
Novel Amidine Derivative K1586 Sensitizes Colorectal Cancer Cells to Ionizing Radiation by Inducing Chk1 Instability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
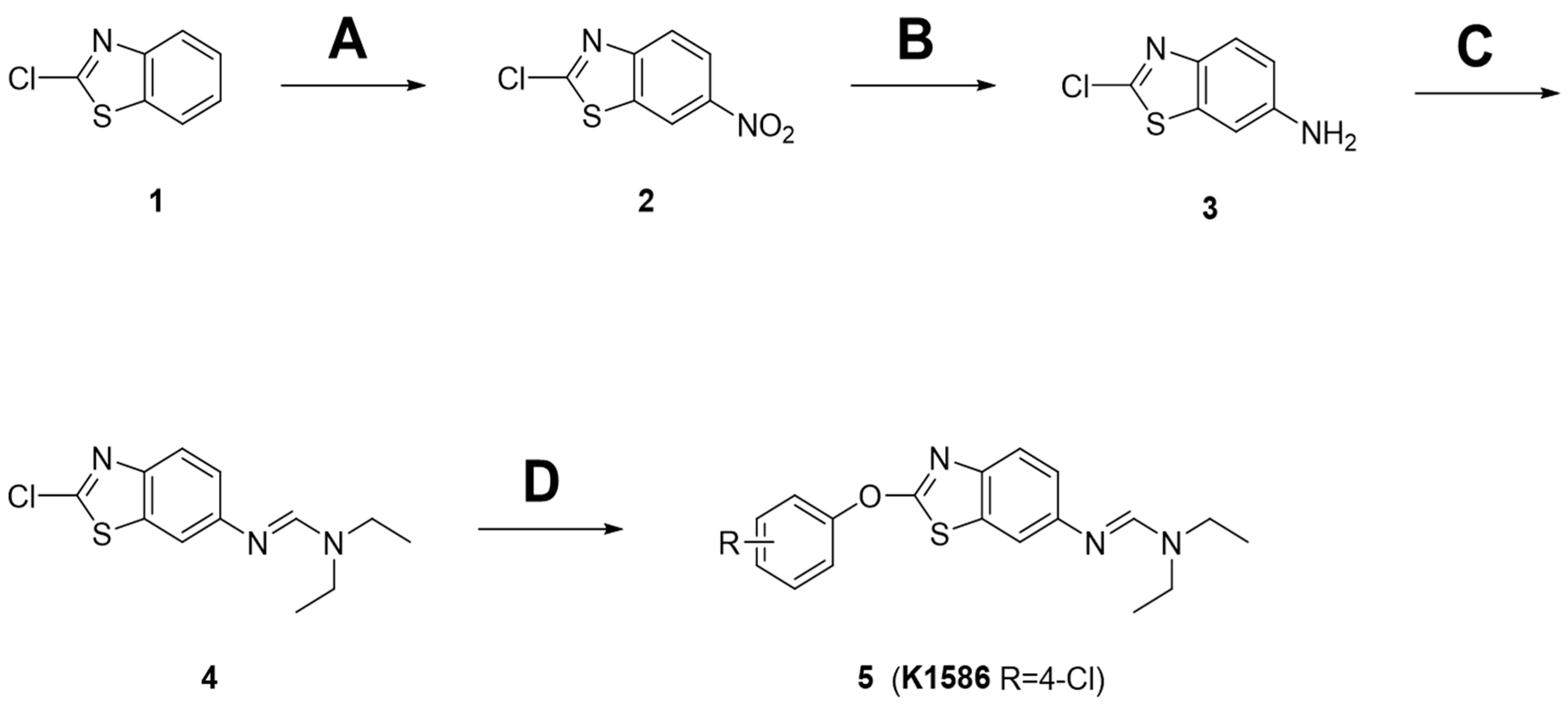
2.1. Preparation of Amidine Derivatives
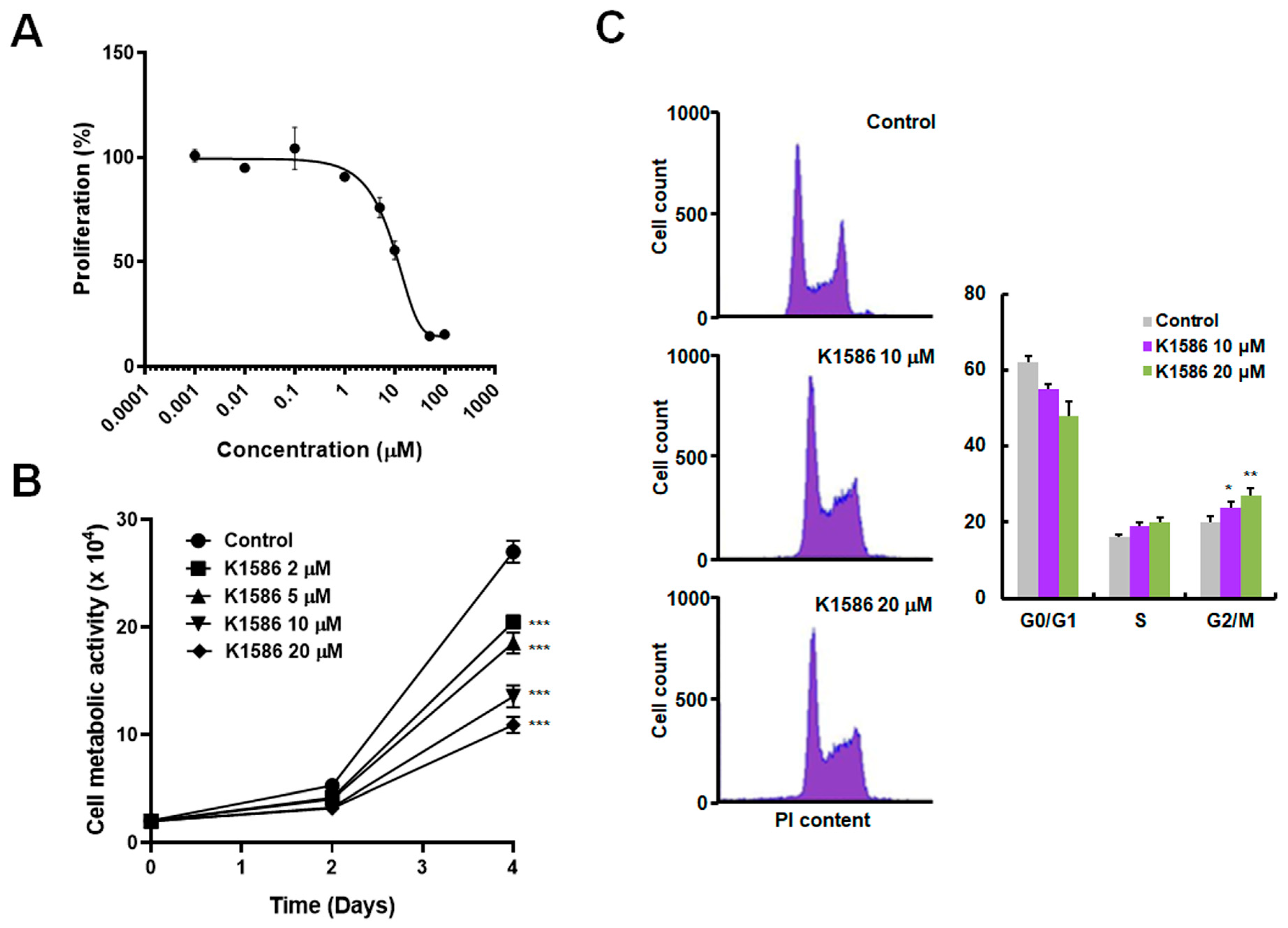
2.2. Anticancer Effects of an Amidine Derivative on Colorectal Cancer Cells
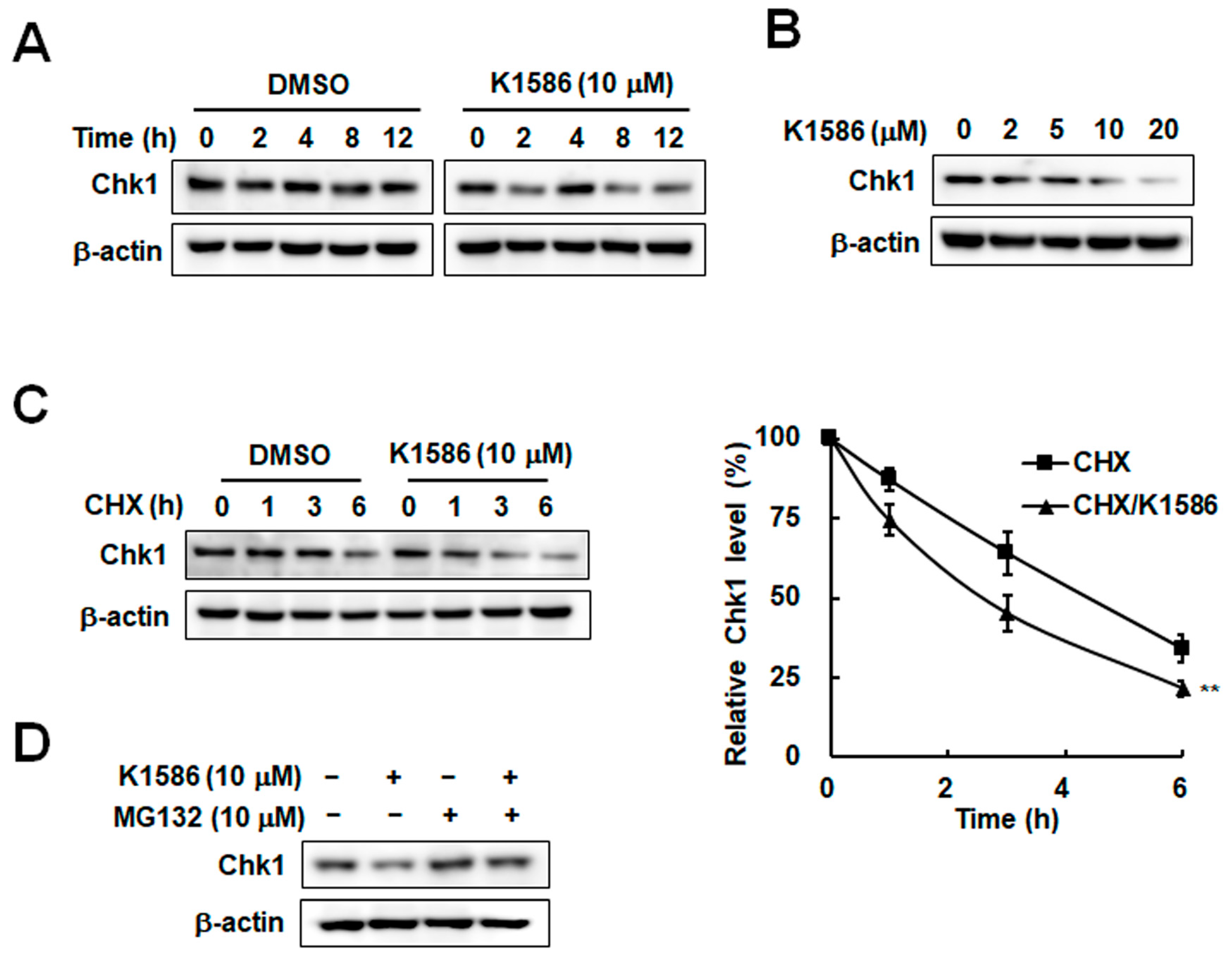
2.3. Chk1 Instability Due to an Amidine Derivative in Colorectal Cancer Cells
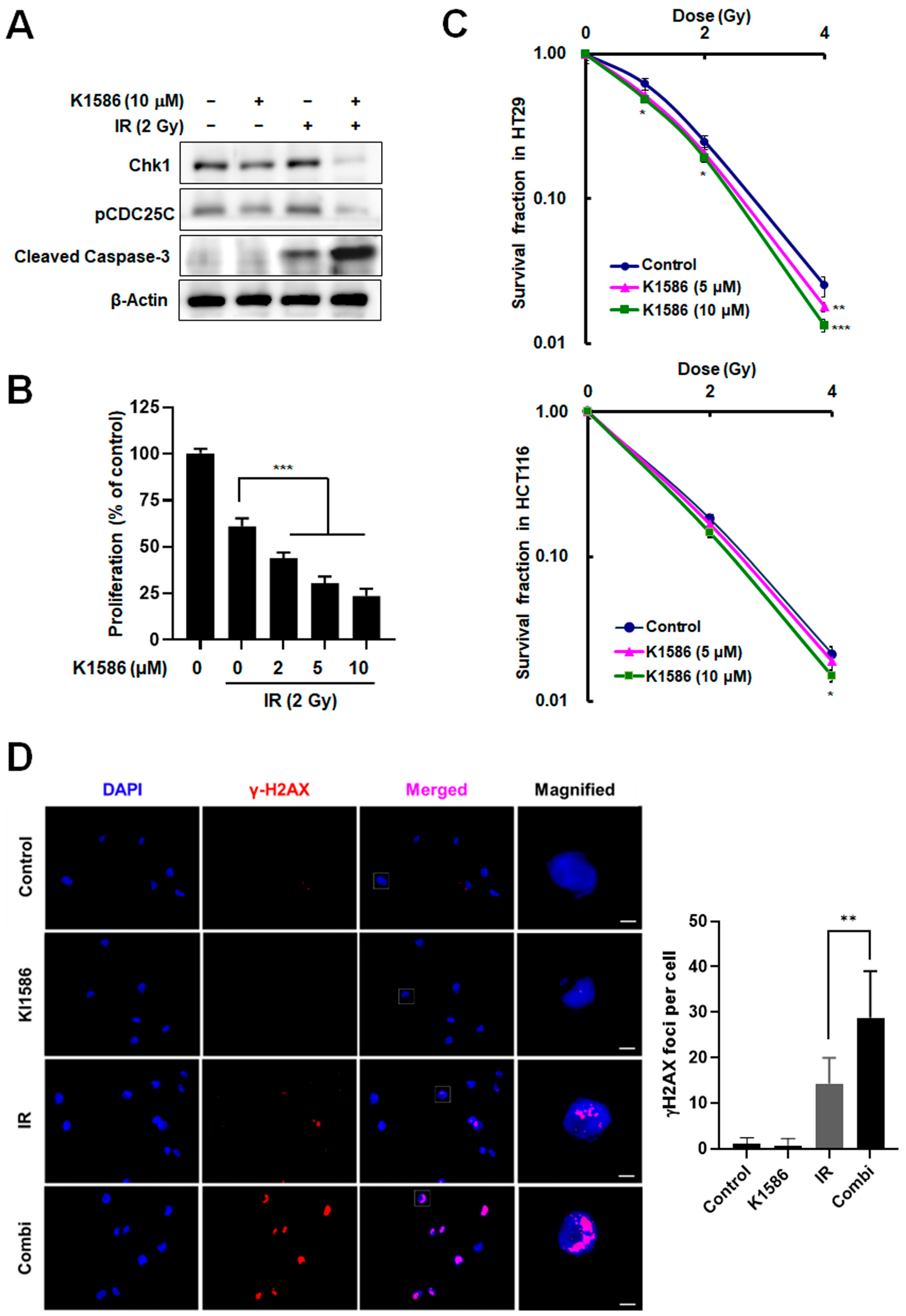
2.4. Induction of Radiosensitization by an Amidine Derivative via Chk1 Degradation
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. Cell Culture
4.3. MTT Assay
4.4. Cell Counting Assay
4.5. Clonogenic Cell Survival Assay
4.6. Protein Extraction and Western Blot Analysis
4.7. Fluorescence-Activated Cell Sorting (FACS) Analysis
4.8. γ-H2AX Staining
4.9. Statistical Analysis and Graphical Representation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petermann, E.; Maya-Mendoza, A.; Zachos, G.; Gillespie, D.A.; Jackson, D.A.; Caldecott, K.W. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol. Cell. Biol. 2006, 26, 3319–3326. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.F.; Walton, M.I.; Cherry, D.L.; Collins, I.; Clarke, P.A.; Garrett, M.D.; Workman, P. CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells. Cancer Res. 2020, 80, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N.J. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, S.; Parrino, B.; Gaggianesi, M.; Pantina, V.D.; Bianca, P.; Nicotra, A.; Mangiapane, L.R.; Lo Iacono, M.; Ganduscio, G.; Veschi, V.; et al. CHK1 inhibitor sensitizes resistant colorectal cancer stem cells to nortopsentin. iScience 2021, 24, 102664. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Diaz, H.; Barnard, D.; Barda, D.; Clawson, D.; Blosser, W.; Cox, K.; Guo, S.; Marshall, M. Characterization and preclinical development of LY2603618: A selective and potent Chk1 inhibitor. Investig. New Drugs 2014, 32, 213–226. [Google Scholar] [CrossRef]
- van Harten, A.M.; Buijze, M.; van der Mast, R.; Rooimans, M.A.; Martens-de Kemp, S.R.; Bachas, C.; Brink, A.; Stigter-van Walsum, M.; Wolthuis, R.M.F.; Brakenhoff, R.H. Targeting the cell cycle in head and neck cancer by Chk1 inhibition: A novel concept of bimodal cell death. Oncogenesis 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Patties, I.; Kallendrusch, S.; Bohme, L.; Kendzia, E.; Oppermann, H.; Gaunitz, F.; Kortmann, R.D.; Glasow, A. The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide, and to decitabine treatment. J. Exp. Clin. Cancer Res. 2019, 38, 420. [Google Scholar] [CrossRef]
- Mattiello, L.; Soliman Abdel Rehim, S.; Musella, M.; Sistigu, A.; Guarracino, A.; Vitale, S.; Corradi, F.; Galassi, C.; Sperati, F.; Manic, G.; et al. The Targeting of MRE11 or RAD51 Sensitizes Colorectal Cancer Stem Cells to CHK1 Inhibition. Cancers 2021, 13, 1957. [Google Scholar] [CrossRef]
- Asif, M.; Srivastava, R.; Fatima, A.; Shakeel, M.; Hassan, F.; Nasibullah, M. A Perspective of the Amide Group Containing FDA Approved Anticancer Drugs from 2021–2022 (A Review). Russ. J. Bioorg. Chem. 2023, 49, 1165–1176. [Google Scholar] [CrossRef]
- Arya, S.; Kumar, N.; Roy, P.; Sondhi, S.M. Synthesis of amidine and bis amidine derivatives and their evaluation for anti-inflammatory and anticancer activity. Eur. J. Med. Chem. 2013, 59, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Carrion, M.D.; Rubio-Ruiz, B.; Franco-Montalban, F.; Amoia, P.; Zuccarini, M.C.; De Simone, C.; Camacho, M.E.; Amoroso, R.; Maccallini, C. New amidine-benzenesulfonamides as iNOS inhibitors for the therapy of the triple negative breast cancer. Eur. J. Med. Chem. 2023, 248, 115112. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, S.M.; Rani, R.; Gupta, P.P.; Agrawal, S.K.; Saxena, A.K. Synthesis, anticancer, and anti-inflammatory activity evaluation of methanesulfonamide and amidine derivatives of 3,4-diaryl-2-imino-4-thiazolines. Mol. Divers. 2009, 13, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Mazur, S.; Tanious, F.A.; Ding, D.; Kumar, A.; Boykin, D.W.; Simpson, I.J.; Neidle, S.; Wilson, W.D. A thermodynamic and structural analysis of DNA minor-groove complex formation. J. Mol. Biol. 2000, 300, 321–337. [Google Scholar] [CrossRef] [PubMed]
- White, E.W.; Tanious, F.; Ismail, M.A.; Reszka, A.P.; Neidle, S.; Boykin, D.W.; Wilson, W.D. Structure-specific recognition of quadruplex DNA by organic cations: Influence of shape, substituents and charge. Biophys. Chem. 2007, 126, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kumar, A.; Depauw, S.; Nhili, R.; David-Cordonnier, M.H.; Lee, M.P.; Ismail, M.A.; Farahat, A.A.; Say, M.; Chackal-Catoen, S.; et al. Water-mediated binding of agents that target the DNA minor groove. J. Am. Chem. Soc. 2011, 133, 10171–10183. [Google Scholar] [CrossRef] [PubMed]
- Bishop, E.P.; Rohs, R.; Parker, S.C.; West, S.M.; Liu, P.; Mann, R.S.; Honig, B.; Tullius, T.D. A map of minor groove shape and electrostatic potential from hydroxyl radical cleavage patterns of DNA. ACS Chem. Biol. 2011, 6, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.A.; Arafa, R.K.; Youssef, M.M.; El-Sayed, W.M. Anticancer, antioxidant activities, and DNA affinity of novel monocationic bithiophenes and analogues. Drug Des. Dev. Ther. 2014, 8, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef]
- Leung-Pineda, V.; Huh, J.; Piwnica-Worms, H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 2009, 69, 2630–2637. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Otterness, D.M.; Chiang, G.G.; Xie, W.; Liu, Y.C.; Mercurio, F.; Abraham, R.T. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 2005, 19, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Hu, K.; Yuan, Y.; Sang, Y.; Bu, Q.; Chen, G.; Yang, L.; Li, B.; Huang, P.; Chen, D.; et al. CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J. Clin. Investig. 2012, 122, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Toouli, C.D.; Fujii, G.H.; Crain, C.; Parry, D. Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle 2005, 4, 131–139. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Wang, F.; Wang, X.; Cheng, B.; Ye, F.; Xie, X.; Zhou, C.; Lu, W. Suppressed miR-424 expression via upregulation of target gene Chk1 contributes to the progression of cervical cancer. Oncogene 2013, 32, 976–987. [Google Scholar] [CrossRef]
- Lundgren, K.; Holm, K.; Nordenskjold, B.; Borg, A.; Landberg, G. Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res. 2008, 10, R81. [Google Scholar] [CrossRef]
- Roe, O.D.; Szulkin, A.; Anderssen, E.; Flatberg, A.; Sandeck, H.; Amundsen, T.; Erlandsen, S.E.; Dobra, K.; Sundstrom, S.H. Molecular resistance fingerprint of pemetrexed and platinum in a long-term survivor of mesothelioma. PLoS ONE 2012, 7, e40521. [Google Scholar] [CrossRef]
- Zhou, Z.R.; Yang, Z.Z.; Wang, S.J.; Zhang, L.; Luo, J.R.; Feng, Y.; Yu, X.L.; Chen, X.X.; Guo, X.M. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Yin, Y.; Dou, X.; Duan, S.; Zhang, L.; Xu, Q.; Li, H.; Li, D. Downregulation of cell division cycle 25 homolog C reduces the radiosensitivity and proliferation activity of esophageal squamous cell carcinoma. Gene 2016, 590, 244–249. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Ma, C.X.; Cai, S.; Li, S.; Ryan, C.E.; Guo, Z.; Schaiff, W.T.; Lin, L.; Hoog, J.; Goiffon, R.J.; Prat, A.; et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Investig. 2012, 122, 1541–1552. [Google Scholar] [CrossRef]
- Kong, X.; Yu, D.; Wang, Z.; Li, S. Relationship between p53 status and the bioeffect of ionizing radiation. Oncol. Lett. 2021, 22, 661. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Choi, K.J.; Bae, S. Interferon-gamma enhances radiation-induced cell death via downregulation of Chk1. Cancer Biol. Ther. 2012, 13, 1018–1025. [Google Scholar] [CrossRef]
- Kim, K.S.; Heo, J.I.; Choi, K.J.; Bae, S. Enhancement of cellular radiation sensitivity through degradation of Chk1 by the XIAP-XAF1 complex. Cancer Biol. Ther. 2014, 15, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes. Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef]
- Michael, W.M.; Ott, R.; Fanning, E.; Newport, J. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science 2000, 289, 2133–2137. [Google Scholar] [CrossRef]
- Taricani, L.; Shanahan, F.; Parry, D. Replication stress activates DNA polymerase alpha-associated Chk1. Cell Cycle 2009, 8, 482–489. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.S.; Park, J.-E.; Lee, W.H.; Kwon, Y.B.; Seu, Y.-B.; Kim, K.S. Novel Amidine Derivative K1586 Sensitizes Colorectal Cancer Cells to Ionizing Radiation by Inducing Chk1 Instability. Int. J. Mol. Sci. 2024, 25, 4396. https://doi.org/10.3390/ijms25084396
Kim HS, Park J-E, Lee WH, Kwon YB, Seu Y-B, Kim KS. Novel Amidine Derivative K1586 Sensitizes Colorectal Cancer Cells to Ionizing Radiation by Inducing Chk1 Instability. International Journal of Molecular Sciences. 2024; 25(8):4396. https://doi.org/10.3390/ijms25084396
Chicago/Turabian StyleKim, Hang Soo, Ji-Eun Park, Won Hyung Lee, Young Bin Kwon, Young-Bae Seu, and Kwang Seok Kim. 2024. "Novel Amidine Derivative K1586 Sensitizes Colorectal Cancer Cells to Ionizing Radiation by Inducing Chk1 Instability" International Journal of Molecular Sciences 25, no. 8: 4396. https://doi.org/10.3390/ijms25084396