
Identification of AURKA as a Biomarker Associated with Cuproptosis and Ferroptosis in HNSCC
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
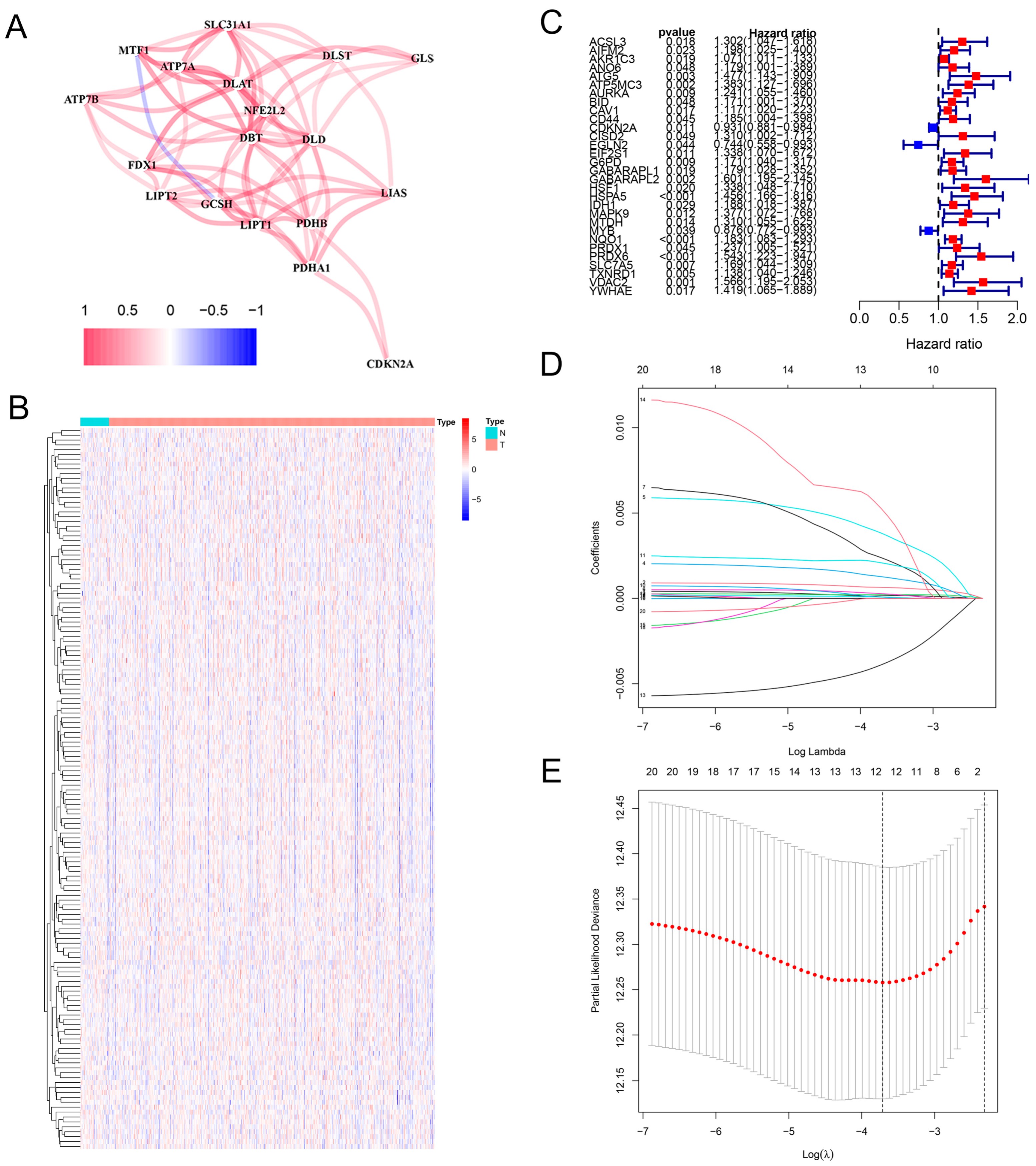
2.1. Construction of the Prognostic Model in HNSCC
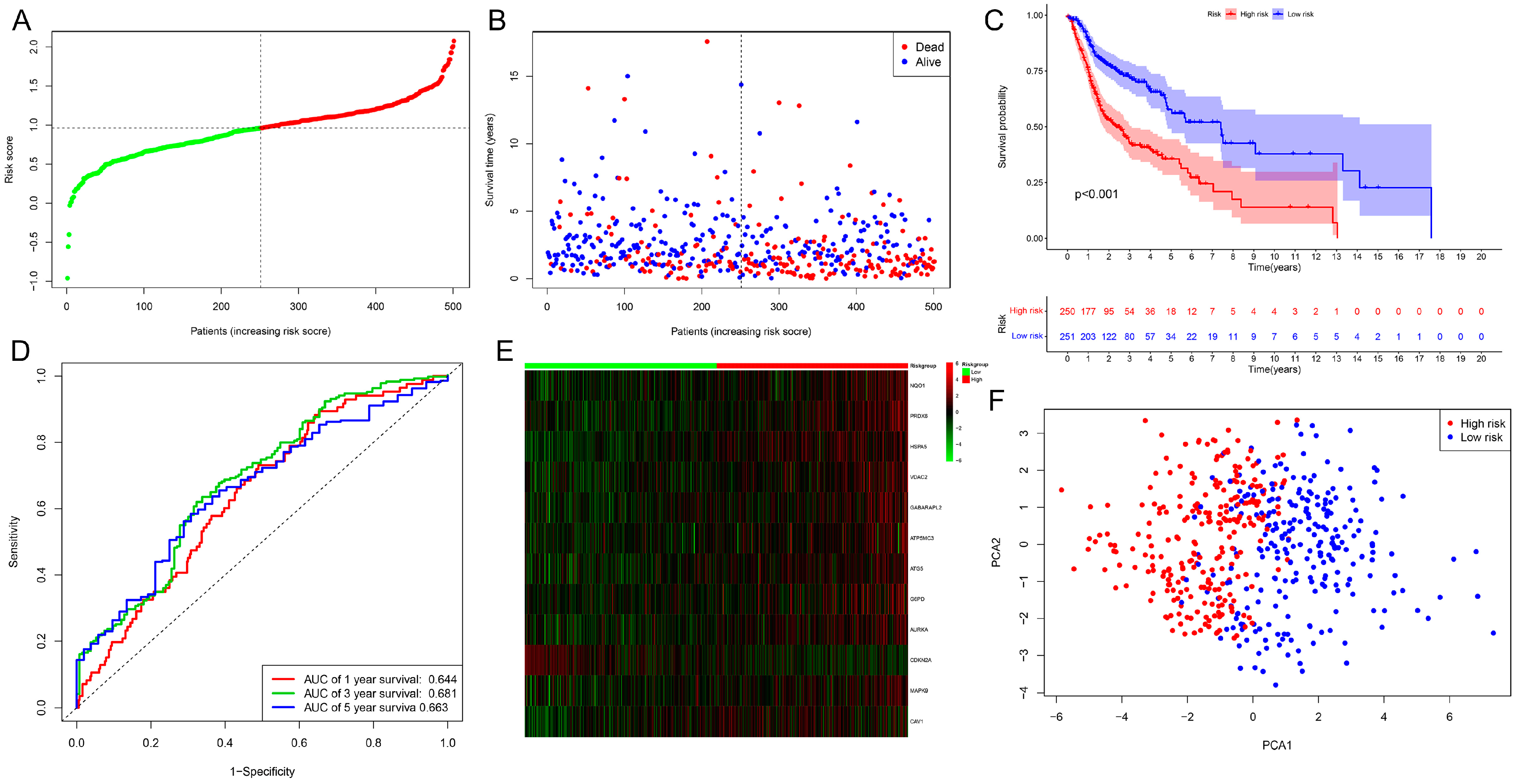
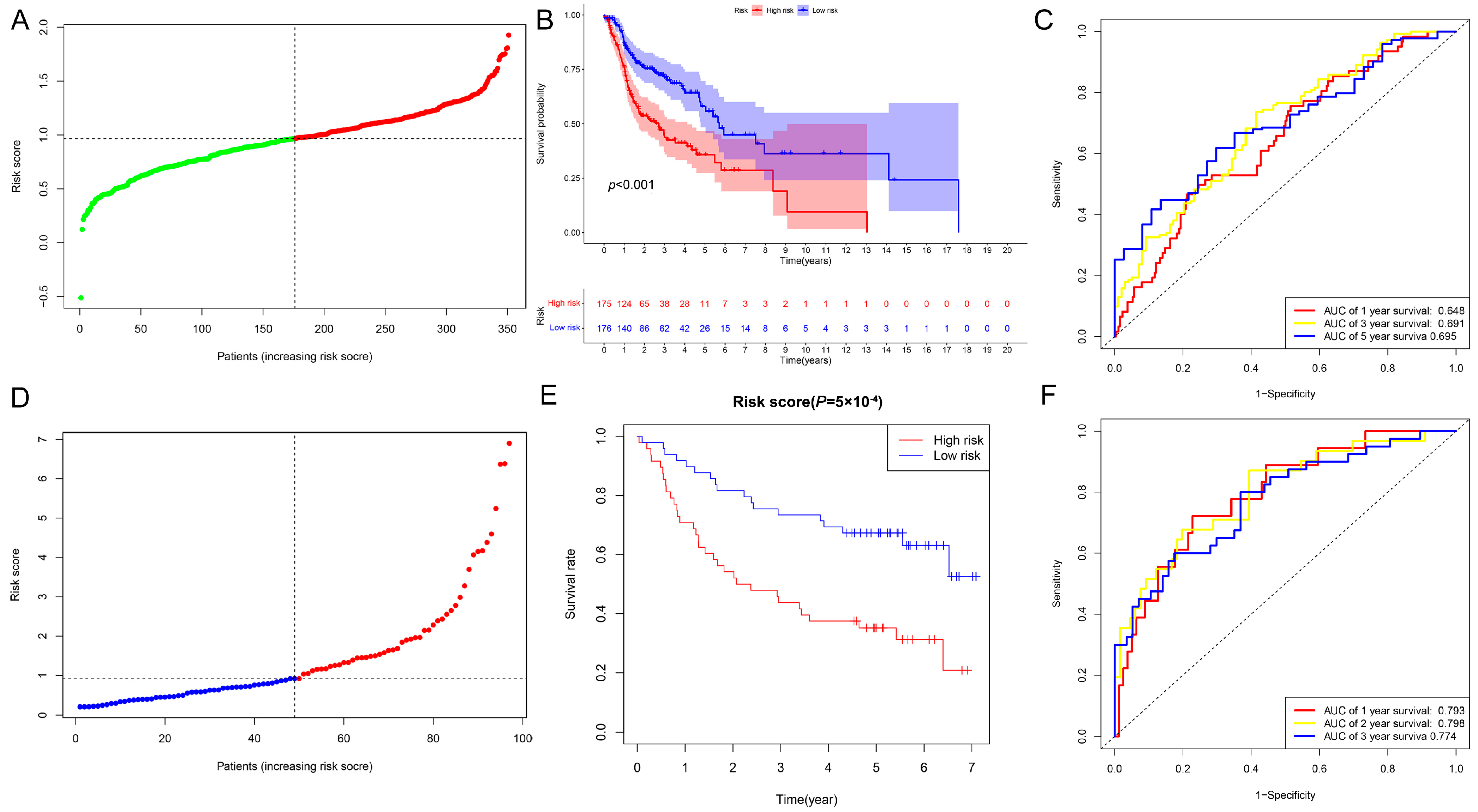
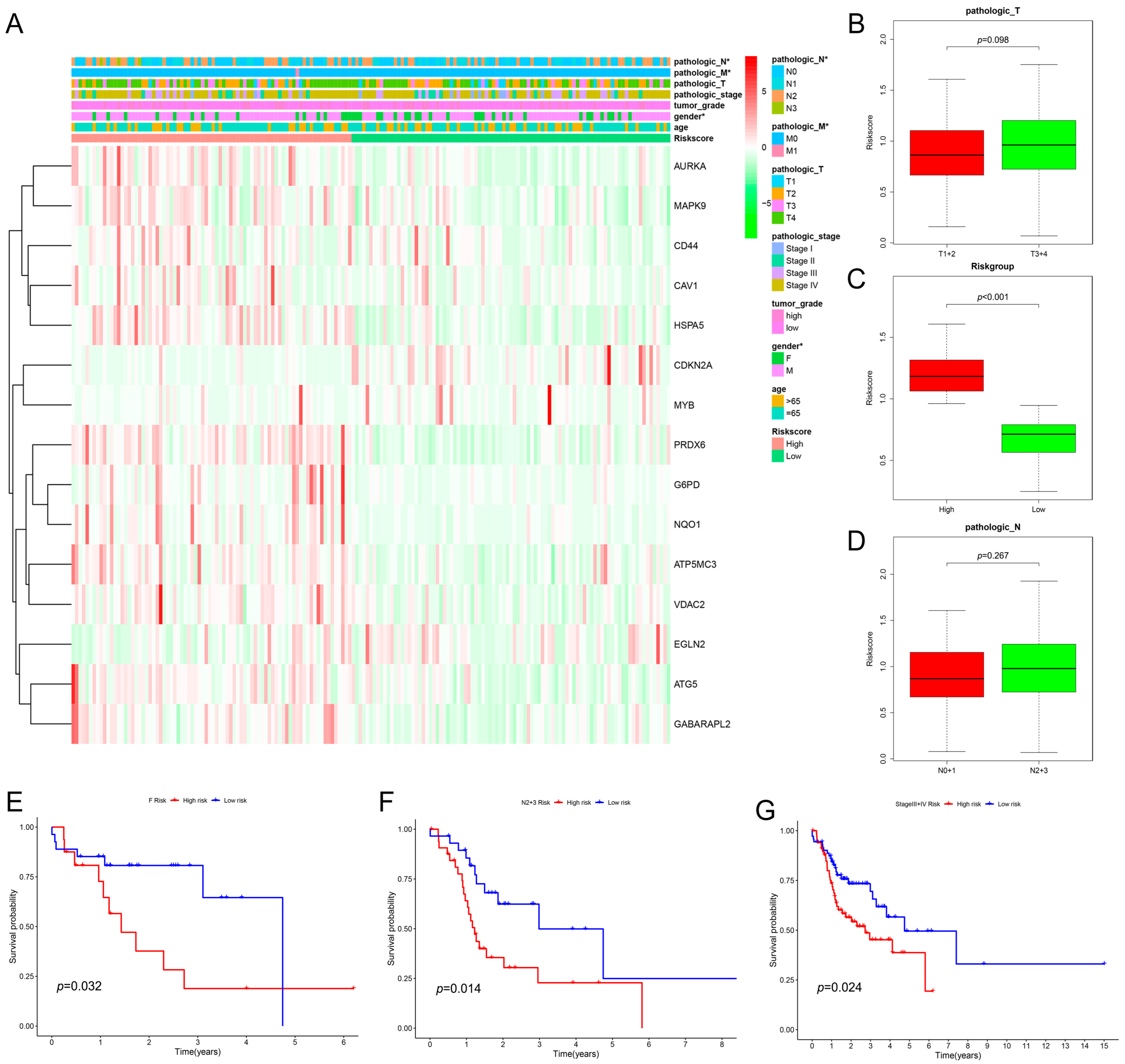
2.2. Evaluation and Validation of the Prognostic Model in HNSCC
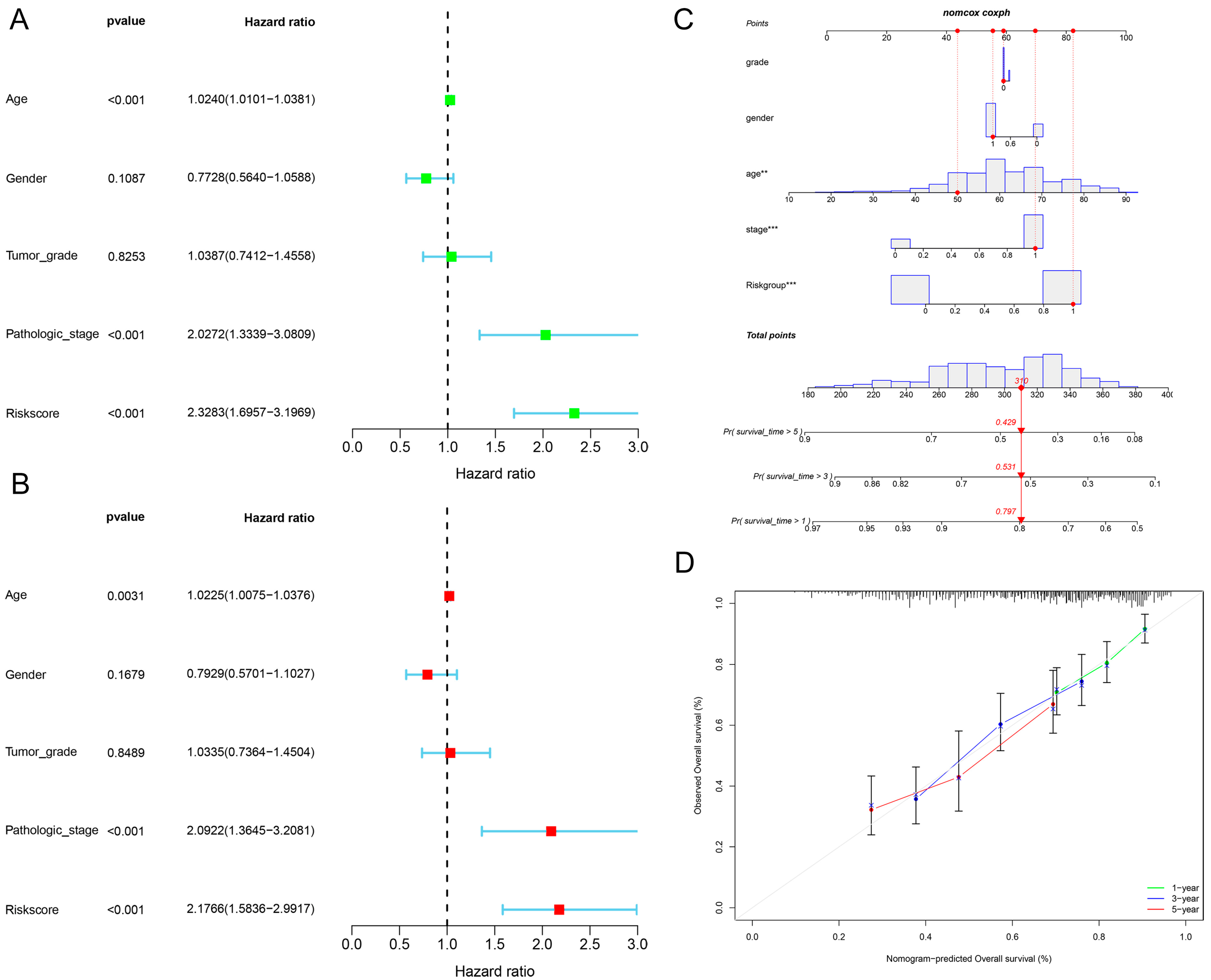
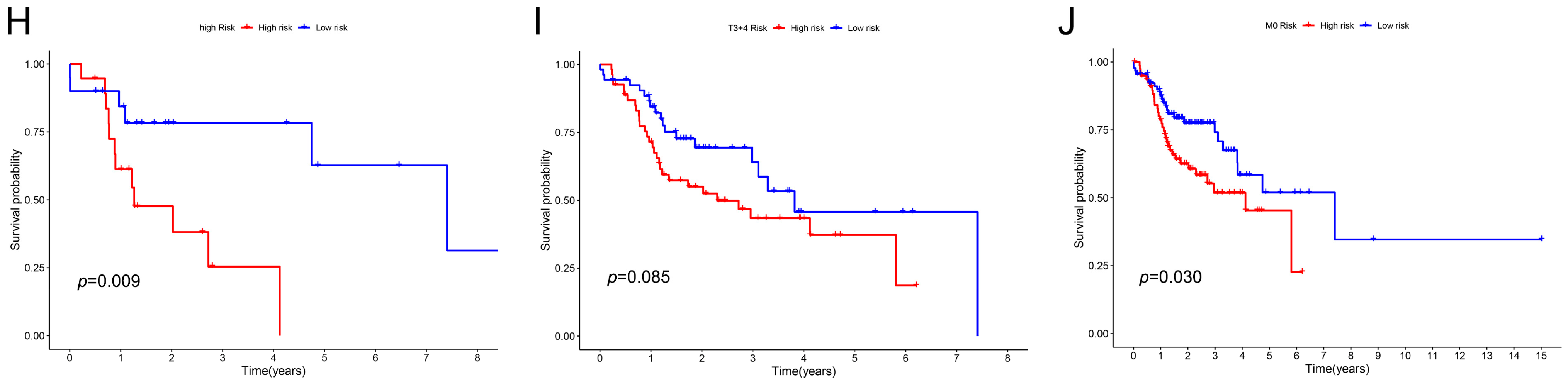
2.3. The Relationship between Clinical Characteristics and the Prognostic Model
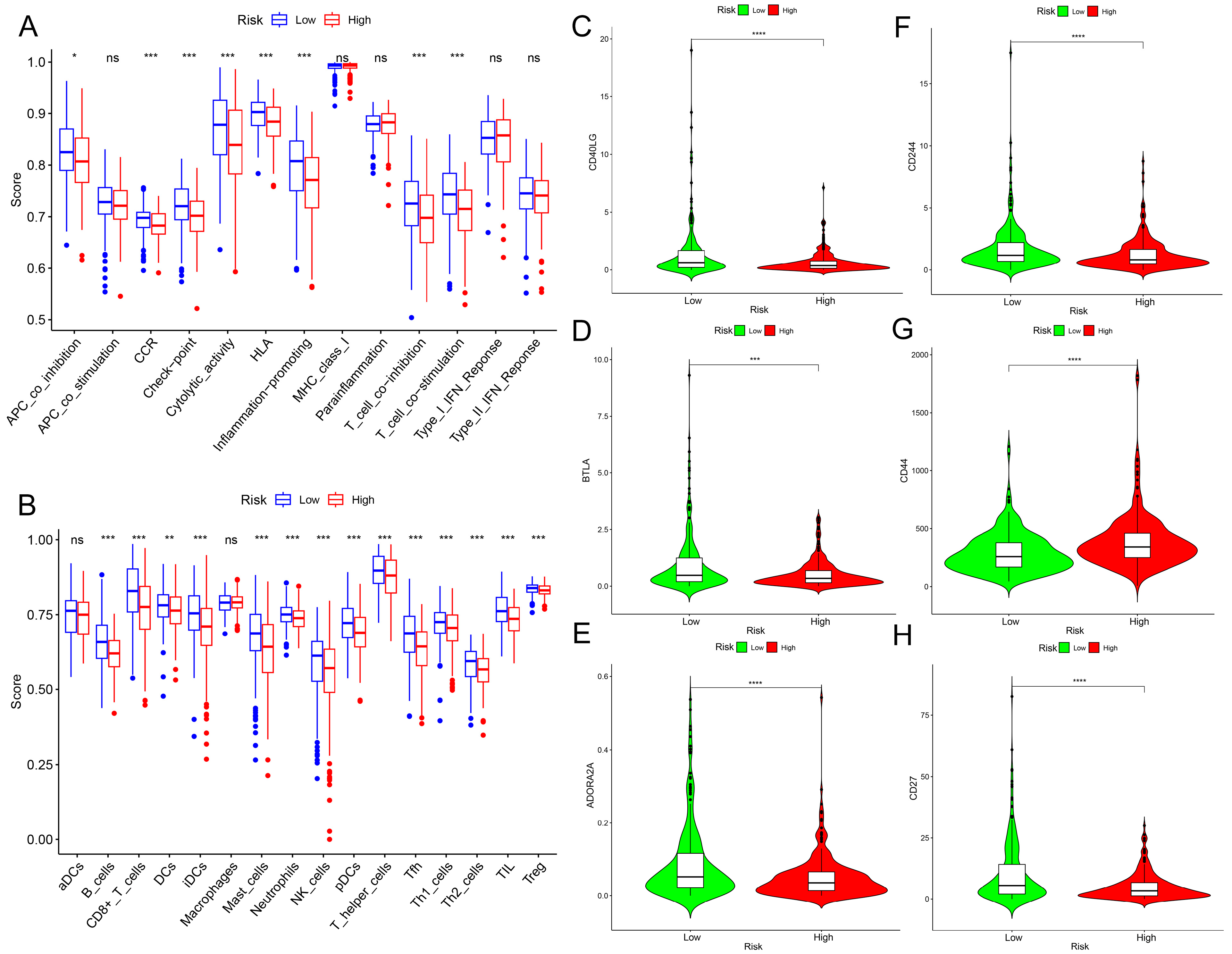
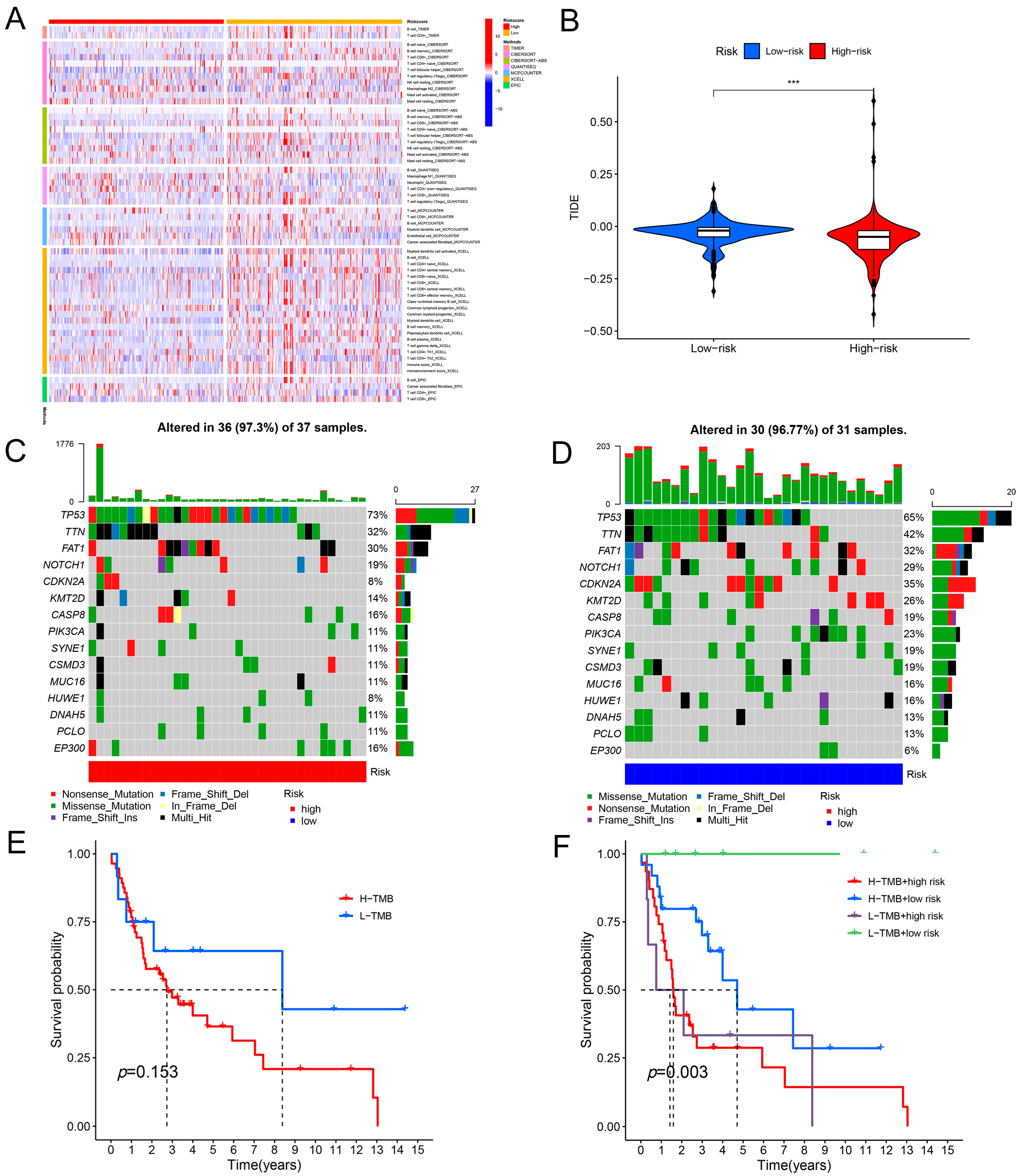
2.4. Immunological Characteristics of the Prognostic Model
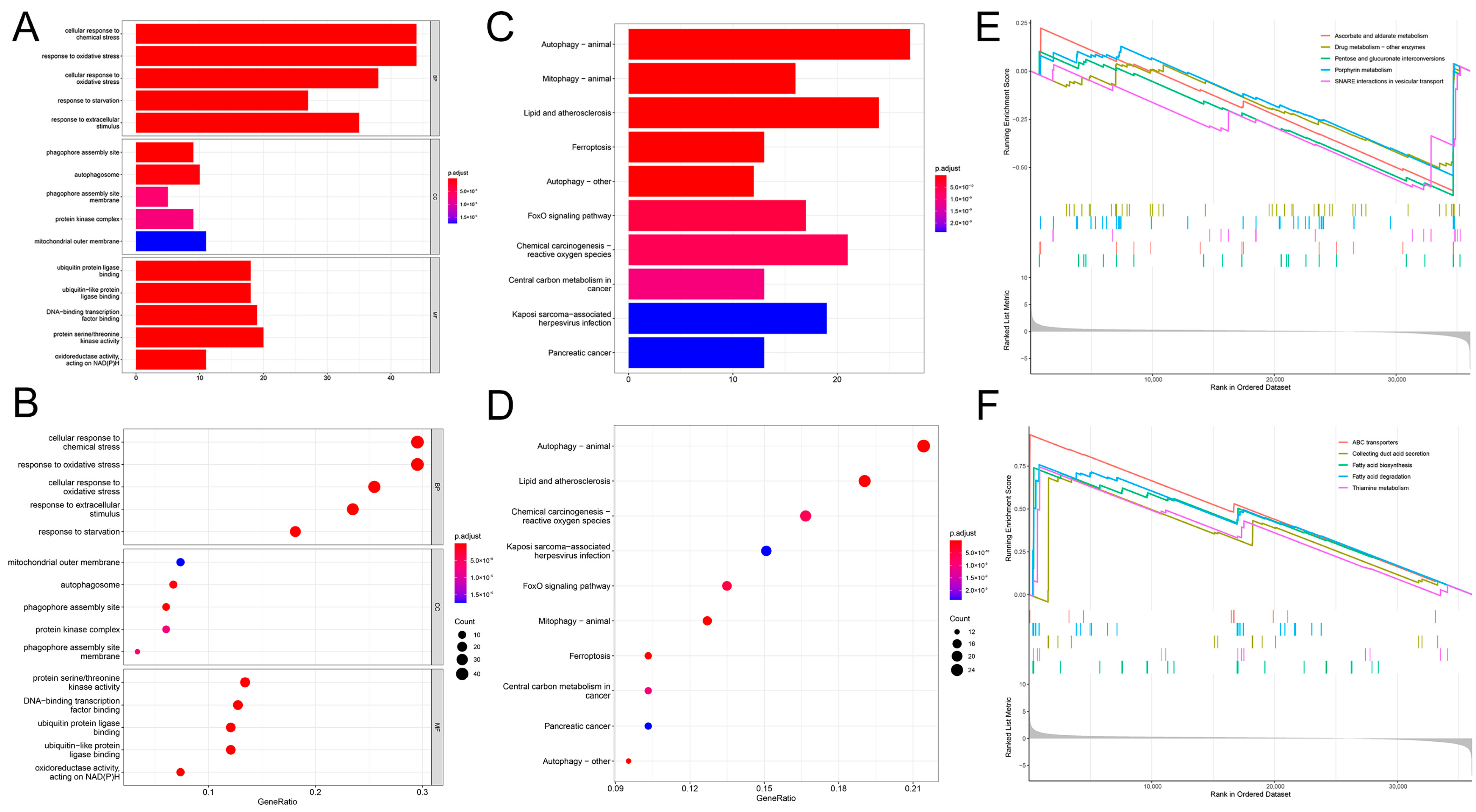
2.5. Functional Enrichment Analysis of the Prognostic Model
2.6. Drug Sensitivity of the Prognostic Model
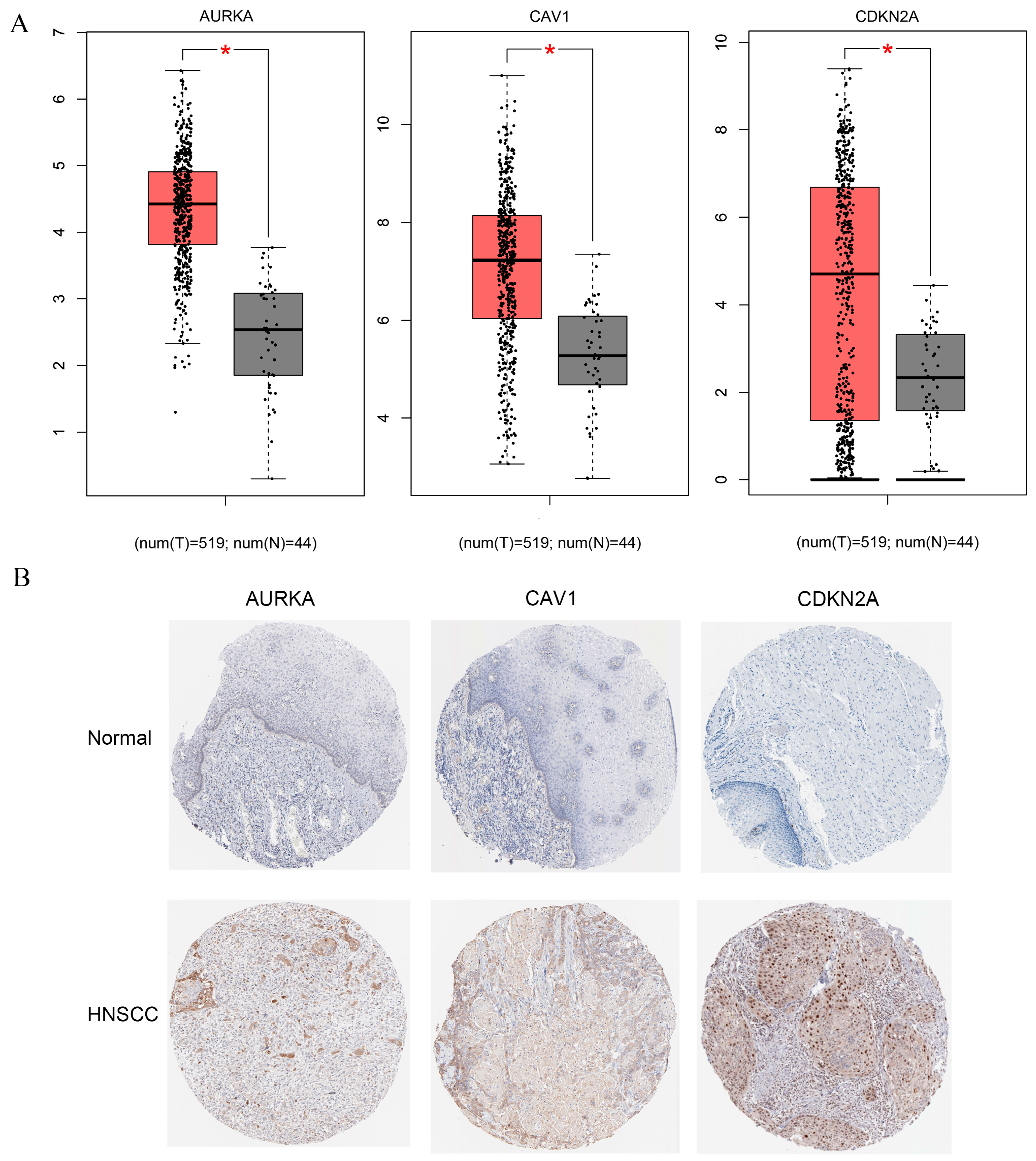
2.7. Identification of Potential Biomarkers in Constructed Model
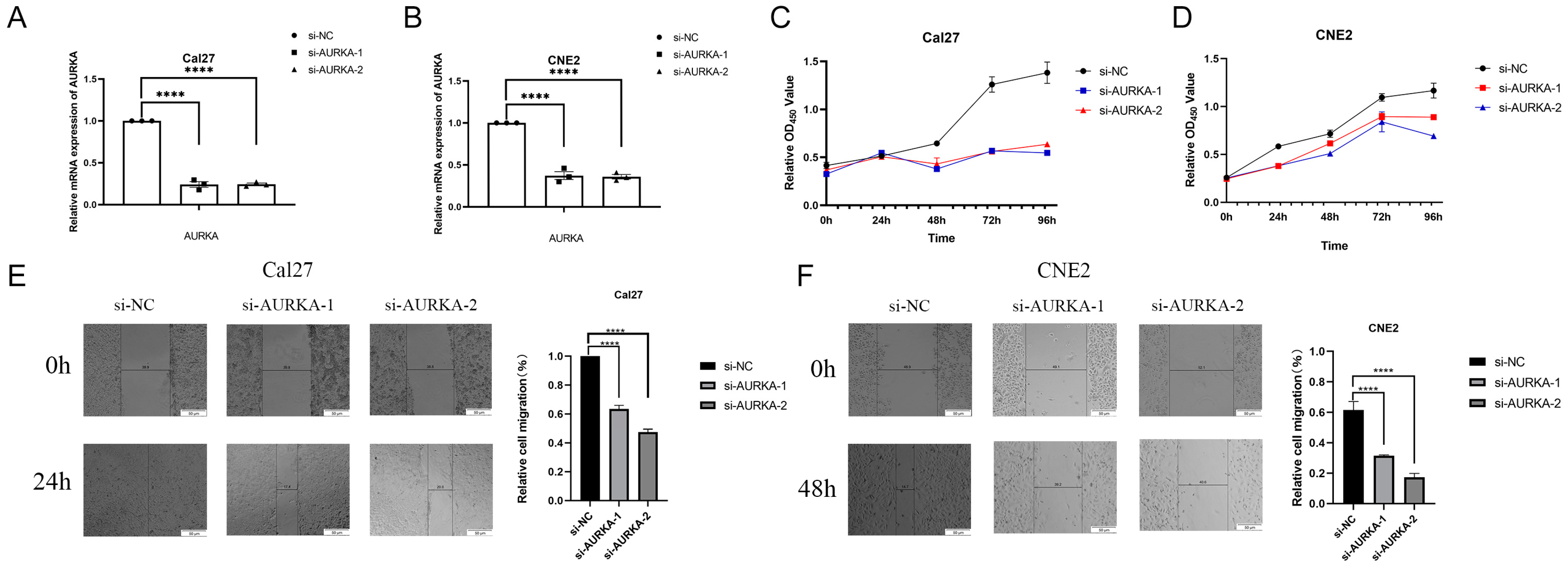
2.8. Inhibition of AURKA Suppresses the Proliferation and Migration of HNSCC Cells
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Identification of Cuproptosis-Associated Ferroptosis Genes in HNSCC
4.3. Construction of a Predictive Model for HNSCC Based on Cuproptosis-Associated Ferroptosis Genes
4.4. Immune Characteristics and Functional Analysis
4.5. Biological Functional Enrichment Analysis and Drug Sensitivity Analysis
4.6. Cell Culture
4.7. siRNA Transfection
4.8. RT-qPCR
4.9. Cell Counting Kit8 Experiment
4.10. Wound Healing
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cramer, J.D.; Burtness, B.; Le, Q.T.; Ferris, R.L. The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 2019, 16, 669–683. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Solomon, B.; Young, R.J.; Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin. Cancer Biol. 2018, 52, 228–240. [Google Scholar] [CrossRef]
- Hu, H.; Xu, Q.; Mo, Z.; Hu, X.; He, Q.; Zhang, Z.; Xu, Z. New anti-cancer explorations based on metal ions. J. Nanobiotechnol. 2022, 20, 457. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Raudenska, M.; Balvan, J.; Masarik, M. Cell death in head and neck cancer pathogenesis and treatment. Cell Death Dis. 2021, 12, 192. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Suver, J.D. Evaluating capital-investment proposals. Health Serv. Manag. 1979, 12, 1–5. [Google Scholar]
- Bennett, W.D.; Scheuch, G.; Zeman, K.L.; Brown, J.S.; Kim, C.; Heyder, J.; Stahlhofen, W. Regional deposition and retention of particles in shallow, inhaled boluses: Effect of lung volume. J. Appl. Physiol. 1999, 86, 168–173. [Google Scholar] [CrossRef]
- Garcia-Morillo, J.S.; Carneado de la Fuente, J.; Stiefel Garcia-Junco, P.; Garcia-Donas, M.A.; Gonzalez Escribano, M.F.; Nunez-Roldan, A. Study of the gene of hemochromatosis in first degree relatives of patient with porphyria cutanea tarda. Med. Clin. 1999, 112, 699–701. [Google Scholar]
- Lv, Y.; Lin, S.Y.; Hu, F.F.; Ye, Z.; Zhang, Q.; Wang, Y.; Guo, A.Y. Landscape of cancer diagnostic biomarkers from specifically expressed genes. Brief. Bioinform. 2020, 21, 2175–2184. [Google Scholar] [CrossRef]
- Wang, T.Y.; Jiang, X.Y.; Lu, Y.; Ruan, Y.M.; Wang, J.M. Identification and integration analysis of a novel prognostic signature associated with cuproptosis-related ferroptosis genes and relevant lncRNA regulatory axis in lung adenocarcinoma. Aging 2023, 15, 33. [Google Scholar] [CrossRef]
- Li, Y.; Fang, T.; Shan, W.Y.; Gao, Q.L. Identification of a Novel Model for Predicting the Prognosis and Immune Response Based on Genes Related to Cuproptosis and Ferroptosis in Ovarian Cancer. Cancers 2023, 15, 579. [Google Scholar] [CrossRef]
- Zhao, C.B.; Zhang, Z.L.; Jing, T. A novel signature of combing cuproptosis- with ferroptosis-related genes for prediction of prognosis, immunologic therapy responses and drug sensitivity in hepatocellular carcinoma. Front. Oncol. 2022, 12, 1000993. [Google Scholar] [CrossRef]
- Lee, J.W.; Parameswaran, J.; Sandoval-Schaefer, T.; Eoh, K.J.; Yang, D.H.; Zhu, F.; Mehra, R.; Sharma, R.; Gaffney, S.G.; Perry, E.B.; et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2019, 25, 3430–3442. [Google Scholar] [CrossRef] [PubMed]
- Lohavanichbutr, P.; Mendez, E.; Holsinger, F.C.; Rue, T.C.; Zhang, Y.; Houck, J.; Upton, M.P.; Futran, N.; Schwartz, S.M.; Wang, P.; et al. A 13-gene signature prognostic of HPV-negative OSCC: Discovery and external validation. Clin. Cancer Res. 2013, 19, 1197–1203. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Q.; Niu, L.; Xu, L.; Guo, Y.; Wang, L.; Guo, C. Suppression of G6PD induces the expression and bisecting GlcNAc-branched N-glycosylation of E-Cadherin to block epithelial-mesenchymal transition and lymphatic metastasis. Br. J. Cancer 2020, 123, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Deneka, A.Y.; Baca, Y.; Serebriiskii, I.G.; Nicolas, E.; Parker, M.I.; Nguyen, T.T.; Xiu, J.; Korn, W.M.; Demeure, M.J.; Wise-Draper, T.; et al. Association of TP53 and CDKN2A Mutation Profile with Tumor Mutation Burden in Head and Neck Cancer. Clin. Cancer Res. 2022, 28, 1925–1937. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef]
- Tang, Y.; Li, C.; Zhang, Y.J.; Wu, Z.H. Ferroptosis-Related Long Non-Coding RNA signature predicts the prognosis of Head and neck squamous cell carcinoma. Int. J. Biol. Sci. 2021, 17, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Jiang, X.; Wang, J.; Zong, Y.; Yuan, Z.; Miao, S.; Mao, X. Effect of regulatory cell death on the occurrence and development of head and neck squamous cell carcinoma. Biomark. Res. 2023, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.P.; Boudreau, M.W.; Selting, K.A.; Chatkewitz, L.E.; Samuelson, J.; Francis, J.M.; Parkinson, E.I.; Barger, A.M.; Hergenrother, P.J.; Fan, T.M. Utilizing feline oral squamous cell carcinoma patients to develop NQO1-targeted therapy. Neoplasia 2021, 23, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Hamada, M.; Inaba, H.; Nishiyama, K.; Yoshida, S.; Yura, Y.; Matsumoto-Nakano, M.; Uzawa, N. Prognostic association of starvation-induced gene expression in head and neck cancer. Sci. Rep. 2021, 11, 19130. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Du, J.N.; Shi, J.J.; Huang, Y.C.X.; Zhao, Y.; Ma, L. Ferroptosis-related gene ATG5 is a novel prognostic biomarker in nasopharyngeal carcinoma and head and neck squamous cell carcinoma. Front. Bioeng. Biotech. 2022, 10, 1006535. [Google Scholar] [CrossRef]
- Ren, Z.Y.; Zhang, L.; Ding, W.; Luo, Y.L.; Shi, Z.Q.; Shrestha, B.; Kan, X.; Zhang, Z.H.; Ding, J.; He, H.J.; et al. Development and validation of a novel survival model for head and neck squamous cell carcinoma based on autophagy-related genes. Genomics 2021, 113, 1166–1175. [Google Scholar] [CrossRef]
- He, J.; Ouyang, S.; Zhao, Y.; Liu, Y.; Liu, Y.; Zhou, B.; Man, Q.; Liu, B.; Wu, T. Prognostic Value of CAV1 and CAV2 in Head and Neck Squamous Cell Carcinoma. Biomolecules 2023, 13, 303. [Google Scholar] [CrossRef] [PubMed]
- Rice, S. Patterns of acquisition in the emerging mental lexicon: The case of to and for in English. Brain Lang. 1999, 68, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Flore, P.; Obert, P.; Courteix, D.; Lecoq, A.M.; Girodon, J.; Lidoreau, M.; Klein, P. Influence of a Biokinergia session on cardiorespiratory and metabolic adaptations of trained subjects. J. Manip. Physiol. Ther. 1998, 21, 621–628. [Google Scholar]
- Ramsay, L.E.; Freestone, S. Xipamide: No advantage over bendrofluazide in hypertension. Br. J. Clin. Pharmacol. 1984, 18, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, X.; Weng, Y.; Wei, J.; Chen, X.; Wang, P.; Wang, T.; Qin, J.; Peng, M. Application of an angiogenesis-related genes risk model in lung adenocarcinoma prognosis and immunotherapy. Front. Genet. 2023, 14, 1092968. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Y.; Li, Y.; Yang, X.; Han, M.; Fan, Q. Construction of a CCL20-centered circadian-signature based prognostic model in cervical cancer. Cancer Cell Int. 2023, 23, 92. [Google Scholar] [CrossRef]
- Liang, Y.; Tan, B.; Du, M.; Wang, B.; Gao, Y.; Wang, M. A tricarboxylic acid cycle-based machine learning model to select effective drug targets for the treatment of esophageal squamous cell carcinoma. Front. Pharmacol. 2023, 14, 1195195. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Zhong, M.; Yang, M.; Sun, X.; Liu, J.; Kroemer, G.; Lotze, M.; Zeh, H.J., 3rd; Kang, R.; et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology 2017, 153, 1429–1443 e1425. [Google Scholar] [CrossRef]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef]
- Donnella, H.J.; Webber, J.T.; Levin, R.S.; Camarda, R.; Momcilovic, O.; Bayani, N.; Shah, K.N.; Korkola, J.E.; Shokat, K.M.; Goga, A.; et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 2018, 14, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Wei, P.; Zhang, H.; Ding, Z.; Yang, L.; Huang, Z.; Zhang, N. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of beta-catenin/Wnt signaling. Mol. Cancer Res. 2013, 11, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Katsha, A.; Soutto, M.; Sehdev, V.; Peng, D.; Washington, M.K.; Piazuelo, M.B.; Tantawy, M.N.; Manning, H.C.; Lu, P.; Shyr, Y.; et al. Aurora kinase A promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology 2013, 145, 1312–1322 e8. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hu, J.; Xiong, X.; Chen, H.; Lin, B.; Chen, Y.; Li, Y.; Cheng, D.; Li, Z. AURKA inhibition induces Ewing’s sarcoma apoptosis and ferroptosis through NPM1/YAP1 axis. Cell Death Dis. 2024, 15, 99. [Google Scholar] [CrossRef]
- Li, L.; Gao, Q.; Wang, J.; Gu, L.; Li, Z.; Zhang, S.; Hu, C.; He, M.; Wang, Y.; Wang, Z.; et al. Induction of Ferroptosis by Ophiopogonin-B Through Regulating the Gene Signature AURKA in NSCLC. Front. Oncol. 2022, 12, 833814. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Chen, X.; Huang, F. Machine learning revealed ferroptosis features and ferroptosis-related gene-based immune microenvironment in lung adenocarcinoma. Chem. Biol. Interact. 2023, 378, 110471. [Google Scholar] [CrossRef] [PubMed]
- Yun, D.; Wang, X.; Wang, W.; Ren, X.; Li, J.; Wang, X.; Liang, J.; Liu, J.; Fan, J.; Ren, X.; et al. A Novel Prognostic Signature Based on Glioma Essential Ferroptosis-Related Genes Predicts Clinical Outcomes and Indicates Treatment in Glioma. Front. Oncol. 2022, 12, 897702. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Huang, X.; Li, Y.; Chen, Y.; Lin, L. m(6)A-related lncRNAs are potential biomarkers for predicting prognoses and immune responses in patients with LUAD. Mol. Ther. Nucleic Acids 2021, 24, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.K.; De, S. An assessment of computational methods for estimating purity and clonality using genomic data derived from heterogeneous tumor tissue samples. Brief. Bioinform. 2015, 16, 232–241. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Erratum to: Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 249. [Google Scholar] [CrossRef] [PubMed]
- Allgauer, M.; Budczies, J.; Christopoulos, P.; Endris, V.; Lier, A.; Rempel, E.; Volckmar, A.L.; Kirchner, M.; von Winterfeld, M.; Leichsenring, J.; et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl. Lung Cancer Res. 2018, 7, 703–715. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, X.; Tian, J.; Fu, Y.; Wang, Y.; Yang, Y.; Zhang, M.; Yang, C.; Liu, Y. Identification of AURKA as a Biomarker Associated with Cuproptosis and Ferroptosis in HNSCC. Int. J. Mol. Sci. 2024, 25, 4372. https://doi.org/10.3390/ijms25084372
Jia X, Tian J, Fu Y, Wang Y, Yang Y, Zhang M, Yang C, Liu Y. Identification of AURKA as a Biomarker Associated with Cuproptosis and Ferroptosis in HNSCC. International Journal of Molecular Sciences. 2024; 25(8):4372. https://doi.org/10.3390/ijms25084372
Chicago/Turabian StyleJia, Xiao, Jiao Tian, Yueyue Fu, Yiqi Wang, Yang Yang, Mengzhou Zhang, Cheng Yang, and Yijin Liu. 2024. "Identification of AURKA as a Biomarker Associated with Cuproptosis and Ferroptosis in HNSCC" International Journal of Molecular Sciences 25, no. 8: 4372. https://doi.org/10.3390/ijms25084372