
The Biologic IRL201805 Alters Immune Tolerance Leading to Prolonged Pharmacodynamics and Efficacy in Rheumatoid Arthritis Patients

 ,
, 
Abstract
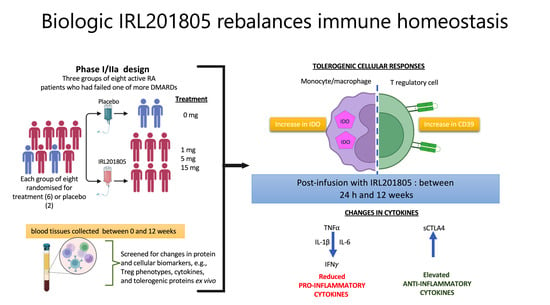
1. Introduction
2. Results
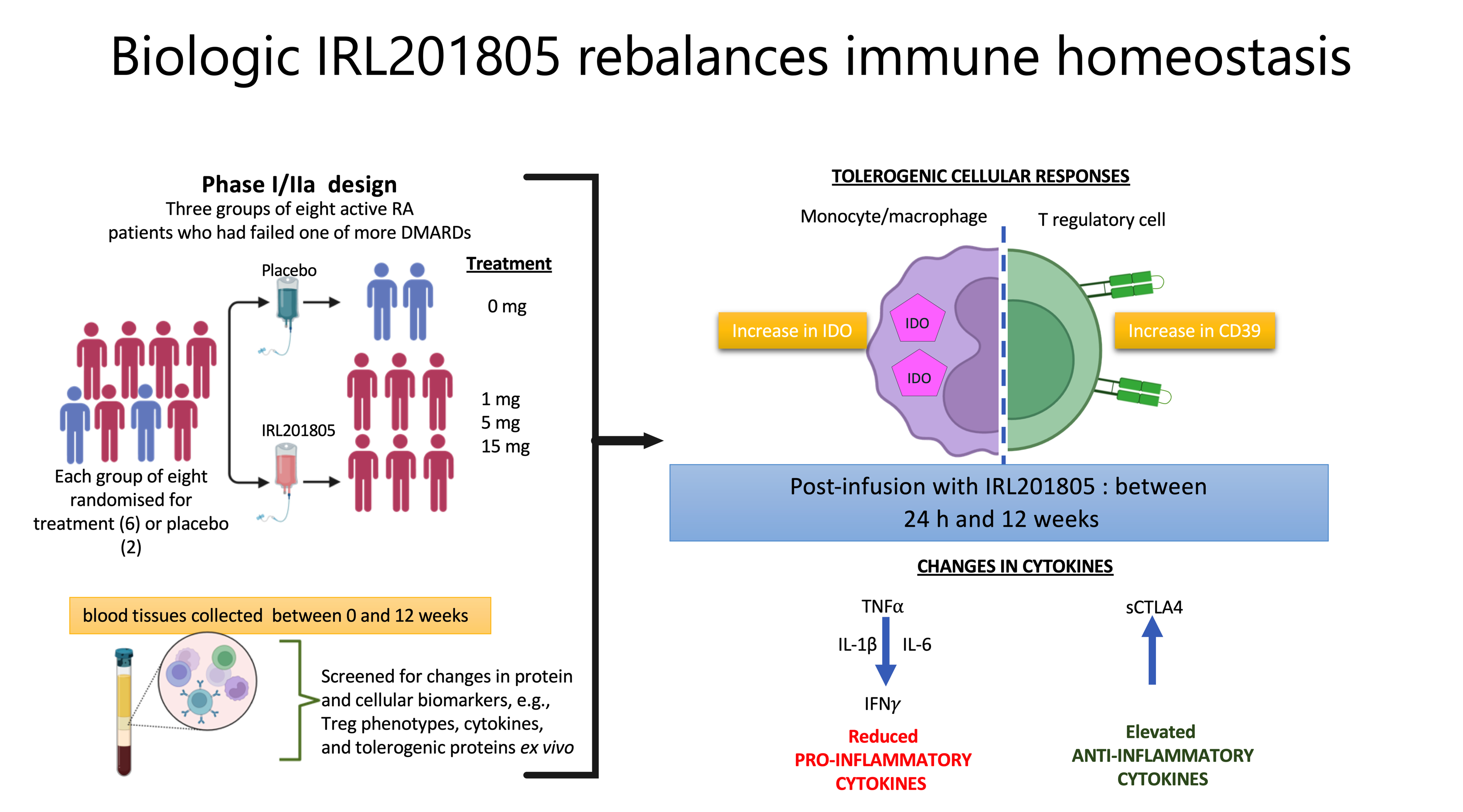
2.1. Disease Activity as Estimated using DAS28 Was Consistently Reduced in IRL201805 Responders
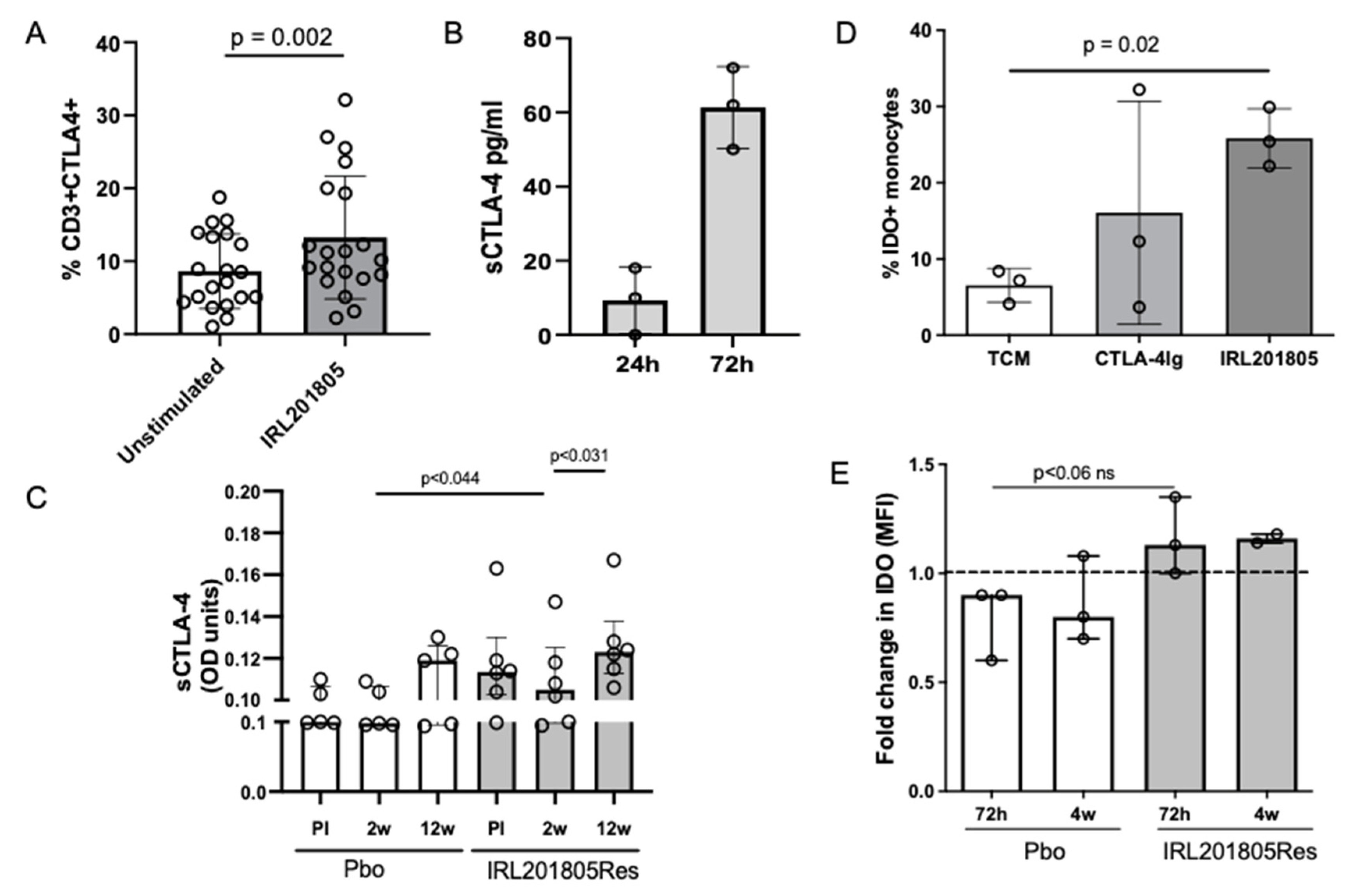
2.2. Serum Endogenous BiP/GRP78 and IRL201805 Concentrations Were Higher in IRL201805 Responders
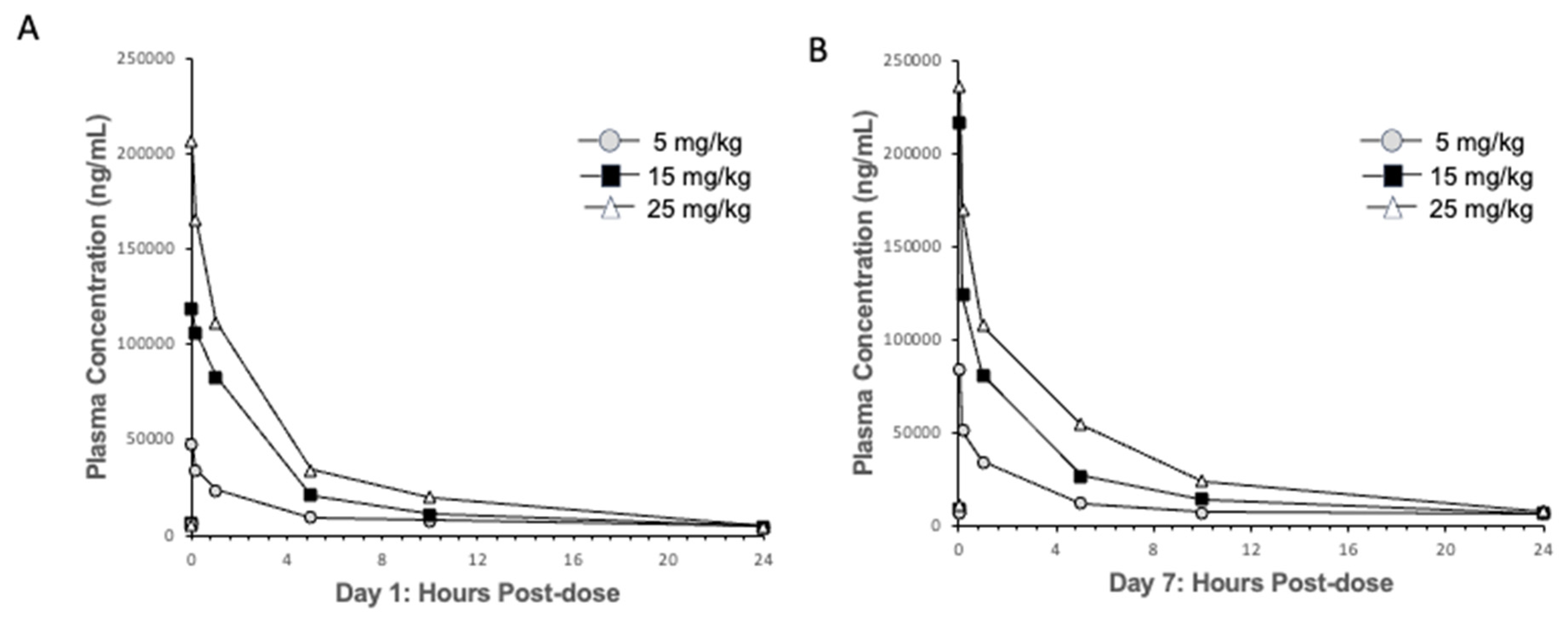
2.3. Pharmacokinetic–Pharmacodynamic (PKPD) Analysis of IRL2028 in Mice
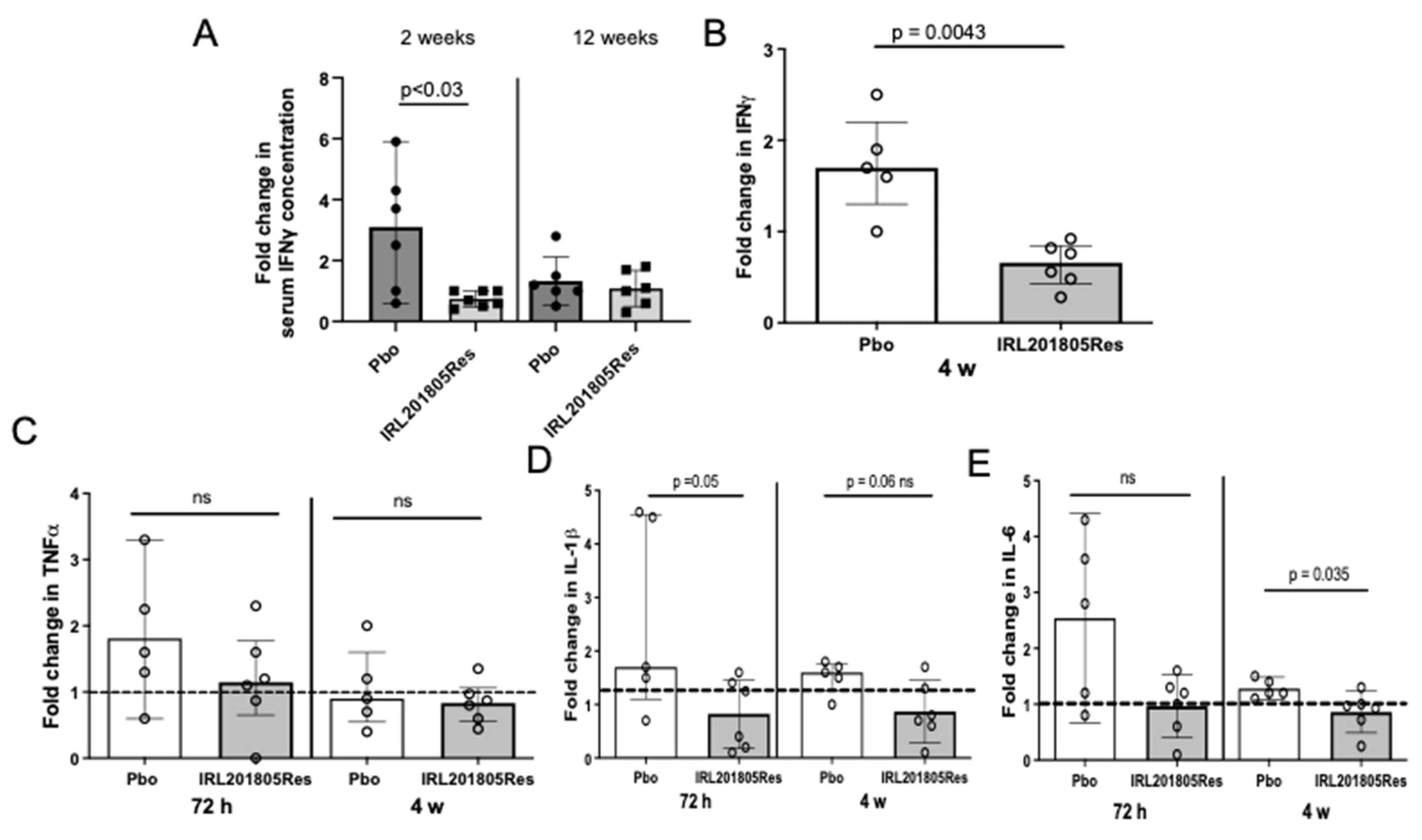
2.4. Detection of Cytokine Changes Post ILR201805 Treatment
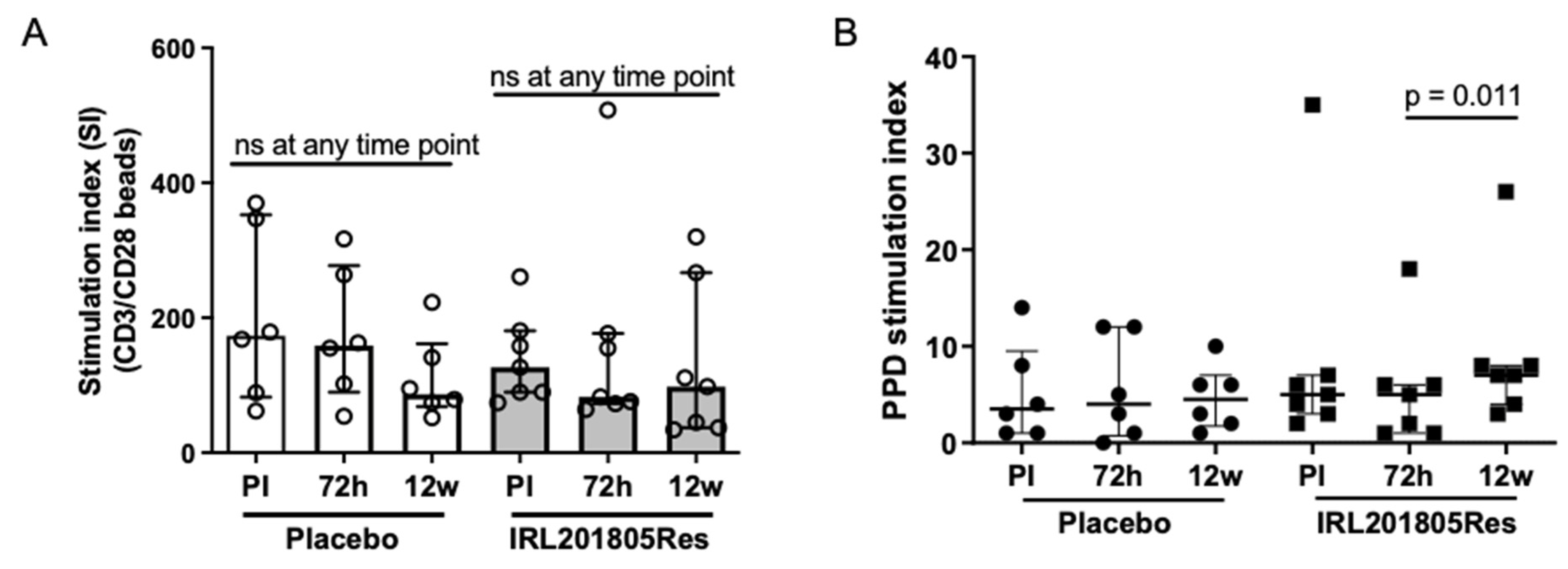
2.5. IRL201805 Is Not a General Immunosuppressive
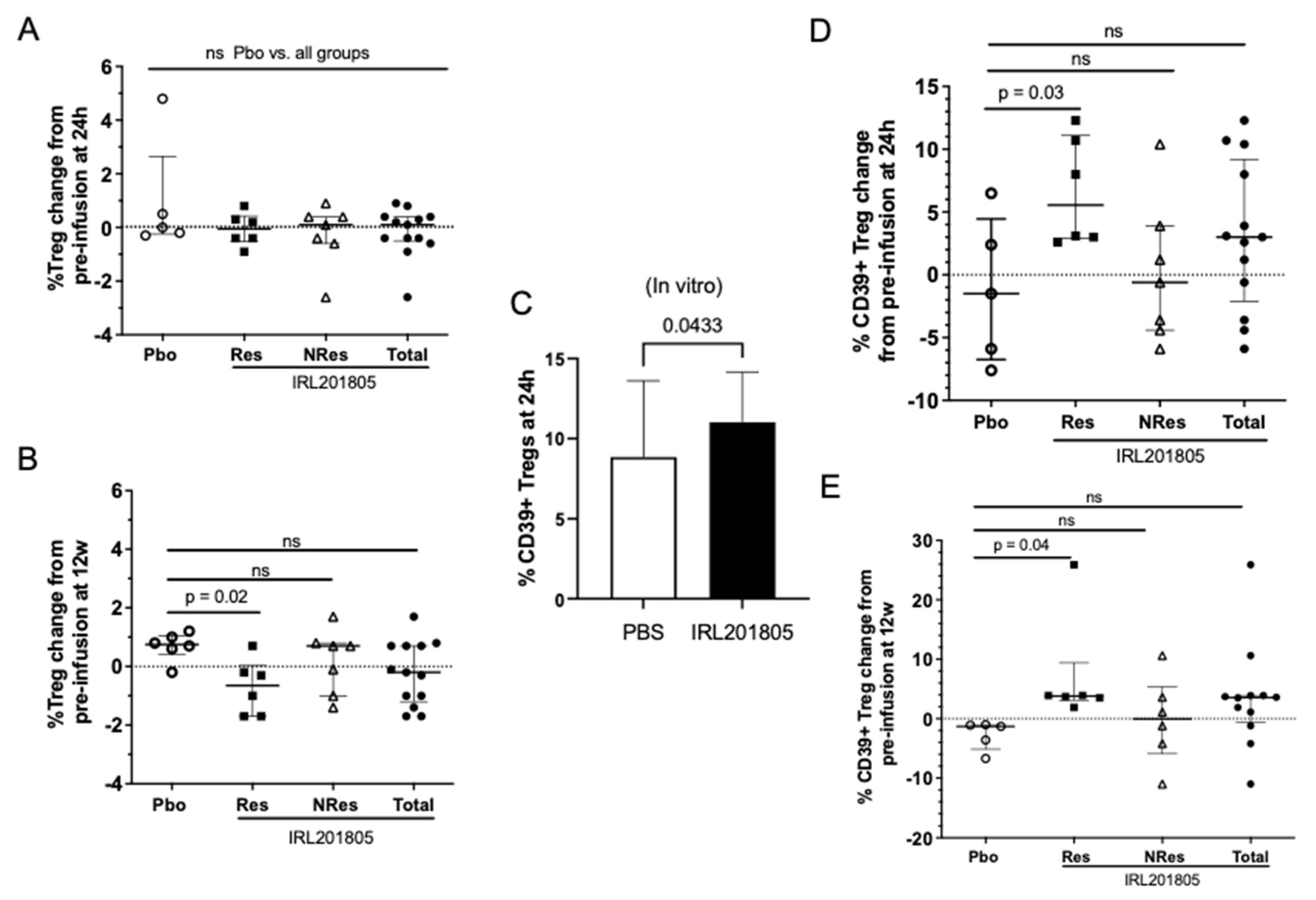
2.6. Frequency of Phenotype Changes of Regulatory T Cells
2.7. IRL201805Res Treatment Confers an Altered Cytokine Profile
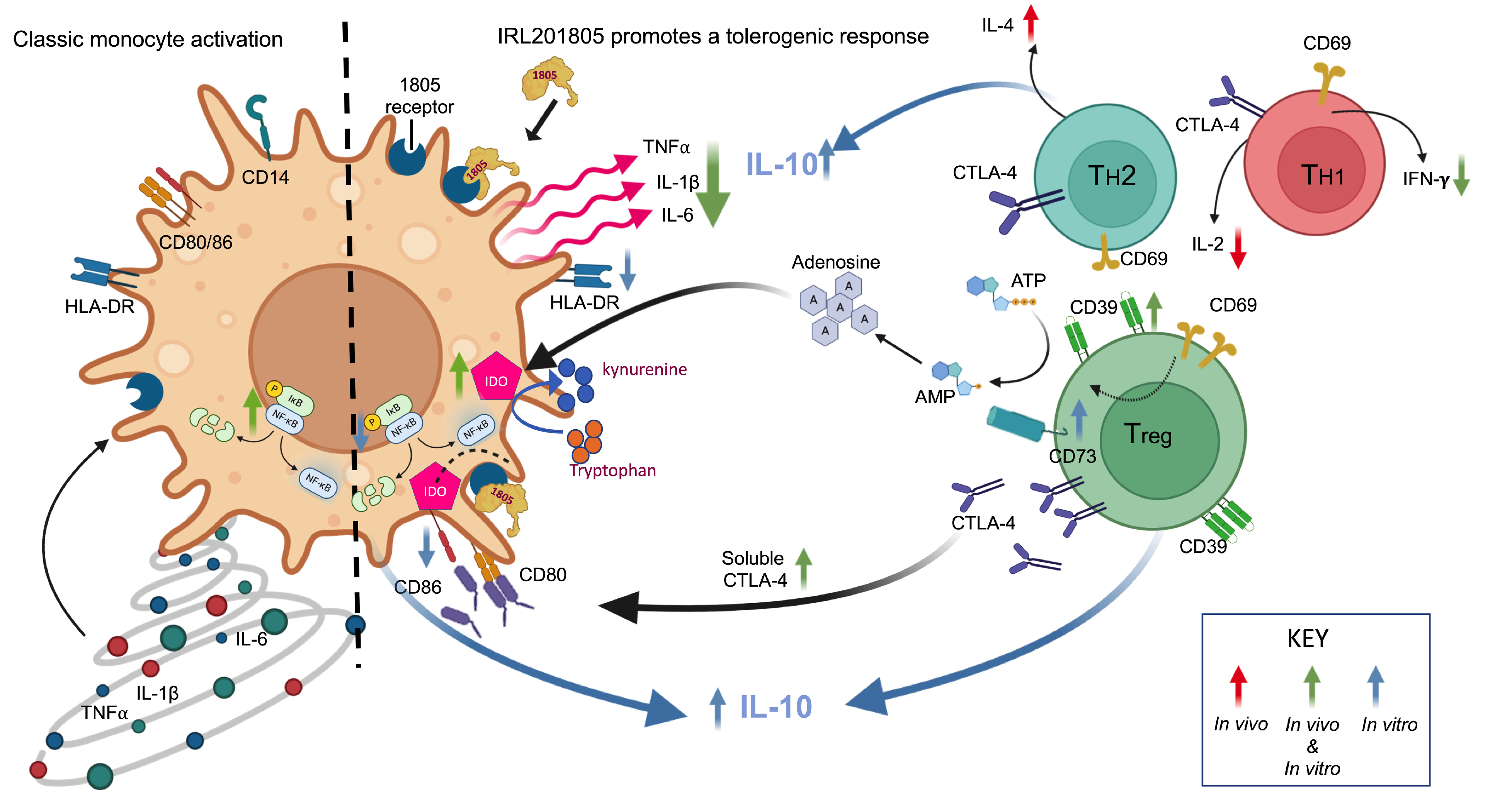
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Study Design and Patient’s Characteristics
4.3. Cell Culture
4.4. Cytokines Production and Detection
4.5. Cell Surface and Intracellular Flow Cytometry Analysis
4.6. Ex Vivo Assessment of Patient Lymphocytes Retention of Antigen-Induced Proliferation and T-Cell Activation
4.7. Pharmacokinetic Determination
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quan, L.D.; Thiele, G.M.; Tian, J.; Wang, D. The Development of Novel Therapies for Rheumatoid Arthritis. Expert Opin. Ther. Pat. 2008, 18, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Choe, Y.H.; Lee, S.I. Lessons From the Success and Failure of Targeted Drugs for Rheumatoid Arthritis: Perspectives for Effective Basic and Translational Research. Immune Netw. 2022, 22, e8. [Google Scholar] [CrossRef]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Eggleton, P.; De Alba, J.; Weinreich, M.; Calias, P.; Foulkes, R.; Corrigall, V.M. The therapeutic mavericks: Potent immunomodulating chaperones capable of treating human diseases. J. Cell Mol. Med. 2023, 27, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, R.J.; Myers, L.K.; Wooley, P.H.; Corrigall, V.M.; Bodman-Smith, M.D.; Panayi, G.S.; Thompson, S.J. Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4-producing regulatory T cells: A novel function for an ancient protein. Arthritis Rheum. 2006, 54, 854–863. [Google Scholar] [CrossRef]
- Corrigall, V.M.; Bodman-Smith, M.D.; Fife, M.S.; Canas, B.; Myers, L.K.; Wooley, P.; Soh, C.; Staines, N.A.; Pappin, D.J.; Berlo, S.E.; et al. The human endoplasmic reticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis and prevents the induction of experimental arthritis. J. Immunol. 2001, 166, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, B.; Chaabo, K.; Hall, C.; Garrood, T.; Mant, T.; Allen, E.; Vincent, A.; Vasconcelos, J.C.; Prevost, A.T.; Panayi, G.S.; et al. Safety and patient response as indicated by biomarker changes to binding immunoglobulin protein in the phase I/IIA RAGULA clinical trial in rheumatoid arthritis. Rheumatology 2016, 55, 1993–2000. [Google Scholar] [CrossRef]
- Mian, A.N.; Ibrahim, F.; Scott, D.L.; Galloway, J.; TITRATE Study Group. Optimal responses in disease activity scores to treatment in rheumatoid arthritis: Is a DAS28 reduction of >1.2 sufficient? Arthritis Res. Ther. 2016, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Morgacheva, O.; Messing, S.P.; Kremer, J.M.; Greenberg, J.D.; Reed, G.W.; Gravallese, E.M.; Furst, D.E. Clinical disease activity and acute phase reactant levels are discordant among patients with active rheumatoid arthritis: Acute phase reactant levels contribute separately to predicting outcome at one year. Arthritis Res. Ther. 2014, 16, R40. [Google Scholar] [CrossRef]
- Meyer, P.W.; Hodkinson, B.; Ally, M.; Musenge, E.; Wadee, A.A.; Fickl, H.; Tikly, M.; Anderson, R. Circulating cytokine profiles and their relationships with autoantibodies, acute phase reactants, and disease activity in patients with rheumatoid arthritis. Mediat. Inflamm. 2010, 2010, 158514. [Google Scholar] [CrossRef]
- Tukaj, S.; Kotlarz, A.; Jozwik, A.; Smolenska, Z.; Bryl, E.; Witkowski, J.M.; Lipinska, B. Cytokines of the Th1 and Th2 type in sera of rheumatoid arthritis patients; correlations with anti-Hsp40 immune response and diagnostic markers. Acta Biochim. Pol. 2010, 57, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Deeks, E.D. Abatacept: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Massalska, M.; Ciechomska, M.; Kuca-Warnawin, E.; Burakowski, T.; Kornatka, A.; Radzikowska, A.; Pawlak, D.; Muz, B.; Loniewska-Lwowska, A.; Palucha, A.; et al. Effectiveness of Soluble CTLA-4-Fc in the Inhibition of Bone Marrow T-Cell Activation in Context of Indoleamine 2.3-Dioxygenase (IDO) and CD4+Foxp3+ Treg Induction. J. Inflamm. Res. 2022, 15, 6813–6829. [Google Scholar] [CrossRef] [PubMed]
- Sucher, R.; Fischler, K.; Oberhuber, R.; Kronberger, I.; Margreiter, C.; Ollinger, R.; Schneeberger, S.; Fuchs, D.; Werner, E.R.; Watschinger, K.; et al. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J. Immunol. 2012, 188, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Timperi, E.; Barnaba, V. CD39 Regulation and Functions in T Cells. Int. J. Mol. Sci. 2021, 22, 8068. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.S.; Lee, H.M.; Lio, C.W. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 2012, 12, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Corrigall, V.M.; Bodman-Smith, M.D.; Brunst, M.; Cornell, H.; Panayi, G.S. Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: Relevance to the treatment of inflammatory arthritis. Arthritis Rheum. 2004, 50, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Ochiai, A.; Matsuno, H.; Panayi, G.S.; Corrigall, V.M. Binding immunoglobulin protein resolves rheumatoid synovitis: A xenogeneic study using rheumatoid arthritis synovial membrane transplants in SCID mice. Arthritis Res. Ther. 2011, 13, R149. [Google Scholar] [CrossRef]
- Cacciapaglia, F.; Buzzulini, F.; Arcarese, L.; Ferraro, E.; Afeltra, A. The use of an interferon-gamma release assay as a biomarker of response to anti-TNF-alpha treatment. Drug Dev. Res. 2014, 75 (Suppl. S1), S50–S53. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Walter, G.J.; Fleskens, V.; Frederiksen, K.S.; Rajasekhar, M.; Menon, B.; Gerwien, J.G.; Evans, H.G.; Taams, L.S. Phenotypic, Functional, and Gene Expression Profiling of Peripheral CD45RA+ and CD45RO+ CD4+CD25+CD127(low) Treg Cells in Patients With Chronic Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Herrath, J.; Chemin, K.; Albrecht, I.; Catrina, A.I.; Malmstrom, V. Surface expression of CD39 identifies an enriched Treg-cell subset in the rheumatic joint, which does not suppress IL-17A secretion. Eur. J. Immunol. 2014, 44, 2979–2989. [Google Scholar] [CrossRef]
- Peres, R.S.; Liew, F.Y.; Talbot, J.; Carregaro, V.; Oliveira, R.D.; Almeida, S.L.; Franca, R.F.; Donate, P.B.; Pinto, L.G.; Ferreira, F.I.; et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 2509–2514. [Google Scholar] [CrossRef]
- Plenge, R.M.; Padyukov, L.; Remmers, E.F.; Purcell, S.; Lee, A.T.; Karlson, E.W.; Wolfe, F.; Kastner, D.L.; Alfredsson, L.; Altshuler, D.; et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4000 samples from North America and Sweden: Association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005, 77, 1044–1060. [Google Scholar] [CrossRef]
- Devarajan, P.; Miska, J.; Lui, J.B.; Swieboda, D.; Chen, Z. Opposing effects of CTLA4 insufficiency on regulatory versus conventional T cells in autoimmunity converge on effector memory in target tissue. J. Immunol. 2014, 193, 4368–4380. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Jury, E.C.; Mauri, C.; Ehrenstein, M.R. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2008, 105, 19396–19401. [Google Scholar] [CrossRef]
- Picchianti Diamanti, A.; Rosado, M.M.; Scarsella, M.; Germano, V.; Giorda, E.; Cascioli, S.; Lagana, B.; D’Amelio, R.; Carsetti, R. Abatacept (cytotoxic T lymphocyte antigen 4-immunoglobulin) improves B cell function and regulatory T cell inhibitory capacity in rheumatoid arthritis patients non-responding to anti-tumour necrosis factor-alpha agents. Clin. Exp. Immunol. 2014, 177, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicz, A.; Boasso, A.; Edghill-Smith, Y.; Vaccari, M.; Fuchs, D.; Venzon, D.; Nacsa, J.; Betts, M.R.; Tsai, W.P.; Heraud, J.M.; et al. CTLA-4 blockade decreases TGF-beta, IDO, and viral RNA expression in tissues of SIVmac251-infected macaques. Blood 2006, 108, 3834–3842. [Google Scholar] [CrossRef]
- Ogbechi, J.; Clanchy, F.I.; Huang, Y.S.; Topping, L.M.; Stone, T.W.; Williams, R.O. IDO activation, inflammation and musculoskeletal disease. Exp. Gerontol. 2020, 131, 110820. [Google Scholar] [CrossRef]
- Bodman-Smith, M.D.; Corrigall, V.M.; Berglin, E.; Cornell, H.R.; Tzioufas, A.G.; Mavragani, C.P.; Chan, C.; Rantapaa-Dahlqvist, S.; Panayi, G.S. Antibody response to the human stress protein BiP in rheumatoid arthritis. Rheumatology 2004, 43, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Bodman-Smith, M.D.; Corrigall, V.M.; Kemeny, D.M.; Panayi, G.S. BiP, a putative autoantigen in rheumatoid arthritis, stimulates IL-10-producing CD8-positive T cells from normal individuals. Rheumatology 2003, 42, 637–644. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Corrigall, V.M.; Vittecoq, O.; Panayi, G.S. Binding immunoglobulin protein-treated peripheral blood monocyte-derived dendritic cells are refractory to maturation and induce regulatory T-cell development. Immunology 2009, 128, 218–226. [Google Scholar] [CrossRef]
- Bell, G.M.; Anderson, A.E.; Diboll, J.; Reece, R.; Eltherington, O.; Harry, R.A.; Fouweather, T.; MacDonald, C.; Chadwick, T.; McColl, E.; et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 2017, 76, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, P.A.; Schinnerling, K.; Sepulveda-Gutierrez, A.; Maggi, J.; Hoyos, L.; Morales, R.A.; Mehdi, A.M.; Nel, H.J.; Soto, L.; Pesce, B.; et al. Treatment with Dexamethasone and Monophosphoryl Lipid A Removes Disease-Associated Transcriptional Signatures in Monocyte-Derived Dendritic Cells from Rheumatoid Arthritis Patients and Confers Tolerogenic Features. Front. Immunol. 2016, 7, 458. [Google Scholar] [CrossRef]
- van Herwijnen, M.J.; Wieten, L.; van der Zee, R.; van Kooten, P.J.; Wagenaar-Hilbers, J.P.; Hoek, A.; den Braber, I.; Anderton, S.M.; Singh, M.; Meiring, H.D.; et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 14134–14139. [Google Scholar] [CrossRef] [PubMed]
- Bechman, K.; Yates, M.; Norton, S.; Cope, A.P.; Galloway, J.B. Placebo Response in Rheumatoid Arthritis Clinical Trials. J. Rheumatol. 2020, 47, 28–34. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Hall, C.; McGowan, N.W.A.; Babb, R.; Devlia, V.; Lucas, S.; Meghji, S.; Henderson, B.; Bozec, A.; Schett, G.; et al. Binding Immunoglobulin Protein (BIP) Inhibits TNF-alpha-Induced Osteoclast Differentiation and Systemic Bone Loss in an Erosive Arthritis Model. ACR Open Rheumatol. 2019, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Woolcock, K.; Corrigall, V.; Ravanetti, L.; De Alba, J.; Foulkes, R.; Eggleton, P.; Goodyear, C. Biologic IRL201805 Drives Differential Cell-contact and Metabolism Transcriptional Profiles in Monocytes from RA Patients Compared to Healthy Donors. Arthritis Rheumatol. 2023, 75 (Suppl. S9), a2149. [Google Scholar]
- Yamamura, Y.; Corrigall, V.; Ravanetti, L.; De Alba, J.; Foulkes, R.; Eggleton, P.; Goodyear, C. Novel Biologic IRL201805 Inhibits Osteoclastogenesis in Monocytic Cells from RA Patients. Arthritis Rheumatol. 2023, 75 (Suppl. S9), a2150. [Google Scholar]
- Shields, A.M.; Panayi, G.S.; Corrigall, V.M. A New-Age for Biologic Therapies: Long-Term Drug-Free Therapy with BiP? Front. Immunol. 2012, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Stack, M.E.; Mishra, S.; Parimala Chelvi Ratnamani, M.; Wang, H.; Gold, L.I.; Wang, H. Biomimetic Extracellular Matrix Nanofibers Electrospun with Calreticulin Promote Synergistic Activity for Tissue Regeneration. ACS Appl. Mater. Interfaces 2022, 14, 51683–51696. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhao, R.; Fan, K.; Iwanowycz, S.; Fan, H.; Li, Z.; Liu, B. Regulation of dendritic cell function improves survival in experimental sepsis through immune chaperone. Innate Immun. 2019, 25, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Riffo-Vasquez, Y.; Kanabar, V.; Keir, S.D.; RR, E.L.; Man, F.; Jackson, D.J.; Corrigall, V.; Coates, A.R.M.; Page, C.P. Modulation of allergic inflammation in the lung by a peptide derived from Mycobacteria tuberculosis chaperonin 60.1. Clin. Exp. Allergy 2020, 50, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Mulyani, W.R.W.; Sanjiwani, M.I.D.; Sandra; Prabawa, I.P.Y.; Lestari, A.A.W.; Wihandani, D.M.; Suastika, K.; Saraswati, M.R.; Bhargah, A.; Manuaba, I. Chaperone-Based Therapeutic Target Innovation: Heat Shock Protein 70 (HSP70) for Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Noori, L.; Saqagandomabadi, V.; Di Felice, V.; David, S.; Caruso Bavisotto, C.; Bucchieri, F.; Cappello, F.; Conway de Macario, E.; Macario, A.J.L.; Scalia, F. Putative Roles and Therapeutic Potential of the Chaperone System in Amyotrophic Lateral Sclerosis and Multiple Sclerosis. Cells 2024, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef]
- Eggleton, P.; Gargan, R.; Fisher, D. Rapid method for the isolation of neutrophils in high yield without the use of dextran or density gradient polymers. J. Immunol. Methods 1989, 121, 105–113. [Google Scholar] [CrossRef]
- Ceeraz, S.; Hall, C.; Choy, E.H.; Spencer, J.; Corrigall, V.M. Defective CD8+CD28+ regulatory T cell suppressor function in rheumatoid arthritis is restored by tumour necrosis factor inhibitor therapy. Clin. Exp. Immunol. 2013, 174, 18–26. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Significance | Comparative Patient Groups |
|---|---|---|
| IFNγ | p = 0.02 | Pbo>Res |
| TNFα | p = 0.085 (ns) | Pbo>Res |
| IFNγ | p = 0.03 | NRes>Res |
| IL1β | p = 0.01 | NRes>Res |
| TNFα | p = 0.054 (ns) | NRes>Res |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, C.; Pleasance, J.; Hickman, O.; Kirkham, B.; Panayi, G.S.; Eggleton, P.; Corrigall, V.M. The Biologic IRL201805 Alters Immune Tolerance Leading to Prolonged Pharmacodynamics and Efficacy in Rheumatoid Arthritis Patients. Int. J. Mol. Sci. 2024, 25, 4394. https://doi.org/10.3390/ijms25084394
Hall C, Pleasance J, Hickman O, Kirkham B, Panayi GS, Eggleton P, Corrigall VM. The Biologic IRL201805 Alters Immune Tolerance Leading to Prolonged Pharmacodynamics and Efficacy in Rheumatoid Arthritis Patients. International Journal of Molecular Sciences. 2024; 25(8):4394. https://doi.org/10.3390/ijms25084394
Chicago/Turabian StyleHall, Christopher, Jill Pleasance, Oliver Hickman, Bruce Kirkham, Gabriel S. Panayi, Paul Eggleton, and Valerie M. Corrigall. 2024. "The Biologic IRL201805 Alters Immune Tolerance Leading to Prolonged Pharmacodynamics and Efficacy in Rheumatoid Arthritis Patients" International Journal of Molecular Sciences 25, no. 8: 4394. https://doi.org/10.3390/ijms25084394
APA StyleHall, C., Pleasance, J., Hickman, O., Kirkham, B., Panayi, G. S., Eggleton, P., & Corrigall, V. M. (2024). The Biologic IRL201805 Alters Immune Tolerance Leading to Prolonged Pharmacodynamics and Efficacy in Rheumatoid Arthritis Patients. International Journal of Molecular Sciences, 25(8), 4394. https://doi.org/10.3390/ijms25084394