass index, diabetes, and the risk of Parkinson’s disease. Mov. Disord. 2020, 35, 236–244. [Google Scholar] [CrossRef]
- Meyer, P.T.; Frings, L.; Hellwig, S. Update on SPECT and PET in parkinsonism—Part 2: Biomarker imaging of cognitive impairment in Lewy-body diseases. Curr. Opin. Neurol. 2014, 27, 398–404. [Google Scholar] [CrossRef]
- Dunn, L.; Allen, G.F.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease. Neurobiol. Aging 2014, 35, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Eggers, C.; Hilker, R.; Burghaus, L.; Schumacher, B.; Heiss, W.D. High resolution positron emission tomography demonstrates basal ganglia dysfunction in early Parkinson’s disease. J. Neurol. Sci. 2009, 276, 27–30. [Google Scholar] [CrossRef]
- Borghammer, P.; Chakravarty, M.; Jonsdottir, K.Y.; Sato, N.; Matsuda, H.; Ito, K.; Arahata, Y.; Kato, T.; Gjedde, A. Cortical hypometabolism and hypoperfusion in Parkinson’s disease is extensive: Probably even at early disease stages. Brain Struct. Funct. 2010, 214, 303–317. [Google Scholar] [CrossRef]
- Szturm, T.; Beheshti, I.; Mahana, B.; Hobson, D.E.; Goertzen, A.; Ko, J.H. Imaging Cerebral Glucose Metabolism during Dual-Task Walking in Patients with Parkinson’s disease. J. Neuroimaging 2021, 31, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Berding, G.; Odin, P.; Brooks, D.J.; Nikkhah, G.; Matthies, C.; Peschel, T.; Shing, M.; Kolbe, H.; van Den Hoff, J.; Fricke, H.; et al. Resting regional cerebral glucose metabolism in advanced Parkinson’s disease studied in the off and on conditions with [(18)F]FDG-PET. Mov. Disord. 2001, 16, 1014–1022. [Google Scholar] [CrossRef]
- Li, D.; Zuo, C.; Guan, Y.; Zhao, Y.; Shen, J.; Zan, S.; Sun, B. FDG-PET study of the bilateral subthalamic nucleus stimulation effects on the regional cerebral metabolism in advanced Parkinson disease. Acta Neurochir. Suppl. 2006, 99, 51–54. [Google Scholar]
- Firbank, M.J.; Yarnall, A.J.; Lawson, R.A.; Duncan, G.W.; Khoo, T.K.; Petrides, G.S.; O’Brien, J.T.; Barker, R.A.; Maxwell, R.J.; Brooks, D.J.; et al. Cerebral glucose metabolism and cognition in newly diagnosed Parkinson’s disease: ICICLE-PD study. J. Neurol. Neurosurg. Psychiatry 2017, 88, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Vander Borght, T.; Minoshima, S.; Giordani, B.; Foster, N.L.; Frey, K.A.; Berent, S.; Albin, R.L.; Koeppe, R.A.; Kuhl, D.E. Cerebral metabolic differences in Parkinson’s and Alzheimer’s diseases matched for dementia severity. J. Nucl. Med. 1997, 38, 797–802. [Google Scholar] [PubMed]
- Peppard, R.F.; Martin, W.R.; Carr, G.D.; Grochowski, E.; Schulzer, M.; Guttman, M.; McGeer, P.L.; Phillips, A.G.; Tsui, J.K.; Calne, D.B. Cerebral glucose metabolism in Parkinson’s disease with and without dementia. Arch. Neurol. 1992, 49, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kasai, T.; Mizuta, I.; Suematsu, M.; Osone, S.; Azuma, Y.; Imamura, T.; Tokuda, T.; Kanno, H.; El-Agnaf, O.M.A.; et al. Early-onset parkinsonism in a pedigree with phosphoglycerate kinase deficiency and a heterozygous carrier: Do PGK-1 mutations contribute to vulnerability to parkinsonism? npj Parkinsons Dis. 2017, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, E.; Greene, P.; Krishna, S.; Hirano, M.; DiMauro, S. Myopathy and parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve 2010, 41, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, C.; Li, X.; Wang, T.; Li, Y.; Cao, C.; Ding, Y.; Dong, M.; Finci, L.; Wang, J.H.; et al. Terazosin activates Pgk1 and Hsp90 to promote stress resistance. Nat. Chem. Biol. 2015, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Simmering, J.E.; Welsh, M.J.; Liu, L.; Narayanan, N.S.; Pottegard, A. Association of Glycolysis-Enhancing alpha-1 Blockers With Risk of Developing Parkinson Disease. JAMA Neurol. 2021, 78, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolanos, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef]
- Barinova, K.; Khomyakova, E.; Semenyuk, P.; Schmalhausen, E.; Muronetz, V. Binding of alpha-synuclein to partially oxidized glyceraldehyde-3-phosphate dehydrogenase induces subsequent inactivation of the enzyme. Arch. Biochem. Biophys. 2018, 642, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, A.; Pozdyshev, D.; Barinova, K.; Kudryavtseva, S.; Muronetz, V.I. alpha-Synuclein Overexpression in SH-SY5Y Human Neuroblastoma Cells Leads to the Accumulation of Thioflavin S-positive Aggregates and Impairment of Glycolysis. Biochemistry 2020, 85, 604–613. [Google Scholar] [PubMed]
- Semenyuk, P.; Barinova, K.; Muronetz, V. Glycation of alpha-synuclein amplifies the binding with glyceraldehyde-3-phosphate dehydrogenase. Int. J. Biol. Macromol. 2019, 127, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Matuda, S.; Yoshino, H.; Mori, H.; Hattori, N.; Ikebe, S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994, 35, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Riederer, P.; Przuntek, H.; Youdim, M.B. MPTP mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur. J. Pharmacol. 1991, 208, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 1994, 36, 876–881. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Elibol, B.; Dalmizrak, O.; Ercan, A.; Kulaksiz, G.; Ogus, H.; Dalkara, T.; Ozer, N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. 2004, 19, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Heck, R.W.; Tanhauser, S.M.; Manda, R.; Tu, C.; Laipis, P.J.; Silverman, D.N. Catalytic properties of mouse carbonic anhydrase V. J. Biol. Chem. 1994, 269, 24742–24746. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.F.; Frasier, M.; Shreve, N.; Calabrese, V.; Wolozin, B.; Butterfield, D.A. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice--a model of familial Parkinson’s disease. Neurobiol. Dis. 2005, 18, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Fan, C.; Gu, L.; Gao, H.; Liu, Q.; Zhang, T.; Qi, Z.; Zhao, C.; Zhao, H.; Cai, Q.; et al. Silencing of PINK1 induces mitophagy via mitochondrial permeability transition in dopaminergic MN9D cells. Brain Res. 2011, 1394, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Cookson, M.R. Mitochondrial quality control and dynamics in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 869–882. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Schapira, A.H.; Taanman, J.W. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 2009, 4, e4756. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in alpha-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Tu, D.; Gao, Y.; Yang, R.; Guan, T.; Hong, J.S.; Gao, H.M. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J. Neuroinflamm. 2019, 16, 255. [Google Scholar] [CrossRef] [PubMed]
- Fecchio, C.; Palazzi, L.; de Laureto, P.P. alpha-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules 2018, 23, 1531. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P., Jr.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- de Pablo-Fernandez, E.; Courtney, R.; Rockliffe, A.; Gentleman, S.; Holton, J.L.; Warner, T.T. Faster disease progression in Parkinson’s disease with type 2 diabetes is not associated with increased alpha-synuclein, tau, amyloid-beta or vascular pathology. Neuropathol. Appl. Neurobiol. 2021, 47, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yuan, F.; Chen, Z.; Zhu, S.; Chang, Z.; Yang, W.; Deng, B.; Que, R.; Cao, P.; Chao, Y.; et al. Vascular, inflammatory and metabolic risk factors in relation to dementia in Parkinson’s disease patients with type 2 diabetes mellitus. Aging 2020, 12, 15682–15704. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef] [PubMed]
- Laiteerapong, N.; Huang, E.S. Diabetes in Older Adults. In Diabetes in America, 3rd ed.; Cowie, C.C., Casagrande, S.S., Menke, A., Cissell, M.A., Eberhardt, M.S., Meigs, J.B., Gregg, E.W., Knowler, W.C., Barrett-Connor, E., Becker, D.J., et al., Eds.; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2018. [Google Scholar]
- Introduction: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44 (Suppl. 1), S1–S2. [CrossRef] [PubMed]
- Hirtz, D.; Thurman, D.J.; Gwinn-Hardy, K.; Mohamed, M.; Chaudhuri, A.R.; Zalutsky, R. How common are the “common” neurologic disorders? Neurology 2007, 68, 326–337. [Google Scholar] [CrossRef]
- De Rijk, M.C.; Launer, L.J.; Berger, K.; Breteler, M.M.; Dartigues, J.F.; Baldereschi, M.; Fratiglioni, L.; Lobo, A.; Martinez-Lage, J.; Trenkwalder, C.; et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54 (Suppl. 5), S21–S23. [Google Scholar]
- Pezzoli, G.; Cereda, E.; Amami, P.; Colosimo, S.; Barichella, M.; Sacilotto, G.; Zecchinelli, A.; Zini, M.; Ferri, V.; Bolliri, C.; et al. Onset and mortality of Parkinson’s disease in relation to type II diabetes. J. Neurol. 2023, 270, 1564–1572. [Google Scholar] [CrossRef]
- Perruolo, G.; Viggiano, D.; Fiory, F.; Cassese, A.; Nigro, C.; Liotti, A.; Miele, C.; Beguinot, F.; Formisano, P. Parkinson-like phenotype in insulin-resistant PED/PEA-15 transgenic mice. Sci. Rep. 2016, 6, 29967. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Valbuena, I.; Amat-Villegas, I.; Valenti-Azcarate, R.; Carmona-Abellan, M.D.M.; Marcilla, I.; Tunon, M.T.; Luquin, M.R. Interaction of amyloidogenic proteins in pancreatic beta cells from subjects with synucleinopathies. Acta Neuropathol. 2018, 135, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Hogg, E.; Athreya, K.; Basile, C.; Tan, E.E.; Kaminski, J.; Tagliati, M. High Prevalence of Undiagnosed Insulin Resistance in Non-Diabetic Subjects with Parkinson’s Disease. J. Parkinsons Dis. 2018, 8, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Markaki, I.; Ntetsika, T.; Sorjonen, K.; Svenningsson, P.; BioPark Study, G. Euglycemia Indicates Favorable Motor Outcome in Parkinson’s Disease. Mov. Disord. 2021, 36, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Zittel, S.; Uyar, M.; Lezius, S.; Gerloff, C.; Choe, C.U. HbA1c and Motor Outcome in Parkinson’s Disease in the Mark-PD Study. Mov. Disord. 2021, 36, 1991–1992. [Google Scholar] [CrossRef] [PubMed]
- Huxford, B.; Haque, T.; Joseph, A.B.; Simonet, C.; Gallagher, D.; Budu, C.; Dobson, R.; Noyce, A. Parkinson’s Disease and Type 2 Diabetes: HbA1c Is Associated with Motor and Cognitive Severity. Mov. Disord. 2022, 37, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Konig, A.; Vicente Miranda, H.; Outeiro, T.F. Alpha-Synuclein Glycation and the Action of Anti-Diabetic Agents in Parkinson’s Disease. J. Parkinsons Dis. 2018, 8, 33–43. [Google Scholar] [CrossRef]
- Schernhammer, E.; Hansen, J.; Rugbjerg, K.; Wermuth, L.; Ritz, B. Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care 2011, 34, 1102–1108. [Google Scholar] [CrossRef]
- Brauer, R.; Wei, L.; Ma, T.; Athauda, D.; Girges, C.; Vijiaratnam, N.; Auld, G.; Whittlesea, C.; Wong, I.; Foltynie, T. Diabetes medications and risk of Parkinson’s disease: A cohort study of patients with diabetes. Brain 2020, 143, 3067–3076. [Google Scholar] [CrossRef]
- Sanchez-Gomez, A.; Alcarraz-Vizan, G.; Fernandez, M.; Fernandez-Santiago, R.; Ezquerra, M.; Camara, A.; Serrano, M.; Novials, A.; Munoz, E.; Valldeoriola, F.; et al. Peripheral insulin and amylin levels in Parkinson’s disease. Parkinsonism Relat. Disord. 2020, 79, 91–96. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Han, K.D.; Kwon, H.; Park, S.E.; Park, Y.G.; Kim, Y.H.; Yoo, S.J.; Rhee, E.J.; Lee, W.Y. Association Between Glycemic Status and the Risk of Parkinson Disease: A Nationwide Population-Based Study. Diabetes Care 2020, 43, 2169–2175. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, V.; Albin, R.L.; Muller, M.L.; Koeppe, R.A.; Frey, K.A.; Bohnen, N.I. Diabetes is associated with postural instability and gait difficulty in Parkinson disease. Parkinsonism Relat. Disord. 2013, 19, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Lawton, M.A.; Swallow, D.M.; Grosset, K.A.; Marrinan, S.L.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Hardy, J.; Morris, H.R.; et al. Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson’s disease. Mov. Disord. 2016, 31, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Barichella, M.; Cassani, E.; Caccialanza, R.; Pezzoli, G. Clinical features of Parkinson disease when onset of diabetes came first: A case-control study. Neurology 2012, 78, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Mohamed Ibrahim, N.; Ramli, R.; Koya Kutty, S.; Shah, S.A. Earlier onset of motor complications in Parkinson’s patients with comorbid diabetes mellitus. Mov. Disord. 2018, 33, 1967–1968. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Zimmermann, J.; Sixel-Doring, F.; Focke, N.K.; Wicke, T.; Ebentheuer, J.; Schaumburg, M.; Lang, E.; Friede, T.; Trenkwalder, C.; et al. Baseline predictors for progression 4 years after Parkinson’s disease diagnosis in the De Novo Parkinson Cohort (DeNoPa). Mov. Disord. 2019, 34, 67–77. [Google Scholar] [CrossRef]
- Ou, R.; Wei, Q.; Hou, Y.; Zhang, L.; Liu, K.; Lin, J.; Jiang, Z.; Song, W.; Cao, B.; Shang, H. Effect of diabetes control status on the progression of Parkinson’s disease: A prospective study. Ann. Clin. Transl. Neurol. 2021, 8, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Evans, J.; Wernick, A.; Virdi, G.; Choi, M.L.; Lawton, M.; Vijiaratnam, N.; Girges, C.; Ben-Shlomo, Y.; Ismail, K.; et al. The Impact of Type 2 Diabetes in Parkinson’s Disease. Mov. Disord. 2022, 37, 1612–1623. [Google Scholar] [CrossRef]
- Barter, J.D.; Thomas, D.; Ni, L.; Bay, A.A.; Johnson, T.M., 2nd; Prusin, T.; Hackney, M.E. Parkinson’s Disease and Diabetes Mellitus: Individual and Combined Effects on Motor, Cognitive, and Psychosocial Functions. Healthcare 2023, 11, 1316. [Google Scholar] [CrossRef]
- Nair, A.T.; Ramachandran, V.; Joghee, N.M.; Antony, S.; Ramalingam, G. Gut Microbiota Dysfunction as Reliable Non-invasive Early Diagnostic Biomarkers in the Pathophysiology of Parkinson’s Disease: A Critical Review. J. Neurogastroenterol. Motil. 2018, 24, 30–42. [Google Scholar] [CrossRef]
- Cersosimo, M.G.; Benarroch, E.E. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Rahmani, E.; Garcia, E.; Jacobs, J.P. Gastrointestinal symptoms are predictive of trajectories of cognitive functioning in de novo Parkinson’s disease. Parkinsonism Relat. Disord. 2020, 72, 7–12. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Mason, S.L.; Evans, J.R.; Foltynie, T.; Brayne, C.; Robbins, T.W.; Barker, R.A. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Caviness, J.N.; Driver-Dunckley, E.; Connor, D.J.; Sabbagh, M.N.; Hentz, J.G.; Noble, B.; Evidente, V.G.; Shill, H.A.; Adler, C.H. Defining mild cognitive impairment in Parkinson’s disease. Mov. Disord. 2007, 22, 1272–1277. [Google Scholar] [CrossRef]
- Janvin, C.C.; Larsen, J.P.; Aarsland, D.; Hugdahl, K. Subtypes of mild cognitive impairment in Parkinson’s disease: Progression to dementia. Mov. Disord. 2006, 21, 1343–1349. [Google Scholar] [CrossRef]
- Domellof, M.E.; Ekman, U.; Forsgren, L.; Elgh, E. Cognitive function in the early phase of Parkinson’s disease, a five-year follow-up. Acta Neurol. Scand. 2015, 132, 79–88. [Google Scholar] [CrossRef]
- Chapelet, G.; Leclair-Visonneau, L.; Clairembault, T.; Neunlist, M.; Derkinderen, P. Can the gut be the missing piece in uncovering PD pathogenesis? Parkinsonism Relat. Disord. 2019, 59, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wu, X.; Jin, F. Gut-Brain Psychology: Rethinking Psychology From the Microbiota-Gut-Brain Axis. Front. Integr. Neurosci. 2018, 12, 33. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, J.T.; Perepezko, K.; Mills, K.A.; Mari, Z.; Butala, A.; Dawson, T.M.; Pantelyat, A.; Rosenthal, L.S.; Pontone, G.M. Dopamine transporter availability reflects gastrointestinal dysautonomia in early Parkinson disease. Parkinsonism Relat. Disord. 2018, 55, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Gareau, M.G.; Wine, E.; Rodrigues, D.M.; Cho, J.H.; Whary, M.T.; Philpott, D.J.; Macqueen, G.; Sherman, P.M. Bacterial infection causes stress-induced memory dysfunction in mice. Gut 2011, 60, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Hall, S.; Surova, Y.; Nielsen, H.M.; Janelidze, S.; Brundin, L.; Hansson, O. Cerebrospinal fluid inflammatory markers in Parkinson’s disease--associations with depression, fatigue, and cognitive impairment. Brain Behav. Immun. 2013, 33, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Zuo, L.J.; Wang, F.; Chen, Z.J.; Hu, Y.; Wang, Y.J.; Wang, X.M.; Zhang, W. Potential biomarkers relating pathological proteins, neuroinflammatory factors and free radicals in PD patients with cognitive impairment: A cross-sectional study. BMC Neurol. 2014, 14, 113. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kotagal, V.; Muller, M.L.; Koeppe, R.A.; Scott, P.J.; Albin, R.L.; Frey, K.A.; Petrou, M. Diabetes mellitus is independently associated with more severe cognitive impairment in Parkinson disease. Parkinsonism Relat. Disord. 2014, 20, 1394–1398. [Google Scholar] [CrossRef]
- Ong, M.; Foo, H.; Chander, R.J.; Wen, M.C.; Au, W.L.; Sitoh, Y.Y.; Tan, L.; Kandiah, N. Influence of diabetes mellitus on longitudinal atrophy and cognition in Parkinson’s disease. J. Neurol. Sci. 2017, 377, 122–126. [Google Scholar] [CrossRef]
- Petrou, M.; Davatzikos, C.; Hsieh, M.; Foerster, B.R.; Albin, R.L.; Kotagal, V.; Muller, M.L.; Koeppe, R.A.; Herman, W.H.; Frey, K.A.; et al. Diabetes, Gray Matter Loss, and Cognition in the Setting of Parkinson Disease. Acad. Radiol. 2016, 23, 577–581. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Elabi, O.F.; Cunha, J.; Gaceb, A.; Fex, M.; Paul, G. High-fat diet-induced diabetes leads to vascular alterations, pericyte reduction, and perivascular depletion of microglia in a 6-OHDA toxin model of Parkinson disease. J. Neuroinflamm. 2021, 18, 175. [Google Scholar] [CrossRef]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Hyperglycemia and advanced glycation end products disrupt BBB and promote occludin and claudin-5 protein secretion on extracellular microvesicles. Sci. Rep. 2020, 10, 7274. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Yamashita, T.; Ohta, Y.; Tadokoro, K.; Omote, Y.; Morihara, R.; Abe, K. Cerebral Microbleeds in Patients with Parkinson’s Disease and Dementia with Lewy Bodies: Comparison Using Magnetic Resonance Imaging and 99 mTc-ECD SPECT Subtraction Imaging. J. Alzheimers Dis. 2021, 80, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Biondetti, E.; Santin, M.D.; Valabregue, R.; Mangone, G.; Gaurav, R.; Pyatigorskaya, N.; Hutchison, M.; Yahia-Cherif, L.; Villain, N.; Habert, M.O.; et al. The spatiotemporal changes in dopamine, neuromelanin and iron characterizing Parkinson’s disease. Brain 2021, 144, 3114–3125. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Pyatigorskaya, N.; Sharman, M.; Corvol, J.C.; Valabregue, R.; Yahia-Cherif, L.; Poupon, F.; Cormier-Dequaire, F.; Siebner, H.; Klebe, S.; Vidailhet, M.; et al. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov. Disord. 2015, 30, 1077–1084. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, Y.; Li, L.; Melchiorsen, J.U.; Rosenkilde, M.; Holscher, C. The Novel Dual GLP-1/GIP Receptor Agonist DA-CH5 Is Superior to Single GLP-1 Receptor Agonists in the MPTP Model of Parkinson’s Disease. J. Parkinsons Dis. 2020, 10, 523–542. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, L.; Zhao, Q.; Du, X.; Bi, M.; Li, Y.; Jiao, Q.; Jiang, H. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med. 2020, 152, 227–234. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Parkkinen, L.; O’Sullivan, S.S.; Collins, C.; Petrie, A.; Holton, J.L.; Revesz, T.; Lees, A.J. Disentangling the relationship between lewy bodies and nigral neuronal loss in Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 277–286. [Google Scholar] [CrossRef]
- Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 2015, 138 Pt 6, 1642–1657. [Google Scholar] [CrossRef]
- Cheong, J.L.Y.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Parkinsons Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J. Differential permeability of the blood-brain barrier to two pancreatic peptides: Insulin and amylin. Peptides 1998, 19, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Wittung-Stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Ly, H.; Liu, M.; Chen, J.; Zhu, H.; Chow, M.; Hersh, L.B.; Despa, F. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1beta Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J. Alzheimers Dis. 2016, 53, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.; Verma, N.; Wu, F.; Liu, M.; Saatman, K.E.; Nelson, P.T.; Slevin, J.T.; Goldstein, L.B.; Biessels, G.J.; Despa, F. Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann. Neurol. 2017, 82, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Byman, E.; Fex, M.; Wennstrom, M. Amylin alters human brain pericyte viability and NG2 expression. J. Cereb. Blood Flow. Metab. 2017, 37, 1470–1482. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Valenti-Azcarate, R.; Amat-Villegas, I.; Riverol, M.; Marcilla, I.; de Andrea, C.E.; Sanchez-Arias, J.A.; Del Mar Carmona-Abellan, M.; Marti, G.; Erro, M.E.; et al. Amylin as a potential link between type 2 diabetes and alzheimer disease. Ann. Neurol. 2019, 86, 539–551. [Google Scholar] [CrossRef]
- Saller, C.F.; Chiodo, L.A. Glucose suppresses basal firing and haloperidol-induced increases in the firing rate of central dopaminergic neurons. Science 1980, 210, 1269–1271. [Google Scholar] [CrossRef]
- Montefusco, O.; Assini, M.C.; Missale, C. Insulin-mediated effects of glucose on dopamine metabolism. Acta Diabetol. Lat. 1983, 20, 71–77. [Google Scholar] [CrossRef]
- Murzi, E.; Contreras, Q.; Teneud, L.; Valecillos, B.; Parada, M.A.; De Parada, M.P.; Hernandez, L. Diabetes decreases limbic extracellular dopamine in rats. Neurosci. Lett. 1996, 202, 141–144. [Google Scholar] [CrossRef]
- Renaud, J.; Bassareo, V.; Beaulieu, J.; Pinna, A.; Schlich, M.; Lavoie, C.; Murtas, D.; Simola, N.; Martinoli, M.G. Dopaminergic neurodegeneration in a rat model of long-term hyperglycemia: Preferential degeneration of the nigrostriatal motor pathway. Neurobiol. Aging 2018, 69, 117–128. [Google Scholar] [CrossRef]
- Perez-Taboada, I.; Alberquilla, S.; Martin, E.D.; Anand, R.; Vietti-Michelina, S.; Tebeka, N.N.; Cantley, J.; Cragg, S.J.; Moratalla, R.; Vallejo, M. Diabetes Causes Dysfunctional Dopamine Neurotransmission Favoring Nigrostriatal Degeneration in Mice. Mov. Disord. 2020, 35, 1636–1648. [Google Scholar] [CrossRef]
- Su, C.J.; Shen, Z.; Cui, R.X.; Huang, Y.; Xu, D.L.; Zhao, F.L.; Pan, J.; Shi, A.M.; Liu, T.; Yu, Y.L. Thioredoxin-Interacting Protein (TXNIP) Regulates Parkin/PINK1-mediated Mitophagy in Dopaminergic Neurons Under High-glucose Conditions: Implications for Molecular Links Between Parkinson’s Disease and Diabetes. Neurosci. Bull. 2020, 36, 346–358. [Google Scholar] [CrossRef]
- Dionisio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef]
- Chegao, A.; Guarda, M.; Alexandre, B.M.; Shvachiy, L.; Temido-Ferreira, M.; Marques-Morgado, I.; Fernandes Gomes, B.; Matthiesen, R.; Lopes, L.V.; Florindo, P.R.; et al. Glycation modulates glutamatergic signaling and exacerbates Parkinson’s disease-like phenotypes. npj Parkinsons Dis. 2022, 8, 51. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Szego, E.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Xie, B.; Lin, F.; Peng, L.; Ullah, K.; Wu, H.; Qing, H.; Deng, Y. Methylglyoxal increases dopamine level and leads to oxidative stress in SH-SY5Y cells. Acta Biochim. Biophys. Sin. 2014, 46, 950–956. [Google Scholar] [CrossRef]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344 Pt 1, 109–116. [Google Scholar] [CrossRef]
- Shaikh, S.; Nicholson, L.F. Advanced glycation end products induce in vitro cross-linking of alpha-synuclein and accelerate the process of intracellular inclusion body formation. J. Neurosci. Res. 2008, 86, 2071–2082. [Google Scholar] [CrossRef]
- Du, X.Y.; Xie, X.X.; Liu, R.T. The Role of alpha-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 86645. [Google Scholar] [CrossRef]
- Uceda, A.B.; Frau, J.; Vilanova, B.; Adrover, M. Glycation of alpha-synuclein hampers its binding to synaptic-like vesicles and its driving effect on their fusion. Cell. Mol. Life Sci. 2022, 79, 342. [Google Scholar] [CrossRef]
- Ambrosi, G.; Cerri, S.; Blandini, F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J. Neural Transm. 2014, 121, 849–859. [Google Scholar] [CrossRef]
- Xie, B.; Lin, F.; Ullah, K.; Peng, L.; Ding, W.; Dai, R.; Qing, H.; Deng, Y. A newly discovered neurotoxin ADTIQ associated with hyperglycemia and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2015, 459, 361–366. [Google Scholar] [CrossRef]
- Castellani, R.; Smith, M.A.; Richey, P.L.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Munch, G.; Luth, H.J.; Wong, A.; Arendt, T.; Hirsch, E.; Ravid, R.; Riederer, P. Crosslinking of alpha-synuclein by advanced glycation endproducts--an early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 2000, 20, 253–257. [Google Scholar] [CrossRef]
- Pearce, R.K.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef]
- Kuhla, B.; Boeck, K.; Luth, H.J.; Schmidt, A.; Weigle, B.; Schmitz, M.; Ogunlade, V.; Munch, G.; Arendt, T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging 2006, 27, 815–822. [Google Scholar] [CrossRef]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural basis of synaptic vesicle assembly promoted by alpha-synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, M.; Iwabuchi, M.; Terawaki, H.; Sato, T.; Kohno, M.; Ito, S. Plasma alpha-oxoaldehyde levels in diabetic and nondiabetic chronic kidney disease patients. Am. J. Nephrol. 2008, 28, 871–878. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Drummond, K.S.; Nelson, R.G.; Howell, S.K.; Szwergold, B.S.; Mauer, M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes 2005, 54, 3274–3281. [Google Scholar] [CrossRef]
- Nemet, I.; Turk, Z.; Duvnjak, L.; Car, N.; Varga-Defterdarovic, L. Humoral methylglyoxal level reflects glycemic fluctuation. Clin. Biochem. 2005, 38, 379–383. [Google Scholar] [CrossRef]
- Yaffe, K.; Lindquist, K.; Schwartz, A.V.; Vitartas, C.; Vittinghoff, E.; Satterfield, S.; Simonsick, E.M.; Launer, L.; Rosano, C.; Cauley, J.A.; et al. Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology 2011, 77, 1351–1356. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Farzadfard, A.; Konig, A.; Petersen, S.V.; Nielsen, J.; Vasili, E.; Dominguez-Meijide, A.; Buell, A.K.; Outeiro, T.F.; Otzen, D.E. Glycation modulates alpha-synuclein fibrillization kinetics: A sweet spot for inhibition. J. Biol. Chem. 2022, 298, 101848. [Google Scholar] [CrossRef]
- Wan, Q.; Xiong, Z.G.; Man, H.Y.; Ackerley, C.A.; Braunton, J.; Lu, W.Y.; Becker, L.E.; MacDonald, J.F.; Wang, Y.T. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 1997, 388, 686–690. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Yao, W.D.; Gainetdinov, R.R.; Arbuckle, M.I.; Sotnikova, T.D.; Cyr, M.; Beaulieu, J.M.; Torres, G.E.; Grant, S.G.; Caron, M.G. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 2004, 41, 625–638. [Google Scholar] [CrossRef]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Victorino, D.B.; Nejm, M.; Guimaraes-Marques, M.; Scorza, F.A.; Scorza, C.A. Repurposing GLP-1 Receptor Agonists for Parkinson’s Disease: Current Evidence and Future Opportunities. Pharm Med. 2021, 35, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sun, T.; He, X.; Wang, Z.; Zhao, K.; An, J.; Wen, L.; Li, J.Y.; Li, W.; Feng, J. Association between Parkinson’s Disease and Diabetes Mellitus: From Epidemiology, Pathophysiology and Prevention to Treatment. Aging Dis. 2022, 13, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Sabari, S.S.; Balasubramani, K.; Iyer, M.; Sureshbabu, H.W.; Venkatesan, D.; Gopalakrishnan, A.V.; Narayanaswamy, A.; Senthil Kumar, N.; Vellingiri, B. Type 2 Diabetes (T2DM) and Parkinson’s Disease (PD): A Mechanistic Approach. Mol. Neurobiol. 2023, 60, 4547–4573. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 2008, 585, 76–87. [Google Scholar] [CrossRef]
- van der Heide, L.P.; Ramakers, G.M.; Smidt, M.P. Insulin signaling in the central nervous system: Learning to survive. Prog. Neurobiol. 2006, 79, 205–221. [Google Scholar] [CrossRef]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Uemura, E.; Greenlee, H.W. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp. Neurol. 2006, 198, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Heidenrich, K.A.; Gilmore, P.R.; Garvey, W.T. Glucose transport in primary cultured neurons. J. Neurosci. Res. 1989, 22, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109 (Suppl. 1), 87–93. [Google Scholar] [CrossRef] [PubMed]
- Arbo, B.D.; Schimith, L.E.; Goulart Dos Santos, M.; Hort, M.A. Repositioning and development of new treatments for neurodegenerative diseases: Focus on neuroinflammation. Eur. J. Pharmacol. 2022, 919, 174800. [Google Scholar] [CrossRef] [PubMed]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Goldin, M.; Segal, M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur. J. Neurosci. 2003, 17, 2529–2539. [Google Scholar] [CrossRef]
- Saravanan, S.; Ramkumar, K.; Adalarasu, K.; Sivanandam, V.; Kumar, S.R.; Stalin, S.; Amirtharajan, R. A Systematic Review of Artificial Intelligence (AI) Based Approaches for the Diagnosis of Parkinson’s Disease. Arch. Comput. Methods Eng. 2022, 29, 3639–3653. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Bassil, F.; Delamarre, A.; Canron, M.H.; Dutheil, N.; Vital, A.; Negrier-Leibreich, M.L.; Bezard, E.; Fernagut, P.O.; Meissner, W.G. Impaired brain insulin signalling in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12760. [Google Scholar] [CrossRef]
- Bassil, F.; Canron, M.H.; Vital, A.; Bezard, E.; Li, Y.; Greig, N.H.; Gulyani, S.; Kapogiannis, D.; Fernagut, P.O.; Meissner, W.G. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain 2017, 140, 1420–1436. [Google Scholar] [CrossRef] [PubMed]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Moroo, I.; Yamada, T.; Makino, H.; Tooyama, I.; McGeer, P.L.; McGeer, E.G.; Hirayama, K. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson’s disease. Acta Neuropathol. 1994, 87, 343–348. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamada, T.; Tooyama, I.; Moroo, I.; Kimura, H.; Yamamoto, T.; Okada, H. Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1996, 204, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.T.; Chen, K.Y.; Wang, W.; Chiu, J.Y.; Wu, D.; Chao, T.Y.; Hu, C.J.; Chau, K.D.; Bamodu, O.A. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of alpha-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020, 9, 740. [Google Scholar] [CrossRef]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Brodacki, B.; Staszewski, J.; Toczylowska, B.; Kozlowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Sawada, H.; Oeda, T.; Umemura, A.; Tomita, S.; Kohsaka, M.; Park, K.; Yamamoto, K.; Sugiyama, H. Baseline C-Reactive Protein Levels and Life Prognosis in Parkinson Disease. PLoS ONE 2015, 10, e0134118. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Sonnen, J.A.; Larson, E.B.; Brickell, K.; Crane, P.K.; Woltjer, R.; Montine, T.J.; Craft, S. Different patterns of cerebral injury in dementia with or without diabetes. Arch. Neurol. 2009, 66, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. Erratum: T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 549, 292. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.M.; Carroll, E.C.; Callender, L.A.; Hood, G.A.; Berryman, V.; Pattrick, M.; Finer, S.; Hitman, G.A.; Ackland, G.L.; Henson, S.M. Type 2 diabetes is associated with the accumulation of senescent T cells. Clin. Exp. Immunol. 2019, 197, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Bahniwal, M.; Little, J.P.; Walker, D.G.; Klegeris, A. Insulin Modulates In Vitro Secretion of Cytokines and Cytotoxins by Human Glial Cells. Curr. Alzheimer Res. 2015, 12, 684–693. [Google Scholar] [CrossRef]
- Hwang, I.K.; Choi, J.H.; Nam, S.M.; Park, O.K.; Yoo, D.Y.; Kim, W.; Yi, S.S.; Won, M.H.; Seong, J.K.; Yoon, Y.S. Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol. Res. 2014, 36, 824–832. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef]
- Gispen, W.H.; Biessels, G.J. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000, 23, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Picconi, B.; De Leonibus, E.; Calabresi, P. Synaptic plasticity and levodopa-induced dyskinesia: Electrophysiological and structural abnormalities. J. Neural. Transm. 2018, 125, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Morgante, F.; Espay, A.J.; Gunraj, C.; Lang, A.E.; Chen, R. Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 2006, 129 Pt 4, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, R.E.; Helwig, M.; Bae, E.J.; Aboutalebi, H.; Lee, S.J.; Ulusoy, A.; Di Monte, D.A. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular alpha-synuclein transfer. J. Clin. Investig. 2019, 129, 3738–3753. [Google Scholar] [CrossRef] [PubMed]
- Scudamore, O.; Ciossek, T. Increased Oxidative Stress Exacerbates alpha-Synuclein Aggregation In Vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Clarke, D.W.; Boyd, F.T., Jr.; Kappy, M.S.; Raizada, M.K. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 1984, 259, 11672–11675. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 2019, 4, e130681. [Google Scholar] [CrossRef]
- Schell, M.; Wardelmann, K.; Kleinridders, A. Untangling the effect of insulin action on brain mitochondria and metabolism. J. Neuroendocrinol. 2021, 33, e12932. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef]
- Moreira, P.I.; Rolo, A.P.; Sena, C.; Seica, R.; Oliveira, C.R.; Santos, M.S. Insulin attenuates diabetes-related mitochondrial alterations: A comparative study. Med. Chem. 2006, 2, 299–308. [Google Scholar] [CrossRef]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Howarth, F.C. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef]
- Santos, M.S.; Santos, D.L.; Palmeira, C.M.; Seica, R.; Moreno, A.J.; Oliveira, C.R. Brain and liver mitochondria isolated from diabetic Goto-Kakizaki rats show different susceptibility to induced oxidative stress. Diabetes Metab. Res. Rev. 2001, 17, 223–230. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Alves, M.G.; Oliveira, P.F.; Cardoso, S.; Carvalho, C.; Seica, R.; Santos, M.S.; Moreira, P.I. Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes. Mol. Cell. Biochem. 2014, 394, 13–22. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Kong, F.J.; Ma, L.L.; Guo, J.J.; Xu, L.H.; Li, Y.; Qu, S. Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin. Sci 2018, 132, 111–125. [Google Scholar] [CrossRef]
- Chen, J.L.; Luo, C.; Pu, D.; Zhang, G.Q.; Zhao, Y.X.; Sun, Y.; Zhao, K.X.; Liao, Z.Y.; Lv, A.K.; Zhu, S.Y.; et al. Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp. Neurol. 2019, 311, 44–56. [Google Scholar] [CrossRef]
- Guan, Z.F.; Tao, Y.H.; Zhang, X.M.; Guo, Q.L.; Liu, Y.C.; Zhang, Y.; Wang, Y.M.; Ji, G.; Wu, G.F.; Wang, N.N.; et al. G-CSF and cognitive dysfunction in elderly diabetic mice with cerebral small vessel disease: Preventive intervention effects and underlying mechanisms. CNS Neurosci. Ther. 2017, 23, 462–474. [Google Scholar] [CrossRef]
- Guan, Z.F.; Zhou, X.L.; Zhang, X.M.; Zhang, Y.; Wang, Y.M.; Guo, Q.L.; Ji, G.; Wu, G.F.; Wang, N.N.; Yang, H.; et al. Beclin-1- mediated autophagy may be involved in the elderly cognitive and affective disorders in streptozotocin-induced diabetic mice. Transl. Neurodegener. 2016, 5, 22. [Google Scholar] [CrossRef]
- Jing, Y.H.; Zhang, L.; Gao, L.P.; Qi, C.C.; Lv, D.D.; Song, Y.F.; Yin, J.; Wang, D.G. Autophagy plays beneficial effect on diabetic encephalopathy in type 2 diabetes: Studies in vivo and in vitro. Neuroendocrinol. Lett. 2017, 38, 27–37. [Google Scholar]
- Li, Y.; Zhang, Y.; Wang, L.; Wang, P.; Xue, Y.; Li, X.; Qiao, X.; Zhang, X.; Xu, T.; Liu, G.; et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 2017, 13, 1145–1160. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Pignalosa, F.C.; Desiderio, A.; Mirra, P.; Nigro, C.; Perruolo, G.; Ulianich, L.; Formisano, P.; Beguinot, F.; Miele, C.; Napoli, R.; et al. Diabetes and Cognitive Impairment: A Role for Glucotoxicity and Dopaminergic Dysfunction. Int. J. Mol. Sci. 2021, 22, 12366. [Google Scholar] [CrossRef]
- Lv, Y.Q.; Yuan, L.; Sun, Y.; Dou, H.W.; Su, J.H.; Hou, Z.P.; Li, J.Y.; Li, W. Long-term hyperglycemia aggravates alpha-synuclein aggregation and dopaminergic neuronal loss in a Parkinson’s disease mouse model. Transl. Neurodegener. 2022, 11, 14. [Google Scholar] [CrossRef]
- Su, C.J.; Feng, Y.; Liu, T.T.; Liu, X.; Bao, J.J.; Shi, A.M.; Hu, D.M.; Liu, T.; Yu, Y.L. Thioredoxin-interacting protein induced alpha-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef]
- Chen, L.; Ding, Y.; Cagniard, B.; Van Laar, A.D.; Mortimer, A.; Chi, W.; Hastings, T.G.; Kang, U.J.; Zhuang, X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 2008, 28, 425–433. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Olanow, C.W. Levodopa is the best symptomatic therapy for PD: Nothing more, nothing less. Mov. Disord. 2019, 34, 812–815. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- International Parkinson’s Disease Genomics Consortium; Wellcome Trust Case Control Consortium 2. A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011, 7, e1002142. [Google Scholar]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef]
- Chen, S.; Yu, S.J.; Li, Y.; Lecca, D.; Glotfelty, E.; Kim, H.K.; Choi, H.I.; Hoffer, B.J.; Greig, N.H.; Kim, D.S.; et al. Post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson’s disease. Sci. Rep. 2018, 8, 10722. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Holscher, C. Semaglutide is Neuroprotective and Reduces alpha-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 318–324. [Google Scholar] [CrossRef]
- AlDakheel, A.; Kalia, L.V.; Lang, A.E. Pathogenesis-targeted, disease-modifying therapies in Parkinson disease. Neurotherapeutics 2014, 11, 6–23. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- National Collaborating Centre for Chronic Conditions. National Collaborating Centre for Chronic, C. National Institute for Health and Clinical Excellence: Guidance. In Parkinson’s Disease: National Clinical Guideline for Diagnosis and Management in Primary and Secondary Care; Royal College of Physicians (UK), Royal College of Physicians of London: London, UK, 2006. [Google Scholar]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Lotti, V.J.; Porter, C.C. Potentiation and inhbition of some central actions of L(-)-dopa by decarboxylase inhibitors. J. Pharmacol. Exp. Ther. 1970, 172, 406–415. [Google Scholar]
- Silva, M.A.; Mattern, C.; Hacker, R.; Tomaz, C.; Huston, J.P.; Schwarting, R.K. Increased neostriatal dopamine activity after intraperitoneal or intranasal administration of L-DOPA: On the role of benserazide pretreatment. Synapse 1997, 27, 294–302. [Google Scholar] [CrossRef]
- Nord, M. Levodopa Pharmacokinetics-From Stomach to Brain: A Study on Patients with Parkinson’s Disease. Ph.D. Thesis, Comprehensive Summary, Linköping University Electronic Press, Linköping, Sweden, 2017. [Google Scholar]
- Oertel, W.H. Recent advances in treating Parkinson’s disease. F1000Research 2017, 6, 260. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Medical management of Parkinson’s disease. Pharm. Ther. 2008, 33, 590–606. [Google Scholar]
- Atmaca, M. Drug-induced impulse control disorders: A review. Curr. Clin. Pharmacol. 2014, 9, 70–74. [Google Scholar] [CrossRef]
- Baumann-Vogel, H.; Valko, P.O.; Eisele, G.; Baumann, C.R. Impulse control disorders in Parkinson’s disease: Don’t set your mind at rest by self-assessments. Eur. J. Neurol. 2015, 22, 603–609. [Google Scholar] [CrossRef]
- Moore, T.J.; Glenmullen, J.; Mattison, D.R. Reports of pathological gambling, hypersexuality, and compulsive shopping associated with dopamine receptor agonist drugs. JAMA Intern. Med. 2014, 174, 1930–1933. [Google Scholar] [CrossRef]
- Saez-Francas, N.; Marti Andres, G.; Ramirez, N.; de Fabregues, O.; Alvarez-Sabin, J.; Casas, M.; Hernandez-Vara, J. Clinical and psychopathological factors associated with impulse control disorders in Parkinson’s disease. Neurologia 2016, 31, 231–238. [Google Scholar] [CrossRef]
- Seeman, P. Parkinson’s disease treatment may cause impulse-control disorder via dopamine D3 receptors. Synapse 2015, 69, 183–189. [Google Scholar] [CrossRef]
- van Eimeren, T.; Ballanger, B.; Pellecchia, G.; Miyasaki, J.M.; Lang, A.E.; Strafella, A.P. Dopamine agonists diminish value sensitivity of the orbitofrontal cortex: A trigger for pathological gambling in Parkinson’s disease? Neuropsychopharmacology 2009, 34, 2758–2766. [Google Scholar] [CrossRef]
- Weintraub, D.; Claassen, D.O. Impulse Control and Related Disorders in Parkinson’s Disease. Int. Rev. Neurobiol. 2017, 133, 679–717. [Google Scholar]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef]
- Sawada, H.; Oeda, T.; Kuno, S.; Nomoto, M.; Yamamoto, K.; Yamamoto, M.; Hisanaga, K.; Kawamura, T.; Amantadine Study, G. Amantadine for dyskinesias in Parkinson’s disease: A randomized controlled trial. PLoS ONE 2010, 5, e15298. [Google Scholar] [CrossRef]
- Shin, M.S.; Kim, T.W.; Lee, J.M.; Ji, E.S.; Lim, B.V. Treadmill exercise alleviates nigrostriatal dopaminergic loss of neurons and fibers in rotenone-induced Parkinson rats. J. Exerc. Rehabil. 2017, 13, 30–35. [Google Scholar] [CrossRef]
- Lau, Y.S.; Patki, G.; Das-Panja, K.; Le, W.D.; Ahmad, S.O. Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson’s disease with moderate neurodegeneration. Eur. J. Neurosci. 2011, 33, 1264–1274. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. eLife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Fang, X.; Han, D.; Cheng, Q.; Zhang, P.; Zhao, C.; Min, J.; Wang, F. Association of Levels of Physical Activity With Risk of Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Netw. Open 2018, 1, e182421. [Google Scholar] [CrossRef]
- Tsukita, K.; Sakamaki-Tsukita, H.; Takahashi, R. Long-term Effect of Regular Physical Activity and Exercise Habits in Patients with Early Parkinson Disease. Neurology 2022, 98, e859–e871. [Google Scholar] [CrossRef]
- van der Kolk, N.M.; de Vries, N.M.; Kessels, R.P.C.; Joosten, H.; Zwinderman, A.H.; Post, B.; Bloem, B.R. Effectiveness of home-based and remotely supervised aerobic exercise in Parkinson’s disease: A double-blind, randomised controlled trial. Lancet Neurol. 2019, 18, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Cameron, I.G.M.; Van der Kolk, N.M.; de Vries, N.M.; Klimars, E.; Toni, I.; Bloem, B.R.; Helmich, R.C. Aerobic Exercise Alters Brain Function and Structure in Parkinson’s Disease: A Randomized Controlled Trial. Ann. Neurol. 2022, 91, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, M.; Moore, C.G.; Kohrt, W.M.; Hall, D.A.; Delitto, A.; Comella, C.L.; Josbeno, D.A.; Christiansen, C.L.; Berman, B.D.; Kluger, B.M.; et al. Effect of High-Intensity Treadmill Exercise on Motor Symptoms in Patients With De Novo Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.; Liu, S.; Solomon, C.G.; Willett, W.C. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N. Engl. J. Med. 2001, 345, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, E.S.; Dryden, D.M.; Vandermeer, B.; Ha, C.; Korownyk, C. Lifestyle interventions for patients with and at risk for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Schleh, M.W.; Ahn, C.; Ludzki, A.C.; Gillen, J.B.; Varshney, P.; Van Pelt, D.W.; Pitchford, L.M.; Chenevert, T.L.; Gioscia-Ryan, R.A.; et al. Moderate-Intensity Exercise and High-Intensity Interval Training Affect Insulin Sensitivity Similarly in Obese Adults. J. Clin. Endocrinol. Metab. 2020, 105, e2941–e2959. [Google Scholar] [CrossRef] [PubMed]
- Holscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert. Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef] [PubMed]
- Labandeira, C.M.; Fraga-Bau, A.; Arias Ron, D.; Munoz, A.; Alonso-Losada, G.; Koukoulis, A.; Romero-Lopez, J.; Rodriguez-Perez, A.I. Diabetes, insulin and new therapeutic strategies for Parkinson’s disease: Focus on glucagon-like peptide-1 receptor agonists. Front. Neuroendocrinol. 2021, 62, 100914. [Google Scholar] [CrossRef]
- Nowell, J.; Blunt, E.; Gupta, D.; Edison, P. Antidiabetic agents as a novel treatment for Alzheimer’s and Parkinson’s disease. Ageing Res. Rev. 2023, 89, 101979. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef] [PubMed]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. alpha-Synuclein promotes IAPP fibril formation in vitro and beta-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Rhen, T.; Schommer, T.; Ghribi, O. The oxysterol 27-hydroxycholesterol regulates alpha-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors--relevance to Parkinson’s disease. J. Neurochem. 2011, 119, 1119–1136. [Google Scholar] [CrossRef]
- Schommer, J.; Marwarha, G.; Schommer, T.; Flick, T.; Lund, J.; Ghribi, O. 27-Hydroxycholesterol increases alpha-synuclein protein levels through proteasomal inhibition in human dopaminergic neurons. BMC Neurosci. 2018, 19, 17. [Google Scholar] [CrossRef] [PubMed]
- García-Sanz, P.; Aerts, J.M.F.G.; Moratalla, R. The Role of Cholesterol in alpha-Synuclein and Lewy Body Pathology in GBA1 Parkinson’s Disease. Mov. Disord. 2021, 36, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.T.; Halliday, G.M.; Kim, W.S. alpha-Synuclein Regulates Neuronal Cholesterol Efflux. Molecules 2017, 22, 1769. [Google Scholar] [CrossRef]
- Jakubec, M.; Barias, E.; Furse, S.; Govasli, M.L.; George, V.; Turcu, D.; Iashchishyn, I.A.; Morozova-Roche, L.A.; Halskau, O. Cholesterol-containing lipid nanodiscs promote an alpha-synuclein binding mode that accelerates oligomerization. FEBS J. 2021, 288, 1887–1905. [Google Scholar] [CrossRef] [PubMed]
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 2016, 101, 393–400. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. alpha-Synuclein-induced synapse damage in cultured neurons is mediated by cholesterol-sensitive activation of cytoplasmic phospholipase A2. Biomolecules 2015, 5, 178–193. [Google Scholar] [CrossRef]
- Schneeberger, A.; Tierney, L.; Mandler, M. Active immunization therapies for Parkinson’s disease and multiple system atrophy. Mov. Disord. 2016, 31, 214–224. [Google Scholar] [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Luhrs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, R.M.; Fraser, K.; Yang, M.; Fox, T.; Hirschhorn, E.; Njingti, E.; Scott, D.; Bedell, B.J.; Kistner, K.M.; Cedarbaum, J.M.; et al. Cinpanemab in Early Parkinson Disease: Evaluation of Biomarker Results from the Phase 2 SPARK Clinical Trial. Neurology 2024, 102, e209137. [Google Scholar] [CrossRef] [PubMed]
- Ionica, L.N.; Gaita, L.; Bina, A.M.; Sosdean, R.; Lighezan, R.; Sima, A.; Malita, D.; Cretu, O.M.; Burlacu, O.; Muntean, D.M.; et al. Metformin alleviates monoamine oxidase-related vascular oxidative stress and endothelial dysfunction in rats with diet-induced obesity. Mol. Cell. Biochem. 2021, 476, 4019–4029. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Nikolajczyk, B.S. The intersection of metformin and inflammation. Am. J. Physiol. Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef]
- Soberanes, S.; Misharin, A.V.; Jairaman, A.; Morales-Nebreda, L.; McQuattie-Pimentel, A.C.; Cho, T.; Hamanaka, R.B.; Meliton, A.Y.; Reyfman, P.A.; Walter, J.M.; et al. Metformin Targets Mitochondrial Electron Transport to Reduce Air-Pollution-Induced Thrombosis. Cell Metab. 2019, 29, 335–347.e5. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, X.; Li, P.; Wang, M.; Yan, L.; Bao, Z.; Liu, Q. Association Between Diabetes Medications and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 678649. [Google Scholar] [CrossRef]
- Kuan, Y.C.; Huang, K.W.; Lin, C.L.; Hu, C.J.; Kao, C.H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 77–83. [Google Scholar] [CrossRef]
- Ping, F.; Jiang, N.; Li, Y. Association between metformin and neurodegenerative diseases of observational studies: Systematic review and meta-analysis. BMJ Open Diabetes Res. Care 2020, 8, e001370. [Google Scholar] [CrossRef]
- Baetta, R.; Corsini, A. Pharmacology of dipeptidyl peptidase-4 inhibitors: Similarities and differences. Drugs 2011, 71, 1441–1467. [Google Scholar] [CrossRef]
- Capuano, A.; Sportiello, L.; Maiorino, M.I.; Rossi, F.; Giugliano, D.; Esposito, K. Dipeptidyl peptidase-4 inhibitors in type 2 diabetes therapy—Focus on alogliptin. Drug Des. Dev. Ther. 2013, 7, 989–1001. [Google Scholar]
- Chen, S.; Zhou, M.; Sun, J.; Guo, A.; Fernando, R.L.; Chen, Y.; Peng, P.; Zhao, G.; Deng, Y. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology 2019, 157, 107668. [Google Scholar] [CrossRef]
- Cheng, Q.; Cheng, J.; Cordato, D.; Gao, J. Can dipeptidyl peptidase-4 inhibitors treat cognitive disorders? Pharmacol. Ther. 2020, 212, 107559. [Google Scholar] [CrossRef]
- Yossef, R.R.; Al-Yamany, M.F.; Saad, M.A.; El-Sahar, A.E. Neuroprotective effects of vildagliptin on drug induced Alzheimer’s disease in rats with metabolic syndrome: Role of hippocampal klotho and AKT signaling pathways. Eur. J. Pharmacol. 2020, 889, 173612. [Google Scholar] [CrossRef]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFkappaB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 2017, 25, 369–382. [Google Scholar] [CrossRef]
- Kabel, A.M.; Omar, M.S.; Alhadhrami, A.; Alharthi, S.S.; Alrobaian, M.M. Linagliptin potentiates the effect of l-dopa on the behavioural, biochemical and immunohistochemical changes in experimentally-induced Parkinsonism: Role of toll-like receptor 4, TGF-beta1, NF-kappaB and glucagon-like peptide 1. Physiol. Behav. 2018, 188, 108–118. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Li, C.; Li, M.; Ma, L. Sitagliptin rescues memory deficits in Parkinsonian rats via upregulating BDNF to prevent neuron and dendritic spine loss. Neurol. Res. 2018, 40, 736–743. [Google Scholar] [CrossRef]
- Nassar, N.N.; Al-Shorbagy, M.Y.; Arab, H.H.; Abdallah, D.M. Saxagliptin: A novel antiparkinsonian approach. Neuropharmacology 2015, 89, 308–317. [Google Scholar] [CrossRef]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and Liraglutide Modulate L-dopa Effect and Attenuate Dyskinetic Movements in Rotenone-Lesioned Rats. Neurotox. Res. 2019, 35, 635–653. [Google Scholar] [CrossRef]
- Svenningsson, P.; Wirdefeldt, K.; Yin, L.; Fang, F.; Markaki, I.; Efendic, S.; Ludvigsson, J.F. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors-A nationwide case-control study. Mov. Disord. 2016, 31, 1422–1423. [Google Scholar] [CrossRef]
- Jeong, S.H.; Chung, S.J.; Yoo, H.S.; Hong, N.; Jung, J.H.; Baik, K.; Lee, Y.H.; Sohn, Y.H.; Lee, P.H. Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson’s disease. Brain 2021, 144, 1127–1137. [Google Scholar] [CrossRef]
- Ates Bulut, E.; Sahin Alak, Z.Y.; Dokuzlar, O.; Kocyigit, S.E.; Soysal, P.; Smith, L.; Isik, A.T. Cognitive and metabolic outcomes of vildagliptin addition to the therapy in patients with type 2 diabetes mellitus: 26 week follow-up study. Arch. Gerontol. Geriatr. 2020, 88, 104013. [Google Scholar] [CrossRef]
- Borzi, A.M.; Condorelli, G.; Biondi, A.; Basile, F.; Vicari, E.S.D.; Buscemi, C.; Luca, S.; Vacante, M. Effects of vildagliptin, a DPP-4 inhibitor, in elderly diabetic patients with mild cognitive impairment. Arch. Gerontol. Geriatr. 2019, 84, 103896. [Google Scholar] [CrossRef]
- Rizzo, M.R.; Barbieri, M.; Boccardi, V.; Angellotti, E.; Marfella, R.; Paolisso, G. Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1122–1131. [Google Scholar] [CrossRef]
- Harati, M.; Tayarani-Najaran, Z.; Javadi, B. Dietary flavonoids: Promising compounds for targeting α-synucleinopathy in Parkinson’s disease. PharmaNutrition 2023, 24, 100334. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [PubMed]
- Grieco, M.; Giorgi, A.; Gentile, M.C.; d’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Bodart-Santos, V.; De Felice, F.G.; Ferreira, S.T. Neuroprotective Actions of Glucagon-Like Peptide-1 (GLP-1) Analogues in Alzheimer’s and Parkinson’s Diseases. CNS Drugs 2019, 33, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Glotfelty, E.J.; Olson, L.; Karlsson, T.E.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert. Opin. Investig. Drugs 2020, 29, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Moon, M.; Park, S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J. Endocrinol. 2009, 202, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Perry, T.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K.; et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef]
- Liu, W.; Jalewa, J.; Sharma, M.; Li, G.; Li, L.; Holscher, C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2015, 303, 42–50. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Holscher, C. Neuroprotective effects of the novel GLP-1 long acting analogue semaglutide in the MPTP Parkinson’s disease mouse model. Neuropeptides 2018, 71, 70–80. [Google Scholar] [CrossRef]
- Bertilsson, G.; Patrone, C.; Zachrisson, O.; Andersson, A.; Dannaeus, K.; Heidrich, J.; Kortesmaa, J.; Mercer, A.; Nielsen, E.; Ronnholm, H.; et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 326–338. [Google Scholar] [CrossRef]
- Lin, T.K.; Lin, K.J.; Lin, H.Y.; Lin, K.L.; Lan, M.Y.; Wang, P.W.; Wang, T.J.; Wang, F.S.; Tsai, P.C.; Liou, C.W.; et al. Glucagon-Like Peptide-1 Receptor Agonist Ameliorates 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Neurotoxicity Through Enhancing Mitophagy Flux and Reducing alpha-Synuclein and Oxidative Stress. Front. Mol. Neurosci. 2021, 14, 697440. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Kahan, J.; Ell, P.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J. Parkinsons Dis. 2014, 4, 337–344. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Budnik, N.; Zampedri, L.; Hibbert, S.; Skene, S.S.; Chowdhury, K.; Aviles-Olmos, I.; Limousin, P.; Foltynie, T. What Effects Might Exenatide have on Non-Motor Symptoms in Parkinson’s Disease: A Post Hoc Analysis. J. Parkinsons Dis. 2018, 8, 247–258. [Google Scholar] [CrossRef]
- Nakamura, K.; Mori, F.; Tanji, K.; Miki, Y.; Yamada, M.; Kakita, A.; Takahashi, H.; Utsumi, J.; Sasaki, H.; Wakabayashi, K. Isopentenyl diphosphate isomerase, a cholesterol synthesizing enzyme, is localized in Lewy bodies. Neuropathology 2015, 35, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Mutez, E.; Duhamel, A.; Defebvre, L.; Bordet, R.; Destee, A.; Kreisler, A. Lipid-lowering drugs are associated with delayed onset and slower course of Parkinson’s disease. Pharmacol. Res. 2009, 60, 41–45. [Google Scholar] [CrossRef]
- Friedman, B.; Lahad, A.; Dresner, Y.; Vinker, S. Long-term statin use and the risk of Parkinson’s disease. Am. J. Manag. Care 2013, 19, 626–632. [Google Scholar] [PubMed]
- Huang, X.; Alonso, A.; Guo, X.; Umbach, D.M.; Lichtenstein, M.L.; Ballantyne, C.M.; Mailman, R.B.; Mosley, T.H.; Chen, H. Statins, plasma cholesterol, and risk of Parkinson’s disease: A prospective study. Mov. Disord. 2015, 30, 552–559. [Google Scholar] [CrossRef]
- Jeong, S.H.; Lee, H.S.; Chung, S.J.; Yoo, H.S.; Jung, J.H.; Baik, K.; Lee, Y.H.; Sohn, Y.H.; Lee, P.H. Effects of statins on dopamine loss and prognosis in Parkinson’s disease. Brain 2021, 144, 3191–3200. [Google Scholar] [CrossRef]
- Liu, G.; Sterling, N.W.; Kong, L.; Lewis, M.M.; Mailman, R.B.; Chen, H.; Leslie, D.; Huang, X. Statins may facilitate Parkinson’s disease: Insight gained from a large, national claims database. Mov. Disord. 2017, 32, 913–917. [Google Scholar] [CrossRef]
- Yan, J.; Qiao, L.; Tian, J.; Liu, A.; Wu, J.; Huang, J.; Shen, M.; Lai, X. Effect of statins on Parkinson’s disease: A systematic review and meta-analysis. Medicine 2019, 98, e14852. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.S.; Cardona, A.E. The IL-1beta phenomena in neuroinflammatory diseases. J. Neural. Transm. 2018, 125, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Luciunaite, A.; McManus, R.M.; Jankunec, M.; Racz, I.; Dansokho, C.; Dalgediene, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Abeta oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. alpha-Synuclein evokes NLRP3 inflammasome-mediated IL-1beta secretion from primary human microglia. Glia 2021, 69, 1413–1428. [Google Scholar] [CrossRef]
- Gonzalez, P.V.; Schioth, H.B.; Lasaga, M.; Scimonelli, T.N. Memory impairment induced by IL-1beta is reversed by alpha-MSH through central melanocortin-4 receptors. Brain Behav. Immun. 2009, 23, 817–822. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Collins, L.; Toulouse, A.; Sullivan, A.M.; Nolan, Y.M. Interleukin-1beta contributes to dopaminergic neuronal death induced by lipopolysaccharide-stimulated rat glia in vitro. J. Neuroimmunol. 2010, 226, 20–26. [Google Scholar] [CrossRef]
- Chakraborty, A.; Tannenbaum, S.; Rordorf, C.; Lowe, P.J.; Floch, D.; Gram, H.; Roy, S. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1beta monoclonal antibody. Clin. Pharmacokinet. 2012, 51, e1–e18. [Google Scholar] [CrossRef]
- Ferrari, F.; Moretti, A.; Villa, R.F. Incretin-based drugs as potential therapy for neurodegenerative diseases: Current status and perspectives. Pharmacol. Ther. 2022, 239, 108277. [Google Scholar] [CrossRef]
- De Iuliis, A.; Montinaro, E.; Fatati, G.; Plebani, M.; Colosimo, C. Diabetes mellitus and Parkinson’s disease: Dangerous liaisons between insulin and dopamine. Neural Regen. Res. 2022, 17, 523–533. [Google Scholar]
- Fiory, F.; Perruolo, G.; Cimmino, I.; Cabaro, S.; Pignalosa, F.C.; Miele, C.; Beguinot, F.; Formisano, P.; Oriente, F. The Relevance of Insulin Action in the Dopaminergic System. Front. Neurosci. 2019, 13, 868. [Google Scholar] [CrossRef]
- Fine, J.M.; Stroebel, B.M.; Faltesek, K.A.; Terai, K.; Haase, L.; Knutzen, K.E.; Kosyakovsky, J.; Bowe, T.J.; Fuller, A.K.; Frey, W.H.; et al. Intranasal delivery of low-dose insulin ameliorates motor dysfunction and dopaminergic cell death in a 6-OHDA rat model of Parkinson’s Disease. Neurosci. Lett. 2020, 714, 134567. [Google Scholar] [CrossRef]
- Iravanpour, F.; Dargahi, L.; Rezaei, M.; Haghani, M.; Heidari, R.; Valian, N.; Ahmadiani, A. Intranasal insulin improves mitochondrial function and attenuates motor deficits in a rat 6-OHDA model of Parkinson’s disease. CNS Neurosci. Ther. 2021, 27, 308–319. [Google Scholar] [CrossRef]
- Galustian, C.; Dalgleish, A. Lenalidomide: A novel anticancer drug with multiple modalities. Expert. Opin. Pharmacother. 2009, 10, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Valera, E.; Mante, M.; Anderson, S.; Rockenstein, E.; Masliah, E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J. Neuroinflamm. 2015, 12, 93. [Google Scholar] [CrossRef]
- Cankara, F.N.; Gunaydin, C.; Bilge, S.S.; Ozmen, O.; Kortholt, A. The neuroprotective action of lenalidomide on rotenone model of Parkinson’s Disease: Neurotrophic and supportive actions in the substantia nigra pars compacta. Neurosci. Lett. 2020, 738, 135308. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, R.; Zenga, J.; Himburg, H.; Wang, L.; Duan, S.; Liu, J.; Bui, D.; Xie, Z.; Du, T.; et al. Comparison of Absolute Expression and Turnover Number of COX-1 and COX-2 in Human and Rodent Cells and Tissues. J. Inflamm. Res. 2022, 15, 4435–4447. [Google Scholar] [CrossRef]
- Kadusevicius, E. Novel Applications of NSAIDs: Insight and Future Perspectives in Cardiovascular, Neurodegenerative, Diabetes and Cancer Disease Therapy. Int. J. Mol. Sci. 2021, 22, 6637. [Google Scholar] [CrossRef] [PubMed]
- Zaminelli, T.; Gradowski, R.W.; Bassani, T.B.; Barbiero, J.K.; Santiago, R.M.; Maria-Ferreira, D.; Baggio, C.H.; Vital, M.A. Antidepressant and antioxidative effect of Ibuprofen in the rotenone model of Parkinson’s disease. Neurotox. Res. 2014, 26, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Hain, E.G.; Sparenberg, M.; Rasinska, J.; Klein, C.; Akyuz, L.; Steiner, B. Indomethacin promotes survival of new neurons in the adult murine hippocampus accompanied by anti-inflammatory effects following MPTP-induced dopamine depletion. J. Neuroinflamm. 2018, 15, 162. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.A.; Thorne, J.E.; Zandi, P.P.; Ek, M.; Messias, E.; Breitner, J.C.; Goodman, S.N. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 2004, 23, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Poly, T.N.; Islam, M.M.R.; Yang, H.C.; Li, Y.J. Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: A meta-analysis. Eur. J. Clin. Pharmacol. 2019, 75, 99–108. [Google Scholar] [CrossRef]
- Kothari, V.; Galdo, J.A.; Mathews, S.T. Hypoglycemic agents and potential anti-inflammatory activity. J. Inflamm. Res. 2016, 9, 27–38. [Google Scholar] [PubMed]
- Zhang, G.; Lin, X.; Zhang, S.; Xiu, H.; Pan, C.; Cui, W. A Protective Role of Glibenclamide in Inflammation-Associated Injury. Mediat. Inflamm. 2017, 2017, 3578702. [Google Scholar] [CrossRef]
- Abdelkader, N.F.; Farid, H.A.; Youness, E.R.; Abdel-Salam, O.M.E.; Zaki, H.F. The role of K(ATP) channel blockade and activation in the protection against neurodegeneration in the rotenone model of Parkinson’s disease. Life Sci. 2020, 257, 118070. [Google Scholar] [CrossRef] [PubMed]
- Ishola, I.O.; Akataobi, O.E.; Alade, A.A.; Adeyemi, O.O. Glimepiride prevents paraquat-induced Parkinsonism in mice: Involvement of oxidative stress and neuroinflammation. Fundam. Clin. Pharmacol. 2019, 33, 277–285. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, Q.; Hou, L.; Zhang, C.; Wang, Q.; Zhao, X. Inhibition of NLRP3 inflammasome by glibenclamide attenuated dopaminergic neurodegeneration and motor deficits in paraquat and maneb-induced mouse Parkinson’s disease model. Toxicol. Lett. 2021, 349, 1–11. [Google Scholar] [CrossRef]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.; Wesolowska, A.; Pawlowski, M.; Chlon-Rzepa, G. Diabetic Theory in Anti-Alzheimer’s Drug Research and Development—Part 1: Therapeutic Potential of Antidiabetic Agents. Curr. Med. Chem. 2020, 27, 6658–6681. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, J.K.; Santiago, R.M.; Persike, D.S.; da Silva Fernandes, M.J.; Tonin, F.S.; da Cunha, C.; Lucio Boschen, S.; Lima, M.M.; Vital, M.A. Neuroprotective effects of peroxisome proliferator-activated receptor alpha and gamma agonists in model of parkinsonism induced by intranigral 1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine. Behav. Brain Res. 2014, 274, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Breidert, T.; Callebert, J.; Heneka, M.T.; Landreth, G.; Launay, J.M.; Hirsch, E.C. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J. Neurochem. 2002, 82, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.R.; Frau, L.; Pisanu, A.; Wardas, J.; Spiga, S.; Carboni, E. Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: New evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 2011, 194, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Nissanka, N.; Peralta, S.; Brambilla, R.; Diaz, F.; Moraes, C.T. Pioglitazone ameliorates the phenotype of a novel Parkinson’s disease mouse model by reducing neuroinflammation. Mol. Neurodegener. 2016, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Schintu, N.; Frau, L.; Ibba, M.; Caboni, P.; Garau, A.; Carboni, E.; Carta, A.R. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.L.; Mounsey, R.B.; Mustafa, S.; Sathe, K.; Teismann, P. Pharmacological manipulation of peroxisome proliferator-activated receptor gamma (PPARgamma) reveals a role for anti-oxidant protection in a model of Parkinson’s disease. Exp. Neurol. 2012, 235, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, W.; Li, G.; Chen, J.; Guan, X.; Chen, X.; Guan, Z. Neuroprotective Effect and Mechanism of Thiazolidinedione on Dopaminergic Neurons In Vivo and In Vitro in Parkinson’s Disease. PPAR Res. 2017, 2017, 4089214. [Google Scholar] [CrossRef]
- Lee, E.Y.; Lee, J.E.; Park, J.H.; Shin, I.C.; Koh, H.C. Rosiglitazone, a PPAR-gamma agonist, protects against striatal dopaminergic neurodegeneration induced by 6-OHDA lesions in the substantia nigra of rats. Toxicol. Lett. 2012, 213, 332–344. [Google Scholar] [CrossRef]
- Machado, M.M.F.; Bassani, T.B.; Coppola-Segovia, V.; Moura, E.L.R.; Zanata, S.M.; Andreatini, R.; Vital, M. PPAR-gamma agonist pioglitazone reduces microglial proliferation and NF-kappaB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson’s disease. Pharmacol. Rep. 2019, 71, 556–564. [Google Scholar] [CrossRef]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar]
- Hussain, S.; Singh, A.; Baxi, H.; Taylor, B.; Burgess, J.; Antony, B. Thiazolidinedione use is associated with reduced risk of Parkinson’s disease in patients with diabetes: A meta-analysis of real-world evidence. Neurol. Sci. 2020, 41, 3697–3703. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef]
- Wang, D.X.; Chen, A.D.; Wang, Q.J.; Xin, Y.Y.; Yin, J.; Jing, Y.H. Protective effect of metformin against rotenone-induced parkinsonism in mice. Toxicol. Mech. Methods 2020, 30, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Park, P.H.; Choi, D.Y. Metformin attenuates rotenone-induced oxidative stress and mitochondrial damage via the AKT/Nrf2 pathway. Neurochem. Int. 2021, 148, 105120. [Google Scholar] [CrossRef]
- Saewanee, N.; Praputpittaya, T.; Malaiwong, N.; Chalorak, P.; Meemon, K. Neuroprotective effect of metformin on dopaminergic neurodegeneration and alpha-synuclein aggregation in C. elegans model of Parkinson’s disease. Neurosci. Res. 2021, 162, 13–21. [Google Scholar] [CrossRef]
- Tayara, K.; Espinosa-Oliva, A.M.; Garcia-Dominguez, I.; Ismaiel, A.A.; Boza-Serrano, A.; Deierborg, T.; Machado, A.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Divergent Effects of Metformin on an Inflammatory Model of Parkinson’s Disease. Front. Cell. Neurosci. 2018, 12, 440. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hamidu, S.; Yang, X.; Yan, Y.; Wang, Q.; Li, L.; Oduro, P.K.; Li, Y. Dietary Supplements and Natural Products: An Update on Their Clinical Effectiveness and Molecular Mechanisms of Action During Accelerated Biological Aging. Front. Genet. 2022, 13, 880421. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Gulyani, S.; Karnati, H.K.; Li, Y.; Tweedie, D.; Mustapic, M.; Chawla, S.; Chowdhury, K.; Skene, S.S.; Greig, N.H.; et al. Utility of Neuronal-Derived Exosomes to Examine Molecular Mechanisms That Affect Motor Function in Patients with Parkinson Disease: A Secondary Analysis of the Exenatide-PD Trial. JAMA Neurol. 2019, 76, 420–429. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Y.; He, X.; Li, H.; Liu, H.; Zhang, X. A systematic review and meta-analysis of serum cholesterol and triglyceride levels in patients with Parkinson’s disease. Lipids Health Dis. 2020, 19, 97. [Google Scholar] [CrossRef]
- Rozani, V.; Gurevich, T.; Giladi, N.; El-Ad, B.; Tsamir, J.; Hemo, B.; Peretz, C. Higher serum cholesterol and decreased Parkinson’s disease risk: A statin-free cohort study. Mov. Disord. 2018, 33, 1298–1305. [Google Scholar] [CrossRef]
- Pike, C.J. Testosterone attenuates beta-amyloid toxicity in cultured hippocampal neurons. Brain Res. 2001, 919, 160–165. [Google Scholar] [CrossRef]
- Fine, J.M.; Kosyakovsky, J.; Baillargeon, A.M.; Tokarev, J.V.; Cooner, J.M.; Svitak, A.L.; Faltesek, K.A.; Frey, W.H., 2nd; Hanson, L.R. Intranasal deferoxamine can improve memory in healthy C57 mice, suggesting a partially non-disease-specific pathway of functional neurologic improvement. Brain Behav. 2020, 10, e01536. [Google Scholar] [CrossRef]
- Novak, P.; Pimentel Maldonado, D.A.; Novak, V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS ONE 2019, 14, e0214364. [Google Scholar] [CrossRef]
- Teismann, P.; Ferger, B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson’s disease. Synapse 2001, 39, 167–174. [Google Scholar] [CrossRef]
- Kurkowska-Jastrzebska, I.; Babiuch, M.; Joniec, I.; Przybylkowski, A.; Czlonkowski, A.; Czlonkowska, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol. 2002, 2, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jacobs, E.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar] [CrossRef]
- Piri, H.; Haghdoost-Yazdi, H.; Fraidouni, N.; Dargahi, T.; Yaghoubidoust, M.; Azadmehr, A. The Anti-Parkinsonism Effects of K(ATP) Channel Blockade in the 6-Hydroxydopamine-Induced Animal Model: The Role of Oxidative Stress. Basic Clin. Neurosci. 2017, 8, 183–192. [Google Scholar]
- Blackburn, J.K.; Curry, D.W.; Thomsen, A.N.; Roth, R.H.; Elsworth, J.D. Pioglitazone activates paraoxonase-2 in the brain: A novel neuroprotective mechanism. Exp. Neurol. 2020, 327, 113234. [Google Scholar] [CrossRef]
- Pisanu, A.; Lecca, D.; Mulas, G.; Wardas, J.; Simbula, G.; Spiga, S.; Carta, A.R. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 2014, 71, 280–291. [Google Scholar] [CrossRef]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson’s Disease: A Multisystem Disorder. Neurosci. Bull. 2023, 39, 113–124. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Junior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Le, W. Biomarkers for Parkinson’s Disease: How Good Are They? Neurosci. Bull. 2020, 36, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Di Lazzaro, G.; Sancesario, G.M.; Summa, S.; Petrucci, S.; Colona, V.L.; Bernardini, S.; Pierantozzi, M.; Stefani, A.; Mercuri, N.B.; et al. Young-onset and late-onset Parkinson’s disease exhibit a different profile of fluid biomarkers and clinical features. Neurobiol. Aging 2020, 90, 119–124. [Google Scholar] [CrossRef]
- Srikanth, V.; Westcott, B.; Forbes, J.; Phan, T.G.; Beare, R.; Venn, A.; Pearson, S.; Greenaway, T.; Parameswaran, V.; Munch, G. Methylglyoxal, cognitive function and cerebral atrophy in older people. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 68–73. [Google Scholar] [CrossRef]
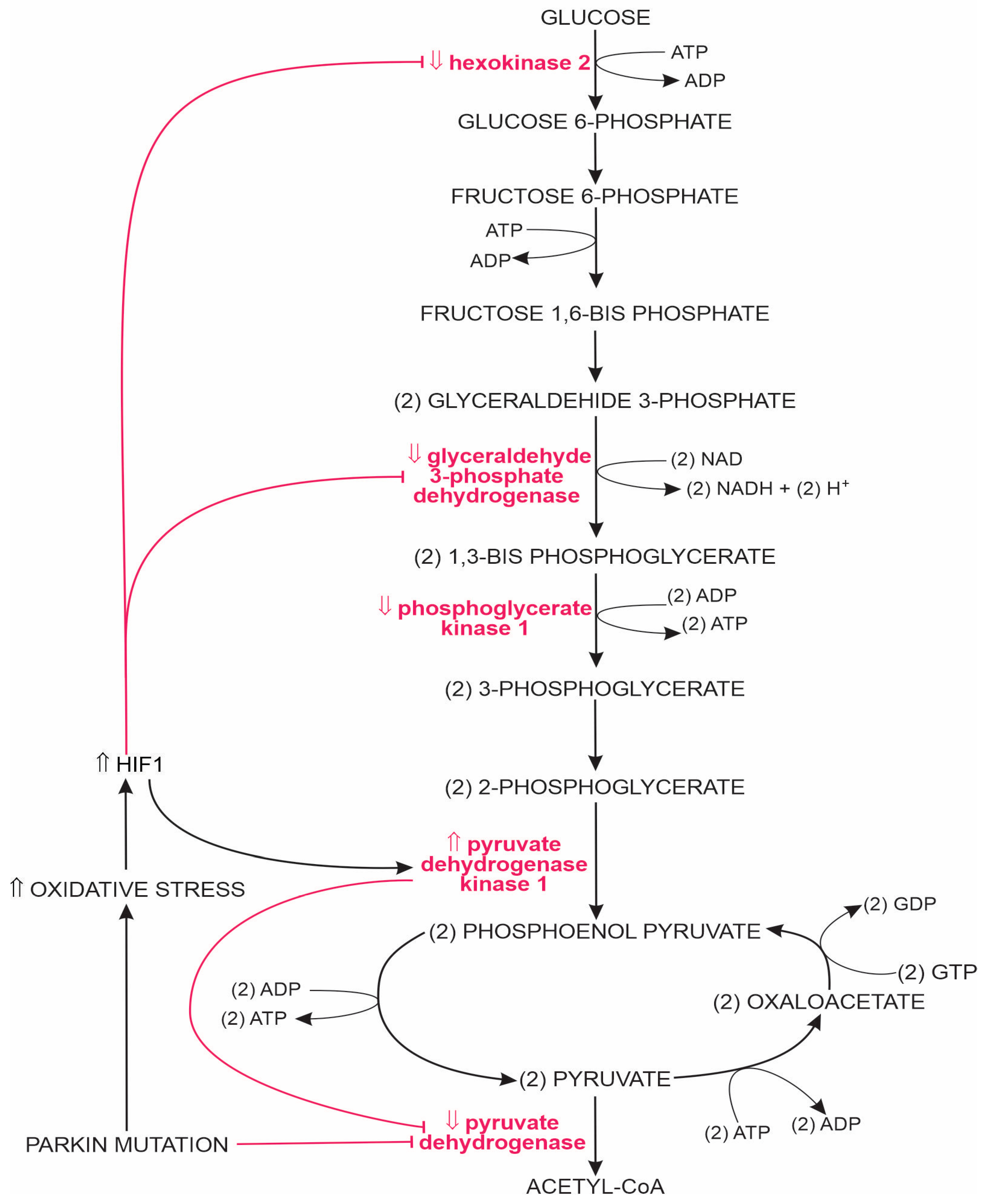
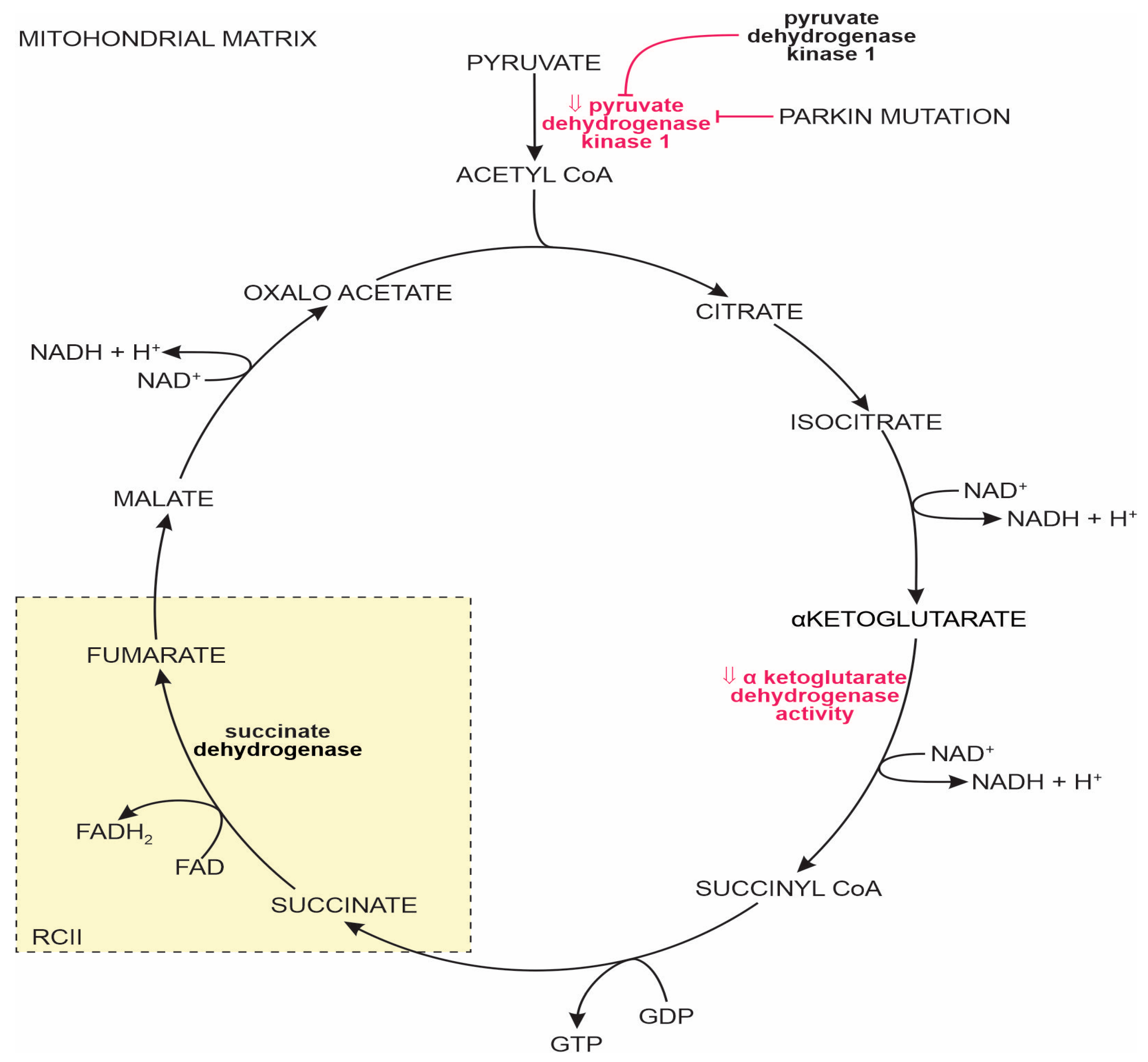


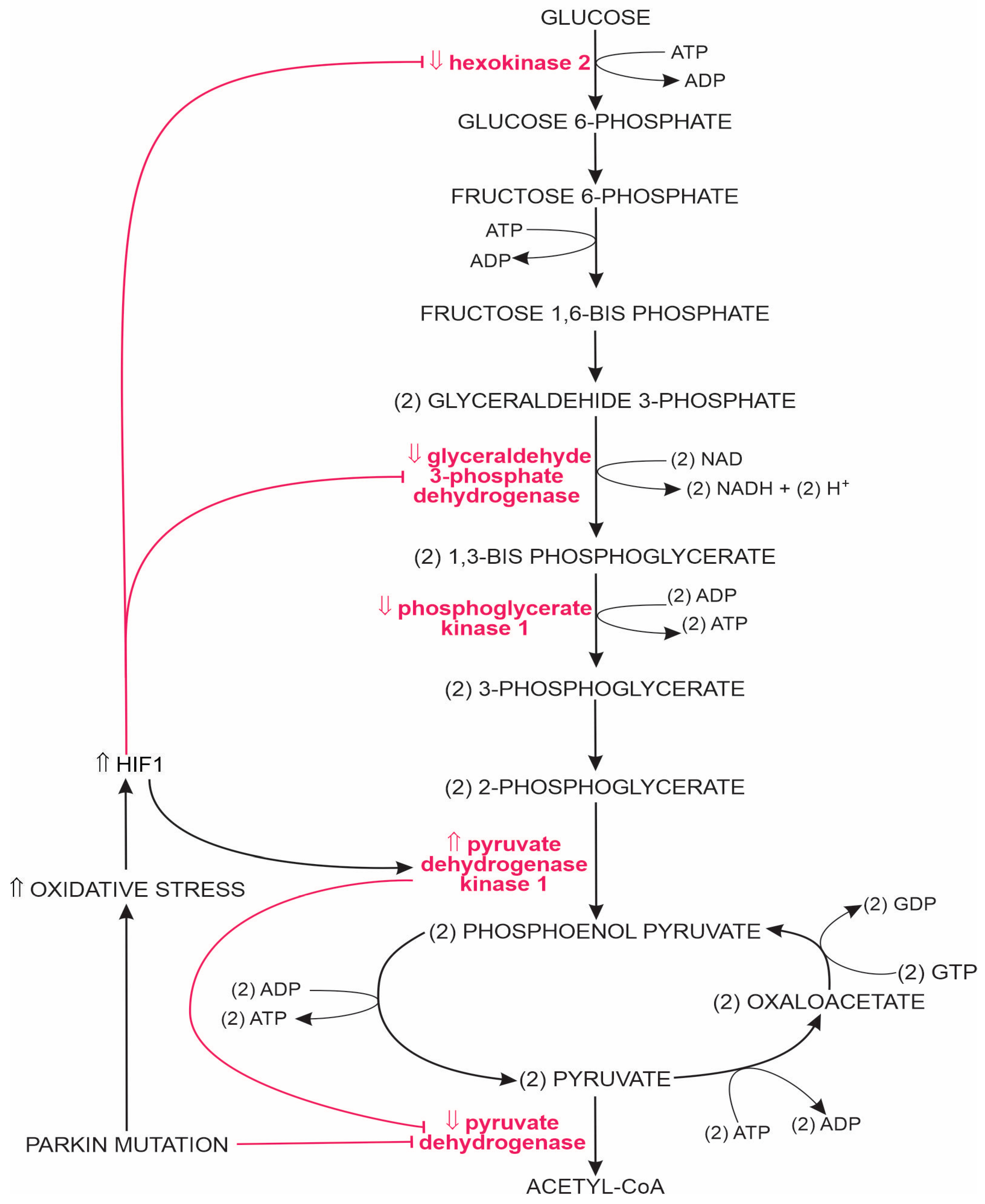{kind=link}
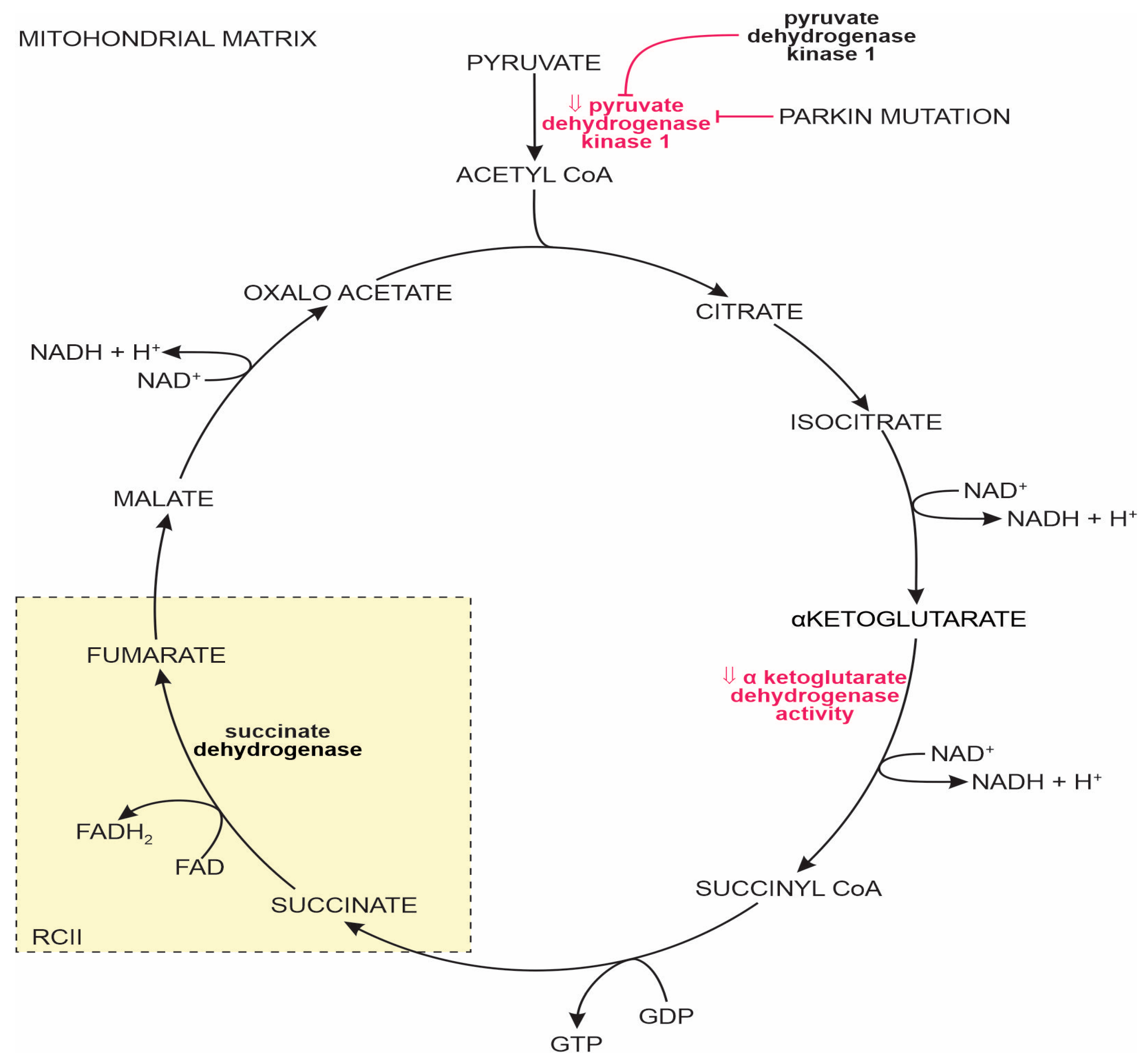{kind=link}
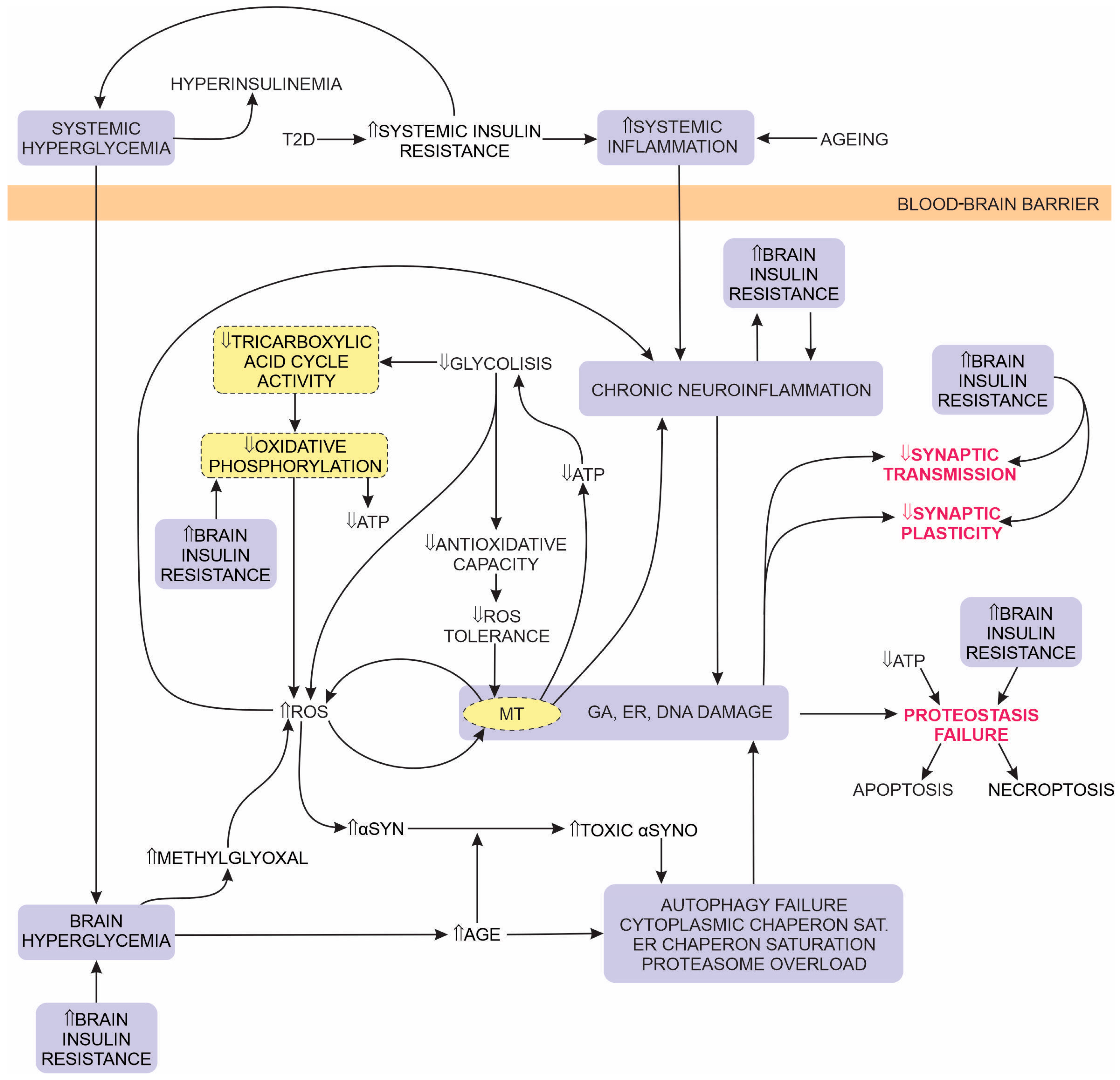{kind=link}
| Change in Clinical Sign | PD vs. HOA | PD vs. PD + T2D |
|---|---|---|
| slower gait | + | ++ |
| worse balance | + | ++ |
| reduced muscle strength | + | ++ |
| reduced motor endurance | + | ++ |
| reduced motor–cognitive function | + | = |
| impaired attention | + | ++ |
| Substance | Mechanism of Action | Observed Effects in Preclinical, PD Animal Model and Cell Studies | Results of PD Epidemiological or Clinical Trial Studies |
|---|---|---|---|
| anti α-synuclein protein monomer (α-syn) vaccination and humanised α-synuclein antibodies | Peptides that elicit an antibody response to oligomeric α-synuclein (e.g., PD01A) or humanised antibody (e.g., PRX002). | The effectiveness of active immunization against Parkinson’s disease (PD) (improved locomotor activity, memory and learning, reduced death of pars compacta of substanca nigra (SNpc) nerve cells) was demonstrated in mouse and rat models [363]. | Phase 1 clinical trials confirmed tolerance, substantial immune response, and dose-dependent effects [364,457,458]. BIIB054 development terminated after Phase 2 clinical trial [245]. |
| biguanides (i.e., metformin) | (1) Inhibit mitochondrial (MT) complex I, which stimulates AMP-activated protein kinase (AMPK) that inhibits (a) gluconeogenesis and hepatic glucose production and increases skeletal muscle glucose uptake by increased glucose transporter (GLUT)4 incorporation into the cell membrane (b). (2) Inhibit nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and reduce reactive oxidative species (ROS) production. | Overall consistent, positive effects. Improved locomotor activity and motor coordination, reduced degeneration of dopaminergic neurons, reversed dopamine depletion, inhibited α-syn phosphorylation and aggregation, decreased MT dysfunction and oxidative stress, inhibited neuroinflammation, increased the production of neurotrophic factors [459,460,461,462,463,464]. | Diverse results of epidemiological studies; either no effect, a decreased risk, or an increased risk of developing PD in type 2 diabetes (T2D) patients [373,374,375,376]. |
| dipeptidyl peptidase-4 (DPP4) inhibitors | Prolong glucagon-like peptide-1 (GLP-1) signalling by inhibiting its degradation. | Improved motor performance; reduced memory deficits, oxidative stress, and dopaminergic degeneration; increased GLP-1 expression in the brain; and reduced neuroinflammation [382,383,384,385,386]. The combination of a DPP4 inhibitor with levodopa was more effective than levodopa alone [384,387]. | Epidemiological studies reported a reduced incidence of PD associated with a record of DPP4-inhibitor intake [140,388]. |
| flavonoids | Multiple actions: (a) modulate the activity/expression of the antioxidant enzymes superoxide dismutase, glutathione peroxidase, and endothelial nitric oxide synthase; (b) reduce ROS damage; and (c) promote autophagy. | Reduced excessive α-syn production, oligomerisation, and aggregation; enhanced α-syn autophagy; reduced oxidative damage and apoptosis of dopaminergic neurons [393,465]. | None. |
| Glucagon-like Peptide-1 receptor (GLP-1) agonists | Promote insulin secretion by pancreatic β cells; in the central nervous system (CNS), they have neuroprotective, antiapoptotic, and anti-inflammatory effects. | Reduced dopaminergic degeneration, α-syn accumulation, and neuroinflammation; restored dopamine levels; attenuated motor dysfunction; improved MT function [316,402,403,405]. | Consistent improvement in motor and cognitive functions in clinical trials [143,406,466,467]. |
| Hydroxymethylglutaryl-CoA (HMG-CoA) reductase inhibitors | Inhibit the conversion of HMG-CoA to mevalonate, the rate-limiting step in cholesterol synthesis. | Consistent reports of a positive association between high levels of cholesterol and PD cholesterol and cholesterol metabolites accelerated α-syn aggregation, inhibited tyrosine hydroxylase expression, and reduced dopamine synthesis; promoted oxidative stress, cell death, synaptic loss, and neuroinflammation in the CNS [356,357,358,359,360,361,362]. | Clinical trials and observational studies report diverse results from a positive association between high cholesterol levels and an increased risk of PD to a reduced risk of PD associated with high cholesterol [410,411,412,413,414,468,469]. |
| interleukin (IL)-1β inhibitors inhibitors | Inhibit α-syn-elicited release of IL-1 β that stimulates additional release of pro-inflammatory cytokines from astrocytes and microglia [415,416,470]. | None. | None. |
| insulin (intranasal insulin (INI) application) | Promotes cell growth and repair, long-term potentiation; reduces apoptosis, oxidative stress, and dopaminergic cell death. | Low-dose, INI application improved motor and MT function and reduced dopaminergic cell death [427,471]. | Randomised, double-blinded, placebo-controlled trial improved verbal fluency and Hoehn-Yahr and unified Parkinson’s disease rating scale-part 3 motor scores with 4 weeks of INI [472]. |
| lenalidomide | Inhibits tumour necrosis factor α (TNF-α), IL-1, IL-6, and IL-12 expression; stimulates T-cell proliferation; and increases production of IL-2 and IFNγ. | Reduced microgliosis, attenuated pro-inflammatory cytokine expression and NF-κB activation, attenuated dopaminergic fibre loss, improved locomotor activity, increased SNpc brain-derived neurotrophic factor expression, and improved neuronal survival [430,431]. | None. |
| nonsteroidal anti-inflammatory drugs (NSAIDs) | Inhibit cyclooxygenase enzymes, thus reducing the conversion of arachidonic acid to prostaglandins, which stimulate the release of inflammatory cytokines. | Reduced loss of nigral neurons, restored dopamine levels, improved locomotor activity, reduced neuroinflammation [434,435,473,474]. | A limited number of epidemiological studies have concluded that NSAIDs may decrease the risk of PD [436,475]. |
| sulfonylureas | Stimulate pancreatic β-cell insulin secretion by closing sulfonylurea receptor (Sur1)-regulated channels that elicit membrane depolarization, the influx of Ca2+, and insulin release from vesicles. Sur1-regulated channels are also expressed in neurons, astrocytes, microglial cells, oligodendrocytes, and endothelial cells. | Attenuated motor and memory impairment; decreased oxidative stress, the inhibition of NF-κB, and NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome activation; attenuated neuroinflammation; reduced α-syn expression, dopaminergic neuronal damage, and apoptosis [441,442,443,476]. | A systematic review and meta-analysis did not identify any association between the use of sulfonylureas and PD risk in T2D patients [374]. |
| thiazolidinediones (TZD) | PPAR-γ agonists that (a) increase adipokine-elicited insulin sensitivity and (b) attenuate inflammation by the inhibition of NF-κB and NLRP3 and the activation of mitogen-activated protein kinase (MAPK) signalling pathways. | Improved motor cognitive functions, reduced dopaminergic neurodegeneration with improved dopamine levels, improved MT function, attenuated microglial and astroglial cytokine inflammatory response [446,448,449,451,452,453,454,477,478]. | A meta-analysis of four observational studies concluded that T2D patients treated with TZD have a reduced risk for PD (Hussain et al., 2020 [456]). A randomized, multicenter, placebo-controlled study reported (a) no effect on disease progression and (b) no change in peripheral biomarkers in patients with PD [455]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribarič, S. The Contribution of Type 2 Diabetes to Parkinson’s Disease Aetiology. Int. J. Mol. Sci. 2024, 25, 4358. https://doi.org/10.3390/ijms25084358
Ribarič S. The Contribution of Type 2 Diabetes to Parkinson’s Disease Aetiology. International Journal of Molecular Sciences. 2024; 25(8):4358. https://doi.org/10.3390/ijms25084358
Chicago/Turabian StyleRibarič, Samo. 2024. "The Contribution of Type 2 Diabetes to Parkinson’s Disease Aetiology" International Journal of Molecular Sciences 25, no. 8: 4358. https://doi.org/10.3390/ijms25084358
APA StyleRibarič, S. (2024). The Contribution of Type 2 Diabetes to Parkinson’s Disease Aetiology. International Journal of Molecular Sciences, 25(8), 4358. https://doi.org/10.3390/ijms25084358