
The Hexokinase 1 5′-UTR Mutation in Charcot–Marie–Tooth 4G Disease Alters Hexokinase 1 Binding to Voltage-Dependent Anion Channel-1 and Leads to Dysfunctional Mitochondrial Calcium Buffering
 , ,
, ,
Abstract
:1. Introduction
2. Results
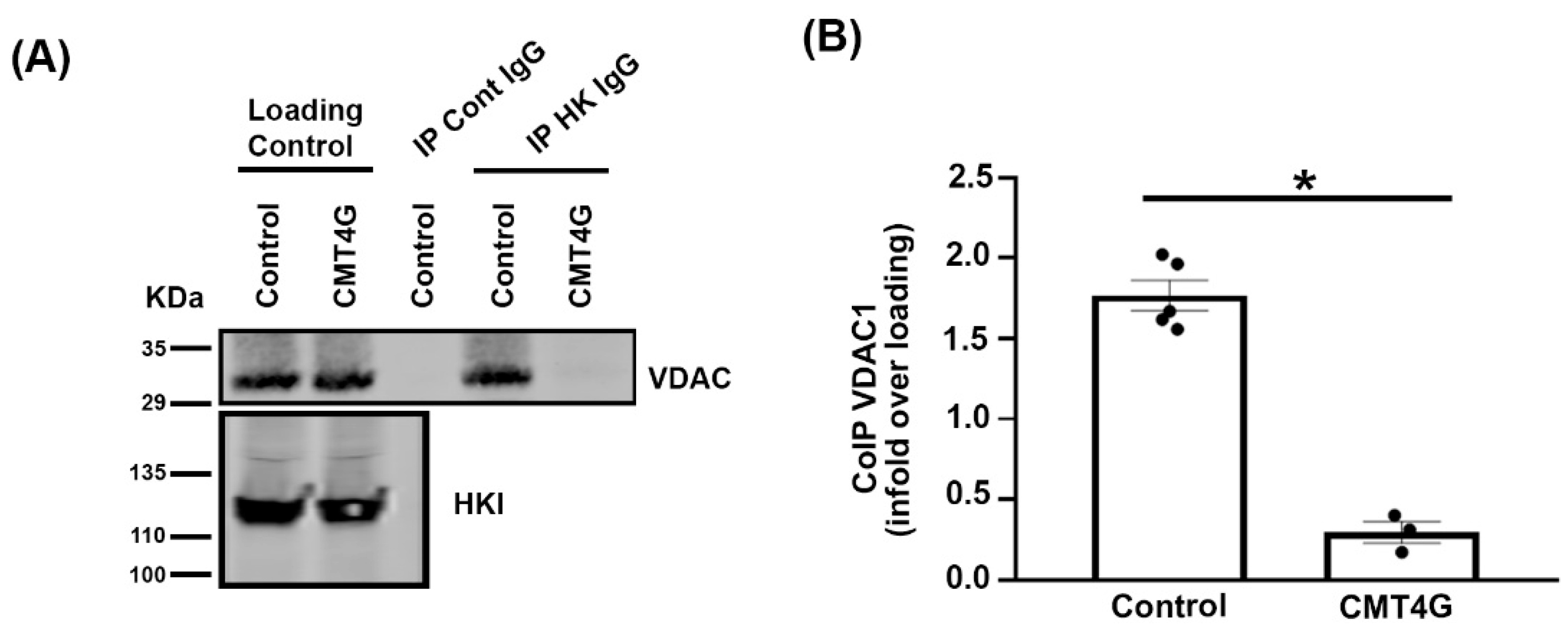
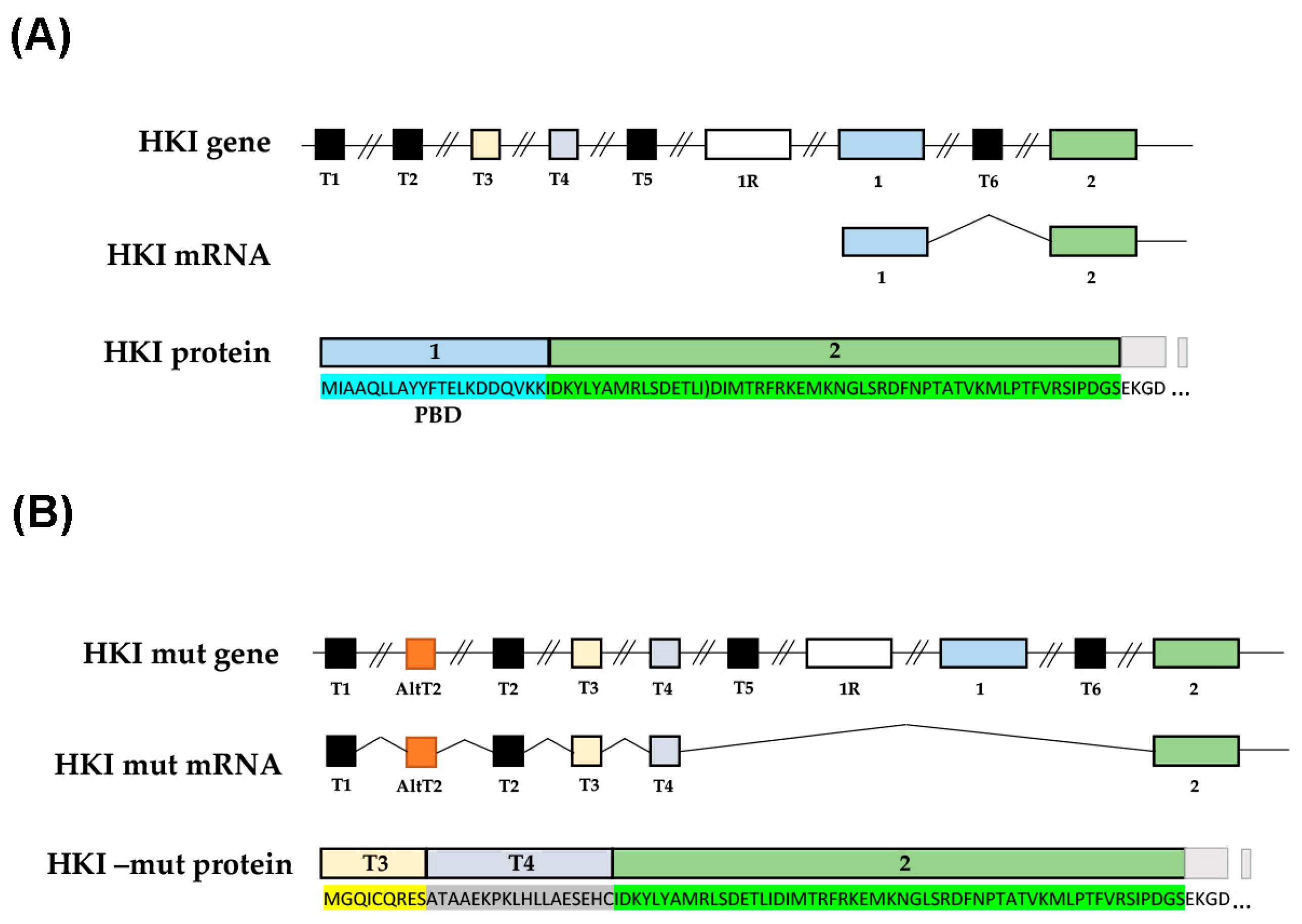
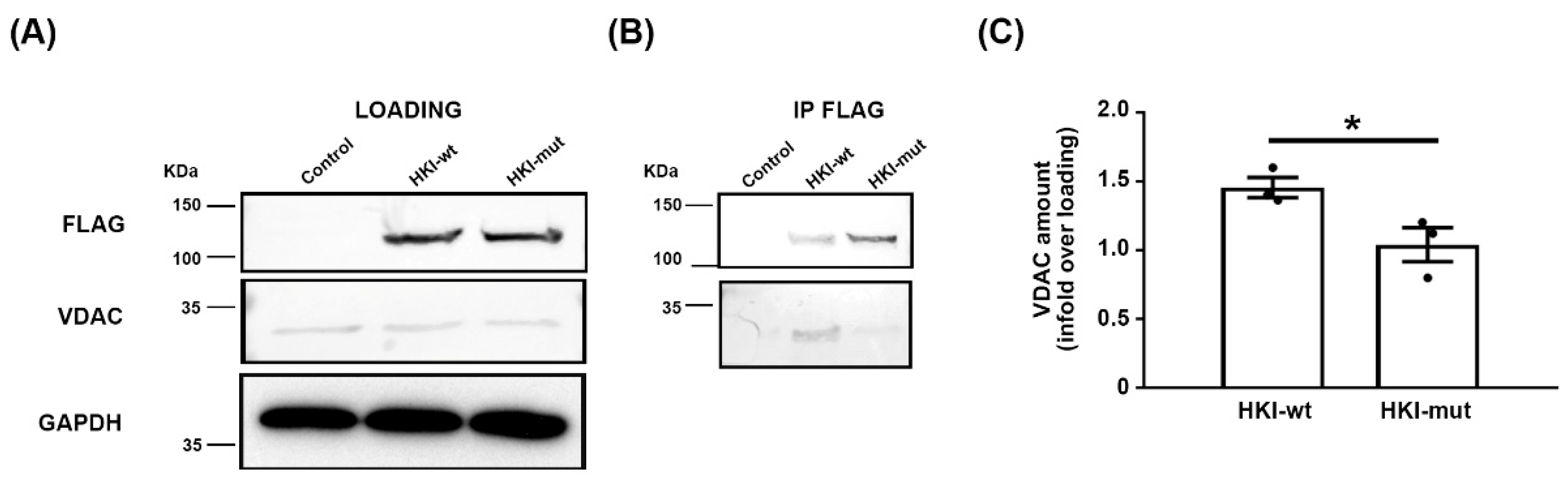
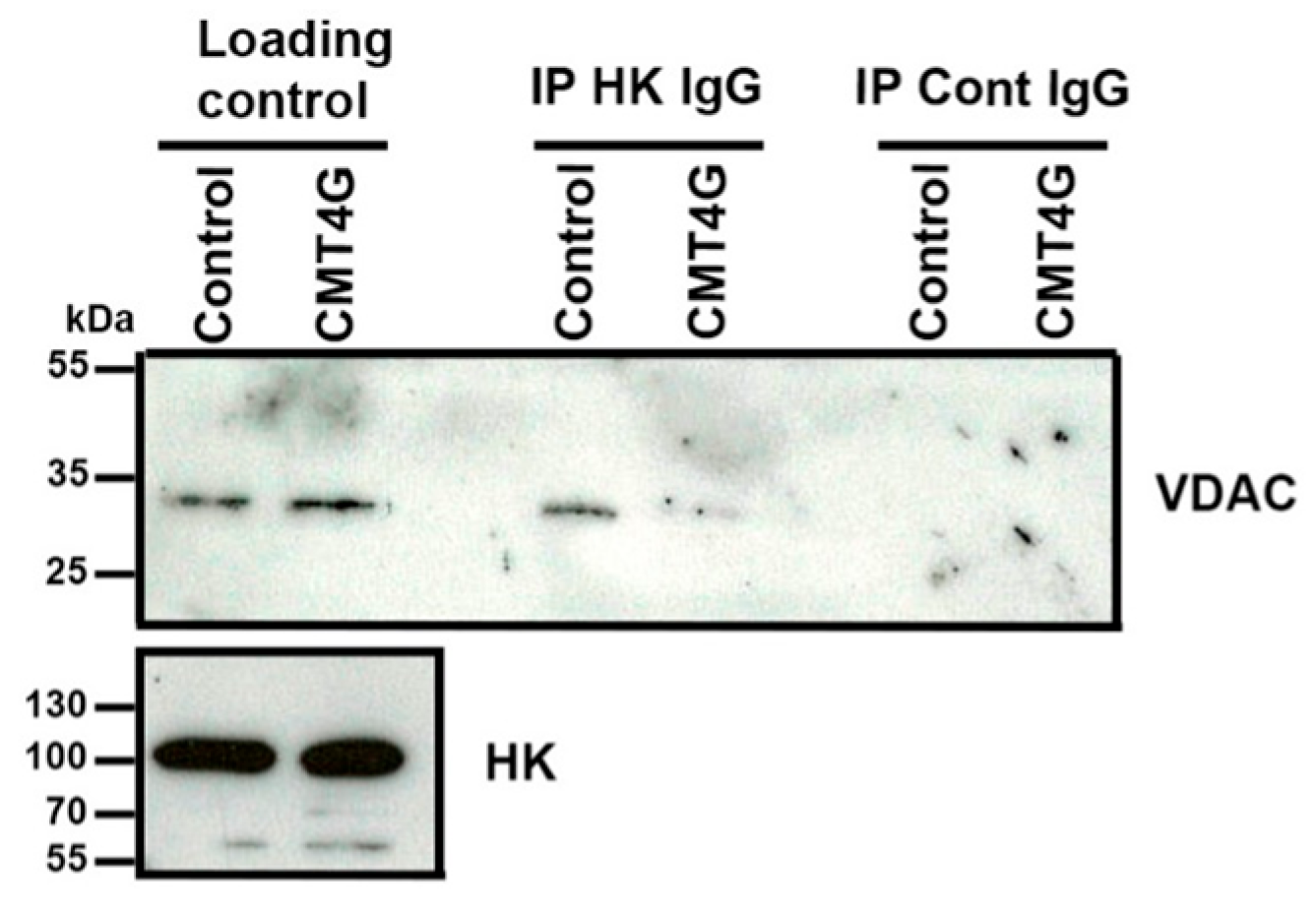
2.1. Mimicking CMT4G in HEK293T Cells Reveals a Defect in HK1/VDAC Interaction
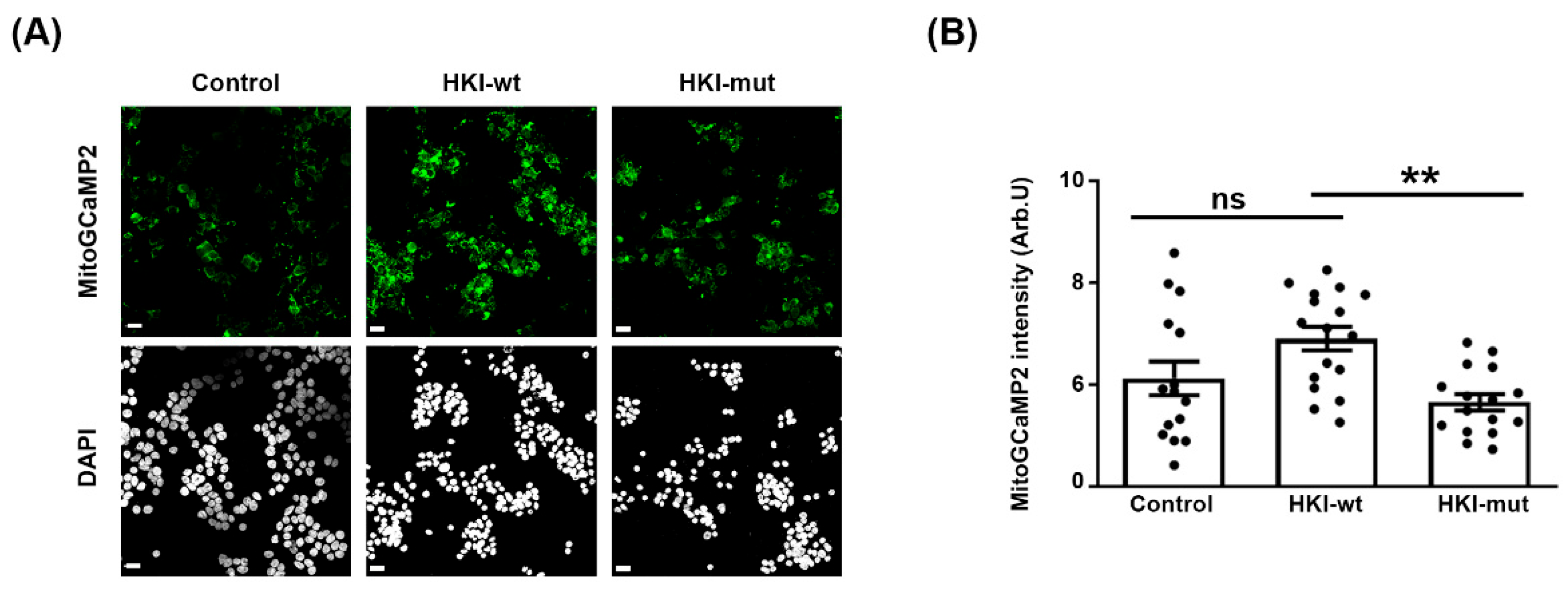
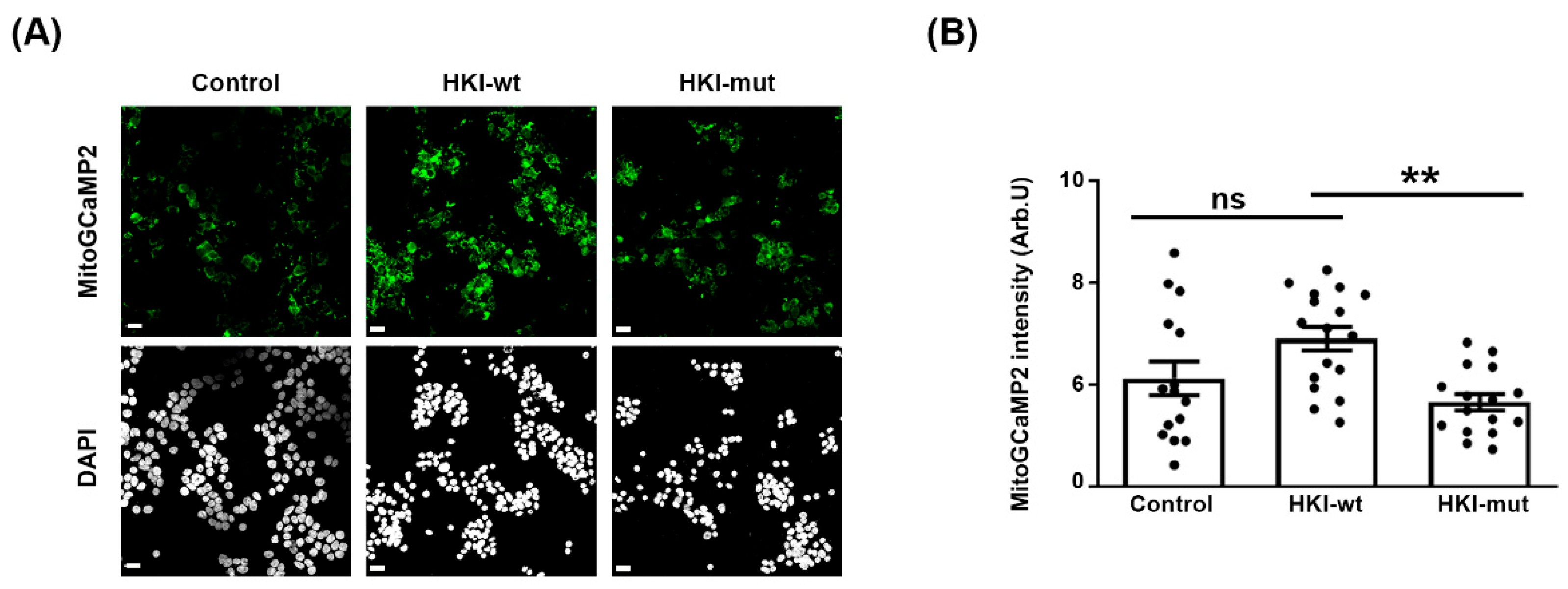
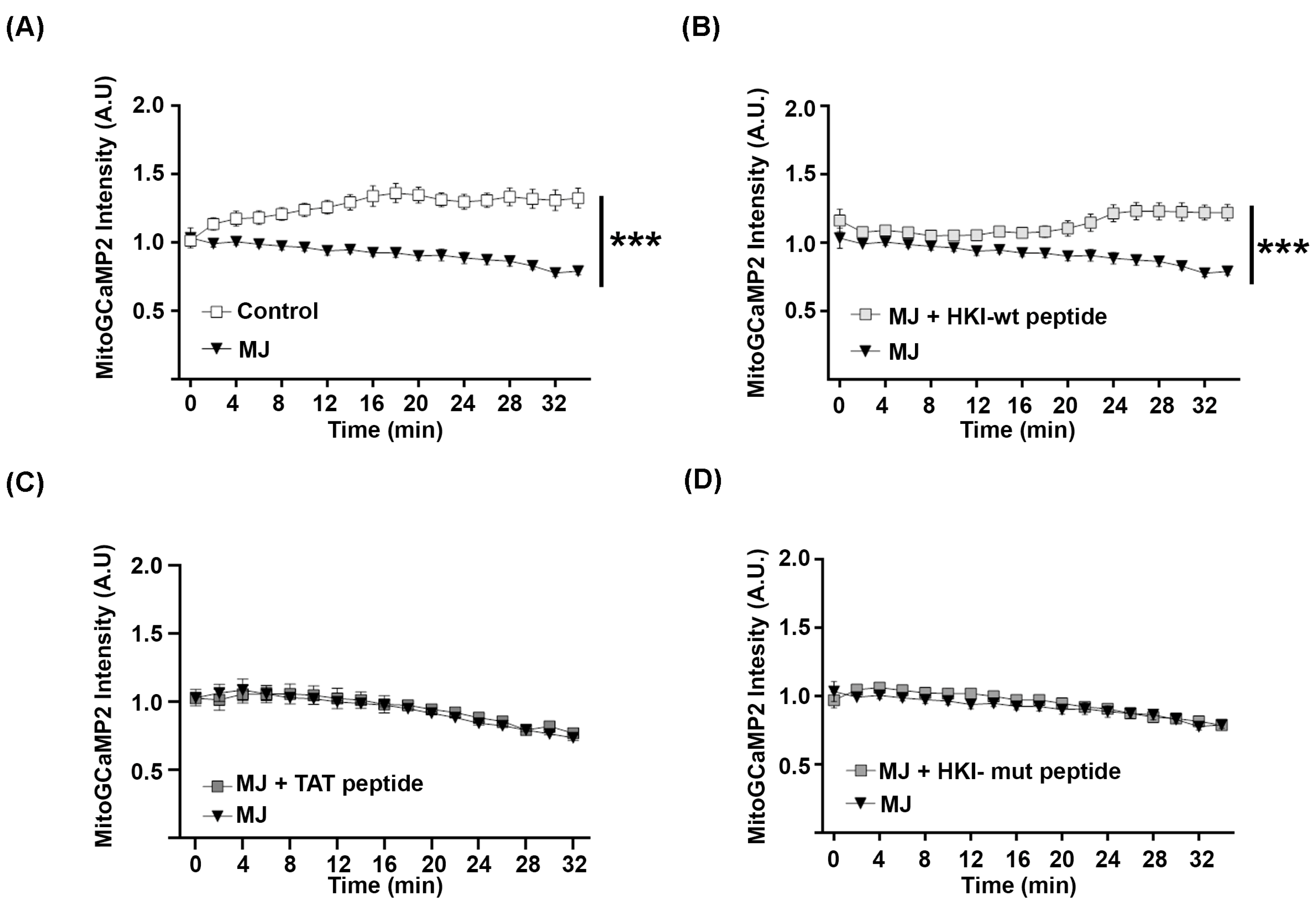
2.2. Defect in HK1/VDAC Interaction Results in the Alteration of Mitochondrial Calcium Homeostasis
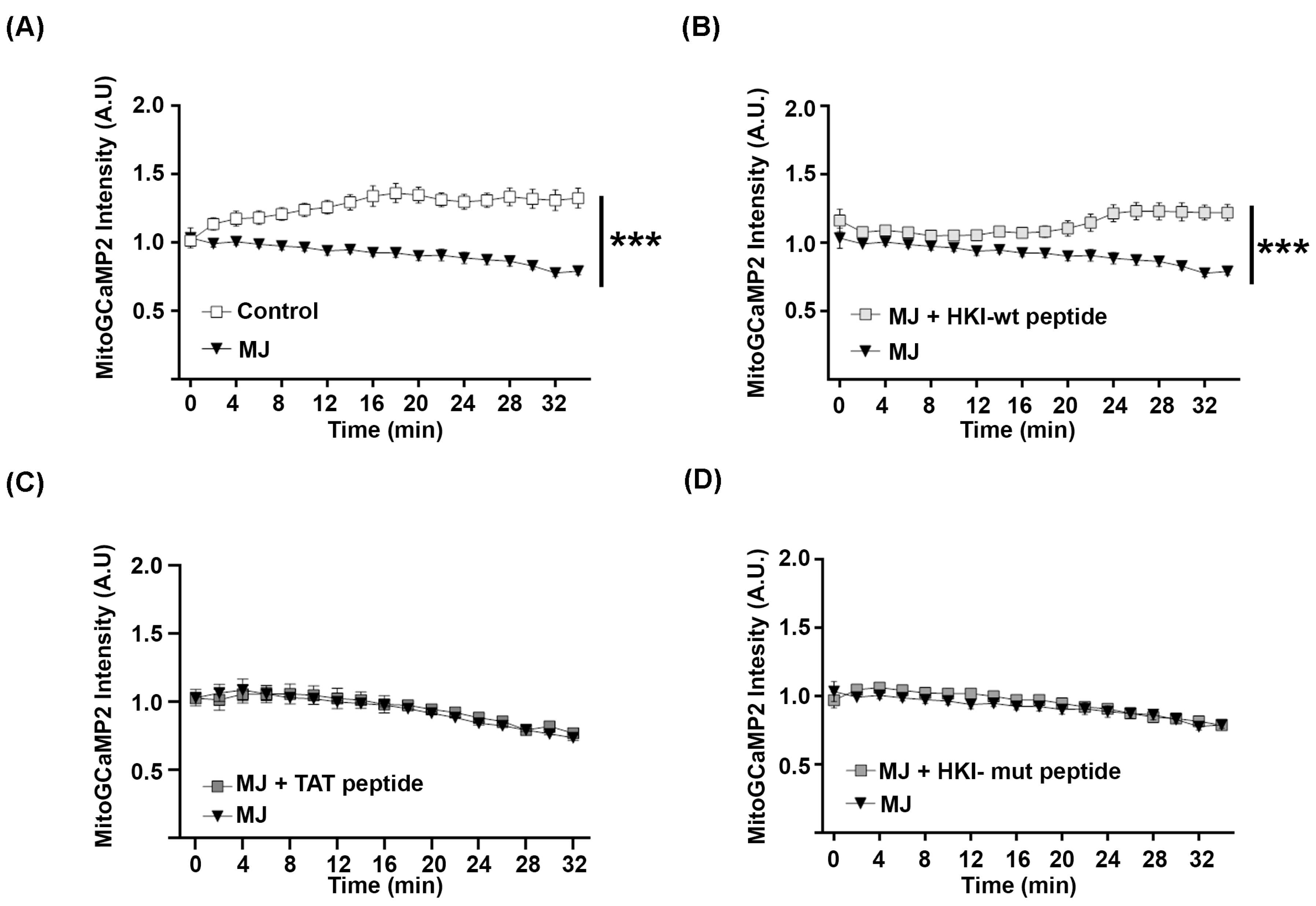
2.3. N-Terminus Peptide of Mutant HK1 Lacks the Ability to Block Mitochondrial Calcium Release through VDAC
2.4. Characterization of a Novel CMT4G Patient Series in the French Gispy Population
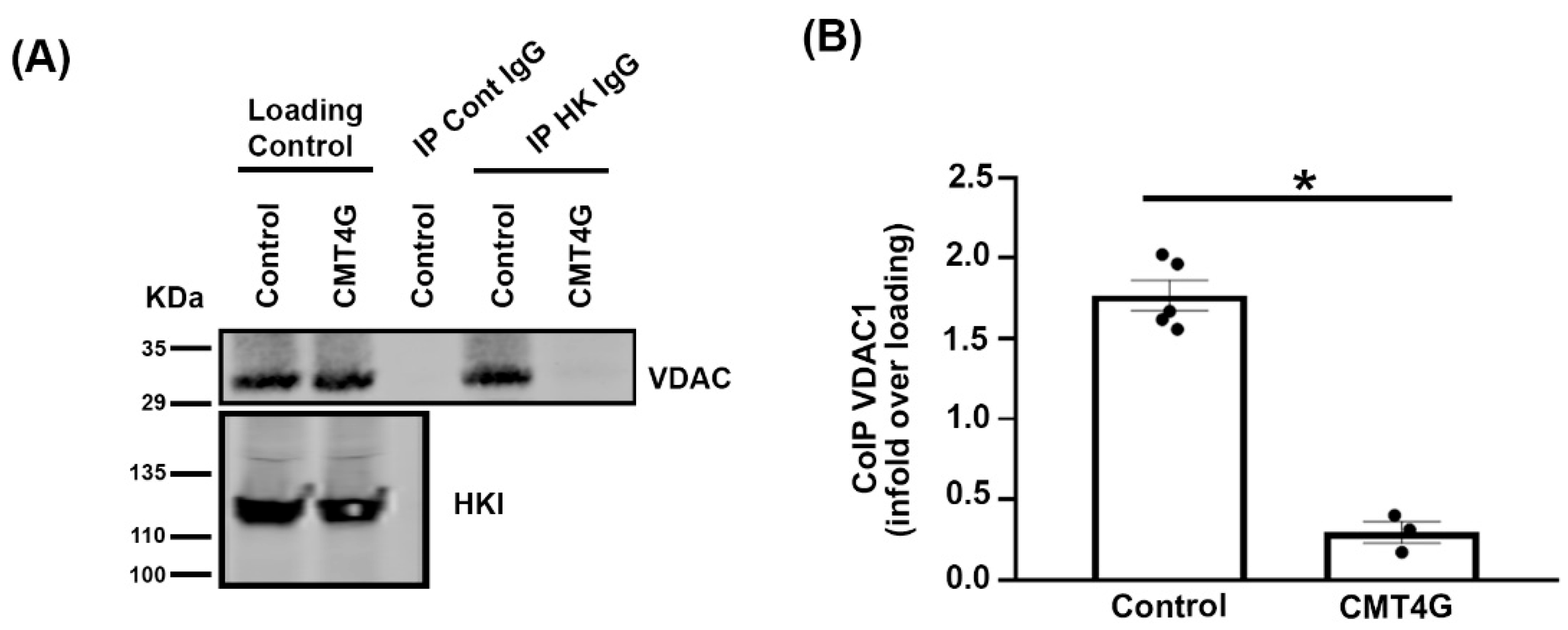
2.5. Mutated HK1 Is Unable to Interact with VDAC in PBMCs of CMT4G Patients
3. Discussion
4. Materials and Methods
4.1. Study Design and Patient Series
4.2. Plasmids and Reagents
4.3. Cell Culture and Transfection
4.4. PBMC Collection and Preparation
4.5. Immunoprecipitation
4.6. Immunocytochemistry
4.7. Videotracking Experiments
4.8. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gabrikova, D.; Mistrik, M.; Bernasovska, J.; Bozikova, A.; Behulova, R.; Tothova, I.; Macekova, S. Founder mutations in NDRG1 and HK1 genes are common causes of inherited neuropathies among Roma/Gypsies in Slovakia. J. Appl. Genet. 2013, 54, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, T.; Martínez-Rubio, D.; Márquez, C.; Paradas, C.; Colomer, J.; Jaijo, T.; Millán, J.; Palau, F.; Espinós, C. Genetics of the Charcot-Marie-Tooth disease in the Spanish Gypsy population: The hereditary motor and sensory neuropathy-Russe in depth. Clin. Genet. 2013, 83, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Brožková, D.; Haberlová, J.; Mazanec, R.; Laštůvková, J.; Seeman, P. HSMNR belongs to the most frequent types of hereditary neuropathy in the Czech Republic and is twice more frequent than HMSNL. Clin. Genet. 2016, 90, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.K.; Kalaydjieva, L.; Youl, B.; Rogers, T.; Angelicheva, D.; King, R.H.M.; Guergueltcheva, V.; Colomer, J.; Lupu, C.; Corches, A.; et al. Hereditary motor and sensory neuropathy-russe: New autosomal recessive neuropathy in balkan gypsies. Ann. Neurol. 2001, 50, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Hantke, J.; Chandler, D.; King, R.; Wanders, R.J.; Angelicheva, D.; Tournev, I.; McNamara, E.; Kwa, M.; Guergueltcheva, V.; Kaneva, R.; et al. A mutation in an alternative untranslated exon of hexokinase 1 associated with hereditary motor and sensory neuropathy—Russe (HMSNR). Eur. J. Hum. Genet. 2009, 17, 1606–1614. [Google Scholar] [CrossRef]
- Rosa, J.C.; César, M.d.C. Role of Hexokinase and VDAC in Neurological Disorders. Curr. Mol. Pharmacol. 2016, 9, 320–331. [Google Scholar] [CrossRef]
- Bianchi, M.; Crinelli, R.; Serafini, G.; Giammarini, C.; Magnani, M. Molecular bases of hexokinase deficiency. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1997, 1360, 211–221. [Google Scholar] [CrossRef]
- Peters, L.L.; Lane, P.W.; Andersen, S.G.; Gwynn, B.; Barker, J.E.; Beutler, E. Downeast Anemia (dea), a New Mouse Model of Severe Nonspherocytic Hemolytic Anemia Caused by Hexokinase (HKI) Deficiency. Blood Cells Mol. Dis. 2001, 27, 850–860. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Tajeddine, N.; Kroemer, G. Disruption of the hexokinase–VDAC complex for tumor therapy. Oncogene 2008, 27, 4633–4635. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Tricaud, N.; Gautier, B.; Berthelot, J.; Gonzalez, S.; Van Hameren, G. Traumatic and Diabetic Schwann Cell Demyelination Is Triggered by a Transient Mitochondrial Calcium Release through Voltage Dependent Anion Channel 1. Biomedicines 2022, 10, 1447. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.; Raisch, K.P. Identification of a mitochondrial-binding site on the N-terminal end of hexokinase II. Biosci. Rep. 2015, 35, e00205. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Belfiore, R.; Reina, S.; Tomasello, M.F.; Di Rosa, M.C.; Guarino, F.; Leggio, L.; De Pinto, V.; Messina, A. Hexokinase I N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability. Sci. Rep. 2016, 6, 34802. [Google Scholar] [CrossRef]
- Gautier, B.; Jacquard, M.F.; Guelfi, S.; Abbou, S.; Gonzalez, E.; Berthelot, J.; Boukhaddaoui, H.; Lebrun, A.; Legrand, B.; Tricaud, N.; et al. Mapping the N-Terminal Hexokinase-I Binding Site onto Voltage-Dependent Anion Channel-1 to Block Peripheral Nerve Demyelination. J. Med. Chem. 2022, 65, 11633–11647. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Flescher, E. Methyl jasmonate: A plant stress hormone as an anti-cancer drug. Phytochemistry 2009, 70, 1600–1609. [Google Scholar] [CrossRef]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef] [PubMed]
- Cesari, I.M.; Carvalho, E.; Rodrigues, M.F.; Mendonça, B.d.S.; Amôedo, N.D.; Rumjanek, F.D. Methyl Jasmonate: Putative Mechanisms of Action on Cancer Cells Cycle, Metabolism, and Apoptosis. Int. J. Cell Biol. 2014, 2014, 572097. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Y.; Hou, T.; Zhang, H.; Qu, A.; Wang, X. Differential mitochondrial calcium responses in different cell types detected with a mitochondrial calcium fluorescent indicator, mito-GCaMP2. Acta Biochim. Biophys. Sin. 2011, 43, 822–830. [Google Scholar] [CrossRef]
- Tallini, Y.N.; Ohkura, M.; Choi, B.R.; Ji, G.; Imoto, K.; Doran, R.; Lee, J.; Plan, P.; Wilson, J.; Xin, H.-B.; et al. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. USA 2006, 103, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Guergueltcheva, V.; Tournev, I.; Bojinova, V.; Hantke, J.; Litvinenko, I.; Ishpekova, B.; Shmarov, A.; Petrova, J.; Jordanova, A.; Kalaydjieva, L. Early Clinical and Electrophysiologic Features of the Two Most Common Autosomal Recessive Forms of Charcot-Marie-Tooth Disease in the Roma (Gypsies). J. Child Neurol. 2006, 21, 20–25. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Id. # Patient | Age at Onset of LL Weakness (Years) | Age at Onset of UL Weakness (Years) | Foot Deformities | CMTNS | MNCV Median (m/s) | CMAP Median, (mV) | SNAP |
|---|---|---|---|---|---|---|---|
| 1 | 6 | 16 | +Arthrodesis | 24 | 16.7 (38) | 0.2 | NR |
| 2 | 6 | 15 | +Arthrodesis | 23 | NR (29) | NR | NR |
| 3 | 9 | 14 | + | 28 | NR (65) | NR | NR |
| 4 | 10 | 11 | + Arthrodesis | 12 | 11.2 (10) | 0.4 | NR |
| 5 | 7 | ND | - | ND | ND | ND | ND |
| 6 | 9 | ND | + | ND | ND | ND | ND |
| 7 | 7 | 16 | +Arthrodesis | ND | ND | ND | ND |
| 8 | 5 | 16 | - | 23 | 29 (9) | 1.5 | NR |
| 9 | 9 | 16 | - | 19 | 16.2 (21) | 1.4 | NR |
| 10 | 8 | ND | +Arthrodesis | 6 | 24.1 (10) | 5.6 | NR |
| 11 | 3 | ND | - | 1 | 28.9 (3) | 4.3 | NR |
| 12 | 4 | ND | + | 8 | 32 (9) | 1.93 | NR |
| 13 | 12 | 35 | - | 25 | ND | ND | ND |
| 14 | 10 | - | ND | - | - | - | - |
| 15 | 3 | - | ND | - | - | - | - |
| 16 | 3 | 16 | +Arthrodesis | 16 | 21 (36) | 0.59 | NR |
| 17 | 3 | - | ND | - | - | - | - |
| 18 | 10 | 10 | + Arthrodesis | 17 | 23.8 (12) | - | NR |
| 19 | 11 | 45 | ND | ND | ND | ND | ND |
| 20 | 13 | 20 | +Arthrodesis | - | - | - | - |
| 21 | 6 | ND | - | 6 | 30.6 (7) | 5.3 | NR |
| 22 | 10 | 14 | +Arthrodesis | ND | 22.2 (8) | 1.19 | NR |
| 23 | 5 | ND | - | ND | 24 (6) | 6.4 | NR |
| 24 | 4 | 7 | - | 20 | 16 (27) | 0.45 | NR |
| 25 | 12 | 8 | - | ND | ND | ND | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceprian, M.; Juntas-Morales, R.; Campbell, G.; Walther-Louvier, U.; Rivier, F.; Camu, W.; Esselin, F.; Echaniz-Laguna, A.; Stojkovic, T.; Bouhour, F.; et al. The Hexokinase 1 5′-UTR Mutation in Charcot–Marie–Tooth 4G Disease Alters Hexokinase 1 Binding to Voltage-Dependent Anion Channel-1 and Leads to Dysfunctional Mitochondrial Calcium Buffering. Int. J. Mol. Sci. 2024, 25, 4364. https://doi.org/10.3390/ijms25084364
Ceprian M, Juntas-Morales R, Campbell G, Walther-Louvier U, Rivier F, Camu W, Esselin F, Echaniz-Laguna A, Stojkovic T, Bouhour F, et al. The Hexokinase 1 5′-UTR Mutation in Charcot–Marie–Tooth 4G Disease Alters Hexokinase 1 Binding to Voltage-Dependent Anion Channel-1 and Leads to Dysfunctional Mitochondrial Calcium Buffering. International Journal of Molecular Sciences. 2024; 25(8):4364. https://doi.org/10.3390/ijms25084364
Chicago/Turabian StyleCeprian, Maria, Raul Juntas-Morales, Graham Campbell, Ulrike Walther-Louvier, François Rivier, William Camu, Florence Esselin, Andoni Echaniz-Laguna, Tanya Stojkovic, Françoise Bouhour, and et al. 2024. "The Hexokinase 1 5′-UTR Mutation in Charcot–Marie–Tooth 4G Disease Alters Hexokinase 1 Binding to Voltage-Dependent Anion Channel-1 and Leads to Dysfunctional Mitochondrial Calcium Buffering" International Journal of Molecular Sciences 25, no. 8: 4364. https://doi.org/10.3390/ijms25084364