
Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension


 , , ,
, , , 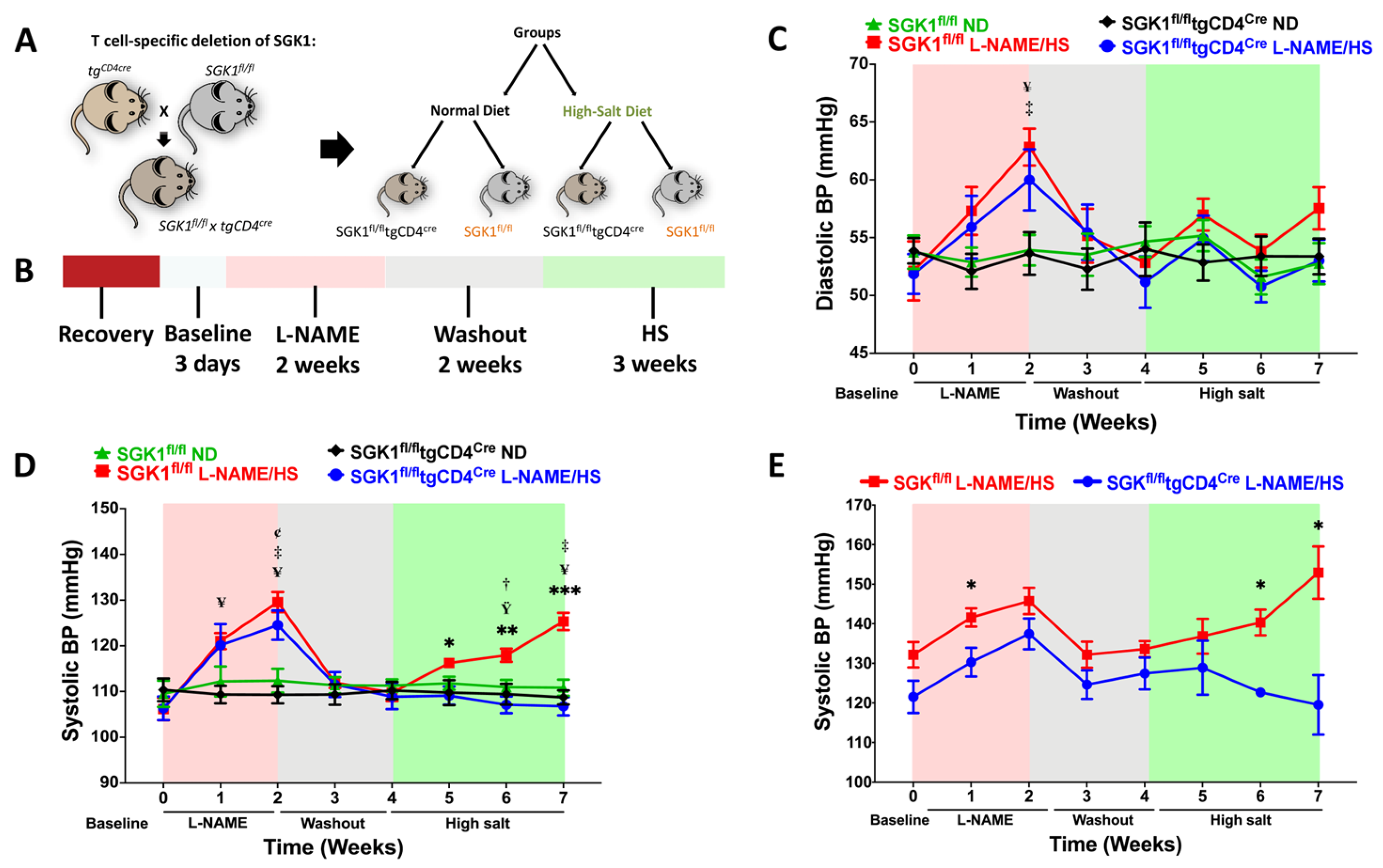{kind=link}
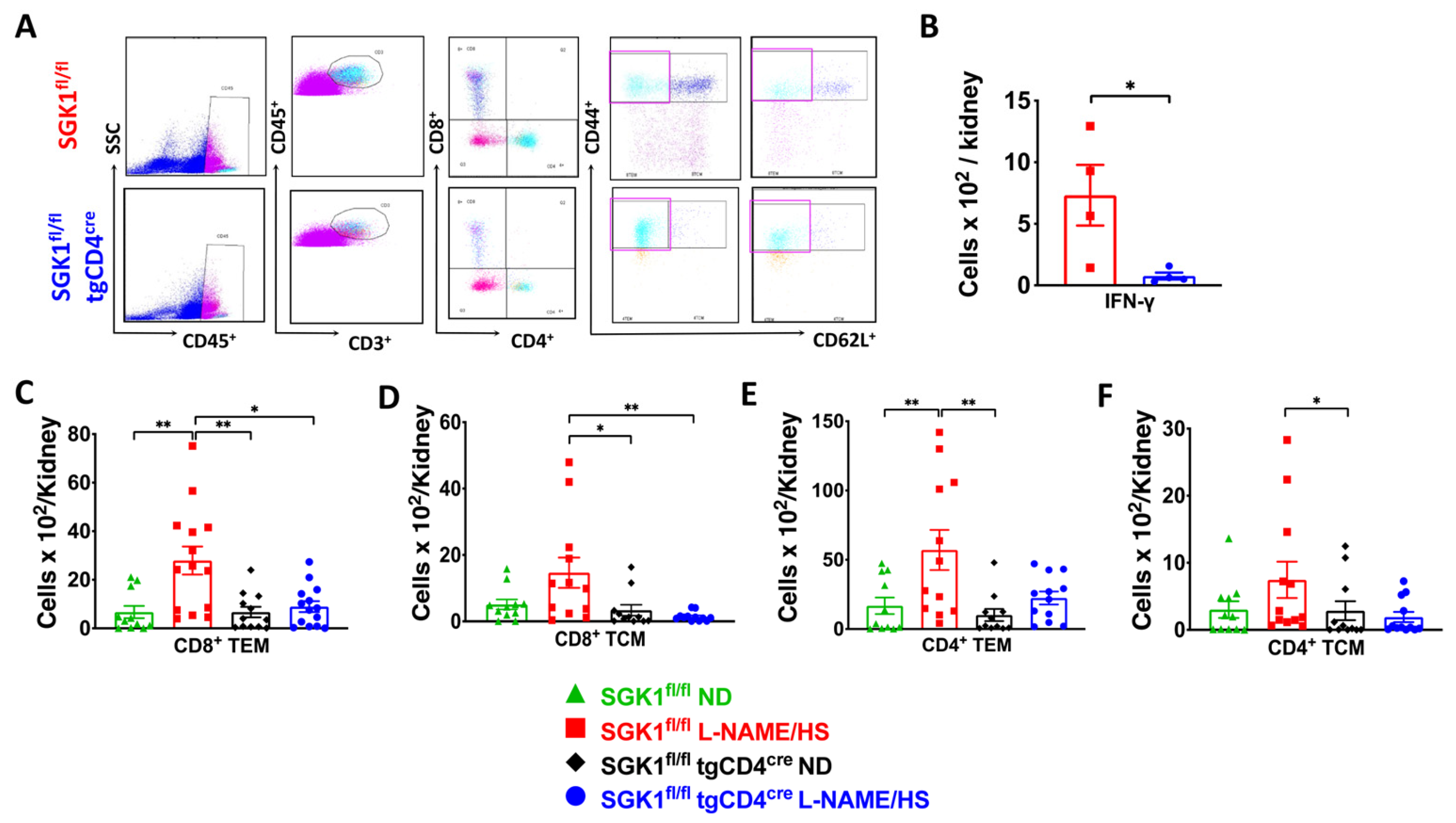{kind=link}
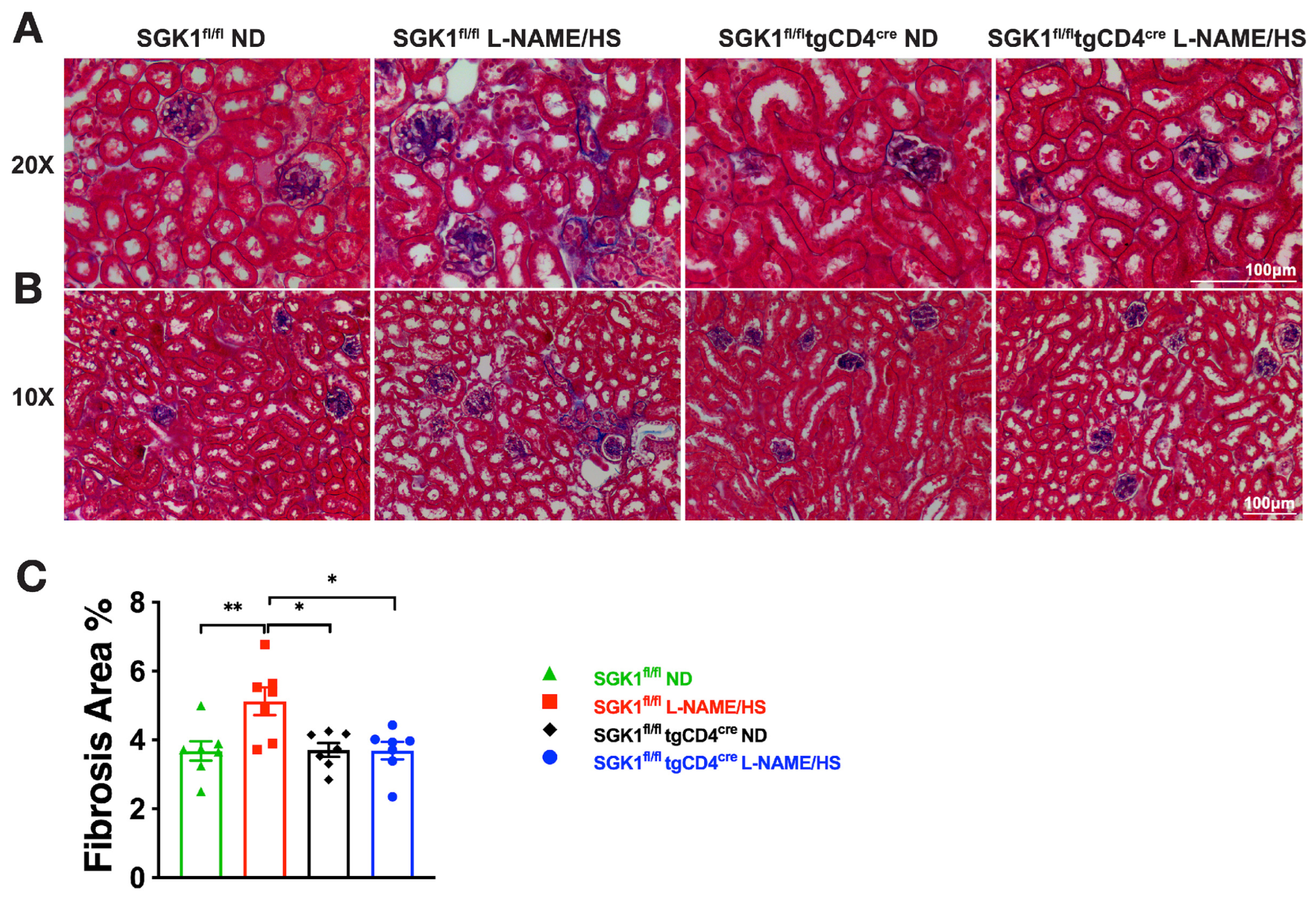{kind=link}
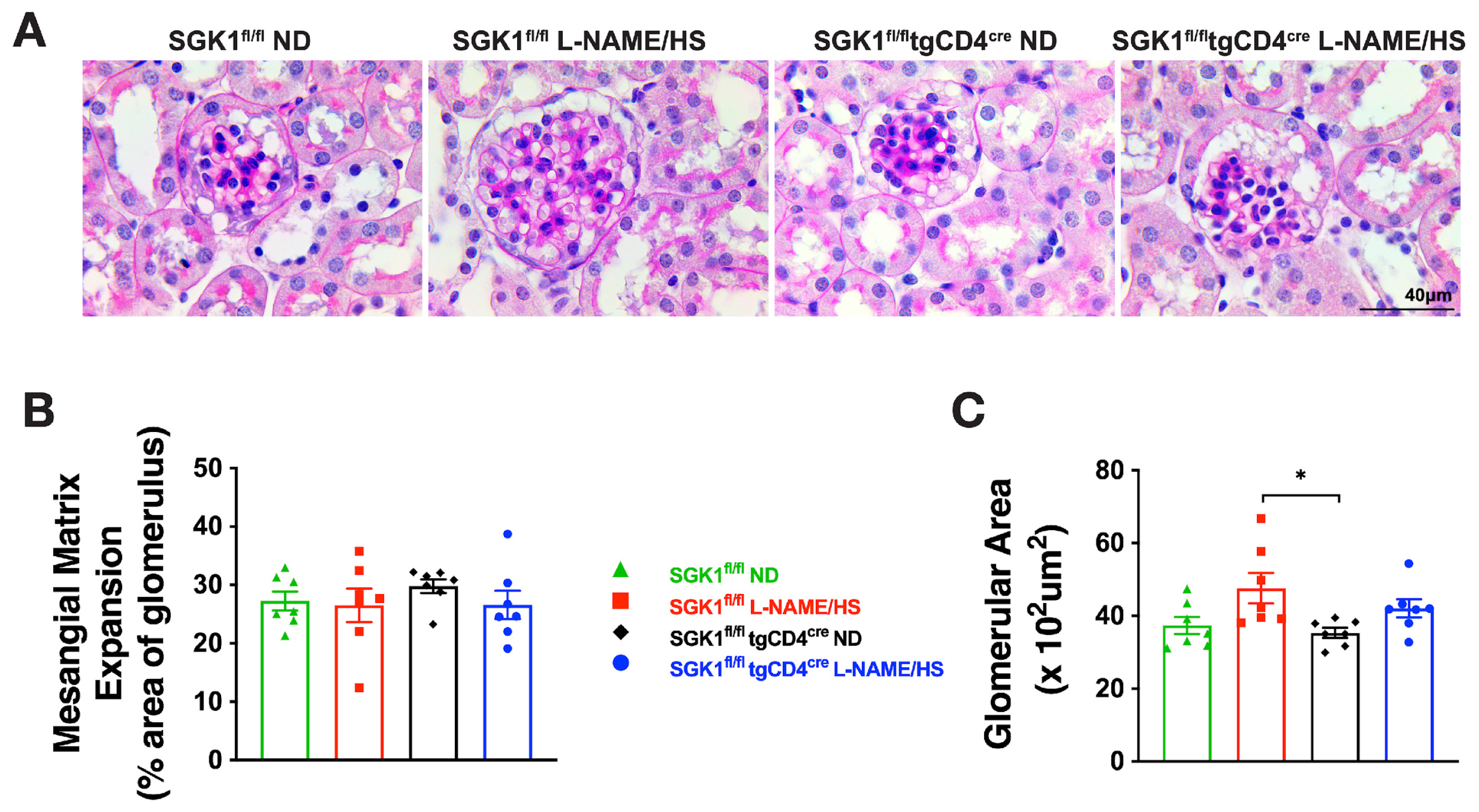{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Maaliki, D.; Itani, M.; Jarrah, H.; El-Mallah, C.; Ismail, D.; El Atie, Y.E.; Obeid, O.; Jaffa, M.A.; Itani, H.A. Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension. Int. J. Mol. Sci. 2024, 25, 4402. https://doi.org/10.3390/ijms25084402
Maaliki D, Itani M, Jarrah H, El-Mallah C, Ismail D, El Atie YE, Obeid O, Jaffa MA, Itani HA. Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension. International Journal of Molecular Sciences. 2024; 25(8):4402. https://doi.org/10.3390/ijms25084402
Chicago/Turabian StyleMaaliki, Dina, Maha Itani, Hala Jarrah, Carla El-Mallah, Diana Ismail, Yara E. El Atie, Omar Obeid, Miran A. Jaffa, and Hana A. Itani. 2024. "Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension" International Journal of Molecular Sciences 25, no. 8: 4402. https://doi.org/10.3390/ijms25084402
APA StyleMaaliki, D., Itani, M., Jarrah, H., El-Mallah, C., Ismail, D., El Atie, Y. E., Obeid, O., Jaffa, M. A., & Itani, H. A. (2024). Dietary High Salt Intake Exacerbates SGK1-Mediated T Cell Pathogenicity in L-NAME/High Salt-Induced Hypertension. International Journal of Molecular Sciences, 25(8), 4402. https://doi.org/10.3390/ijms25084402