
The OTX2 Gene Induces Tumor Growth and Triggers Leptomeningeal Metastasis by Regulating the mTORC2 Signaling Pathway in Group 3 Medulloblastomas

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
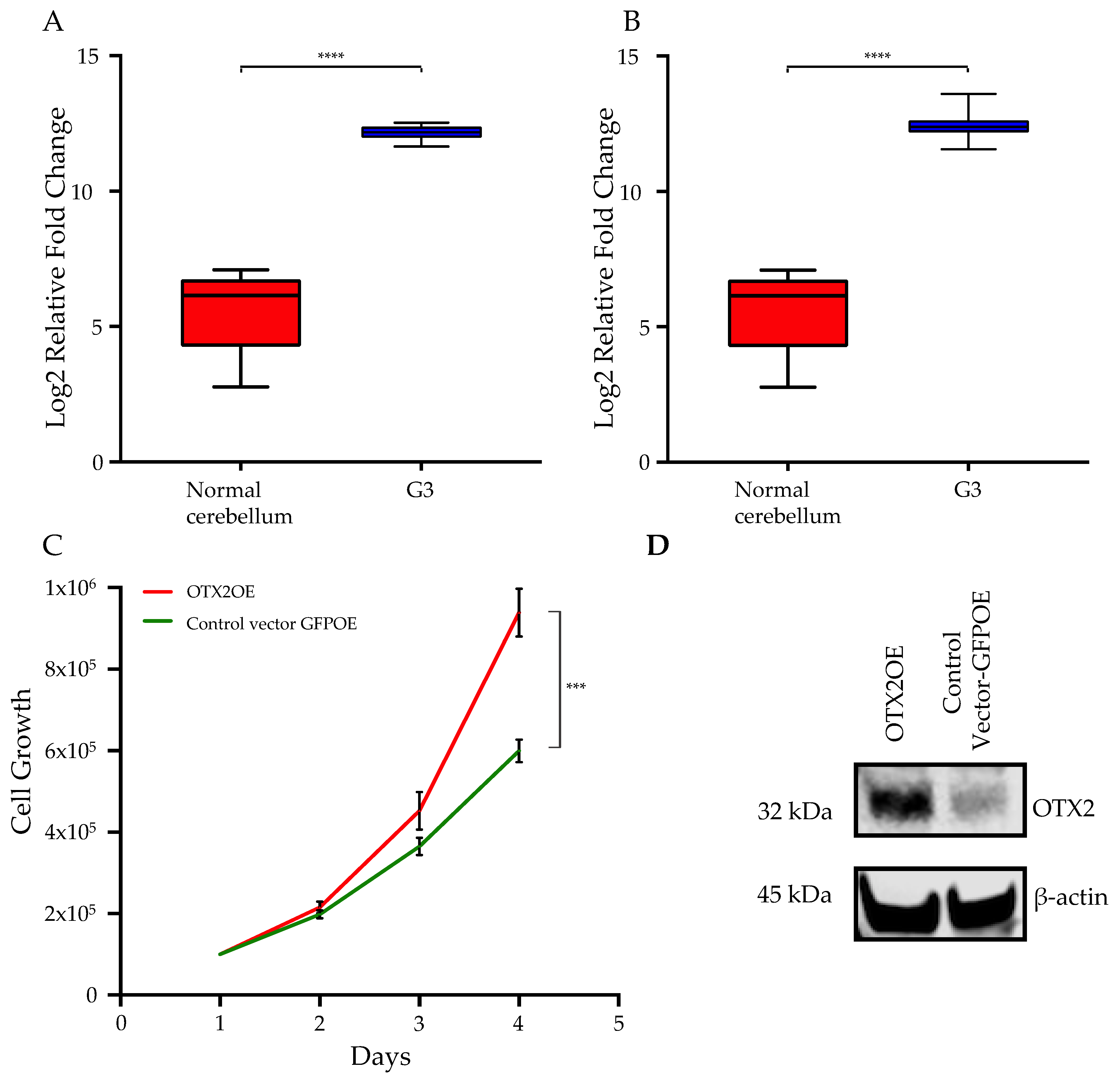
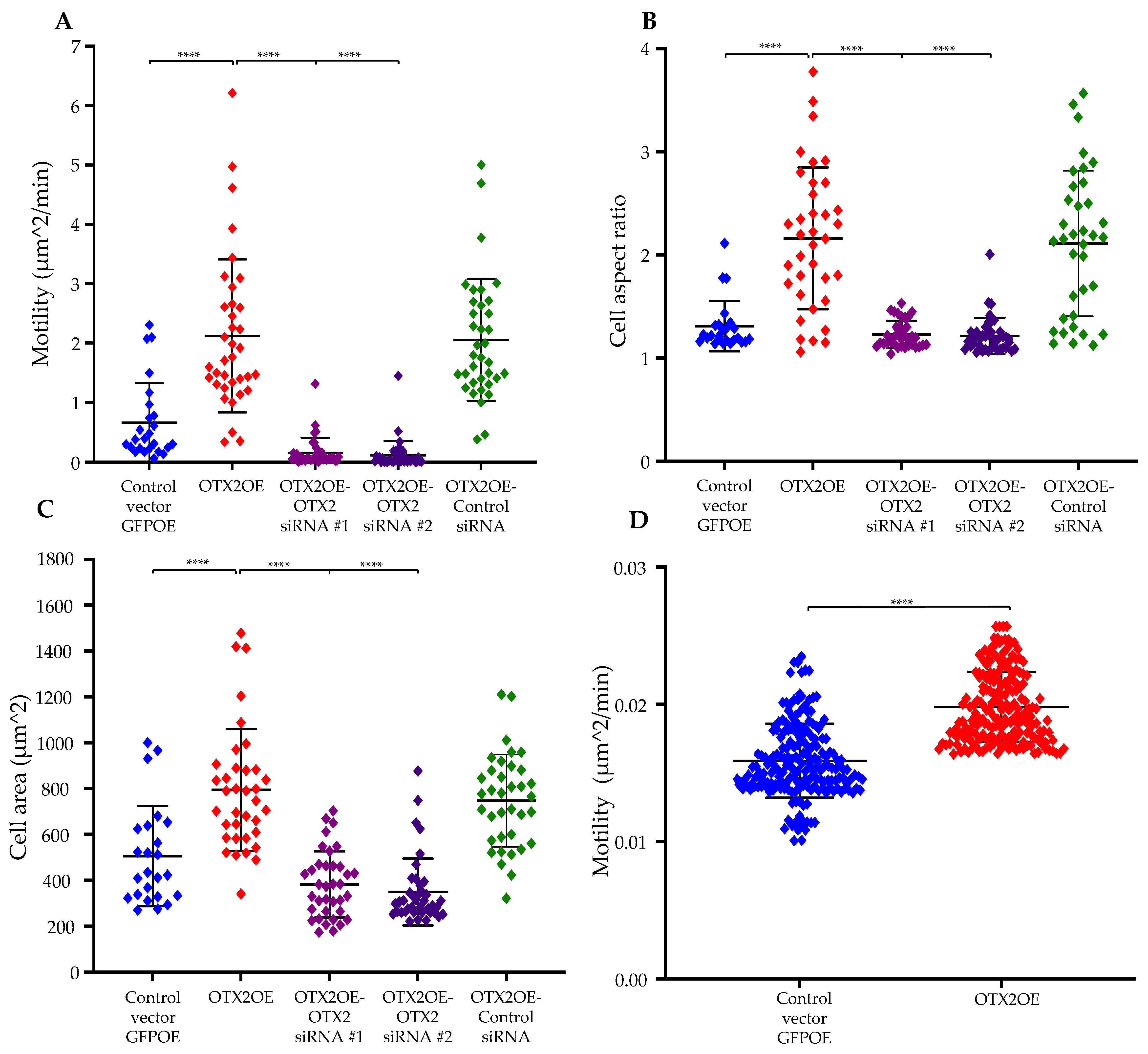
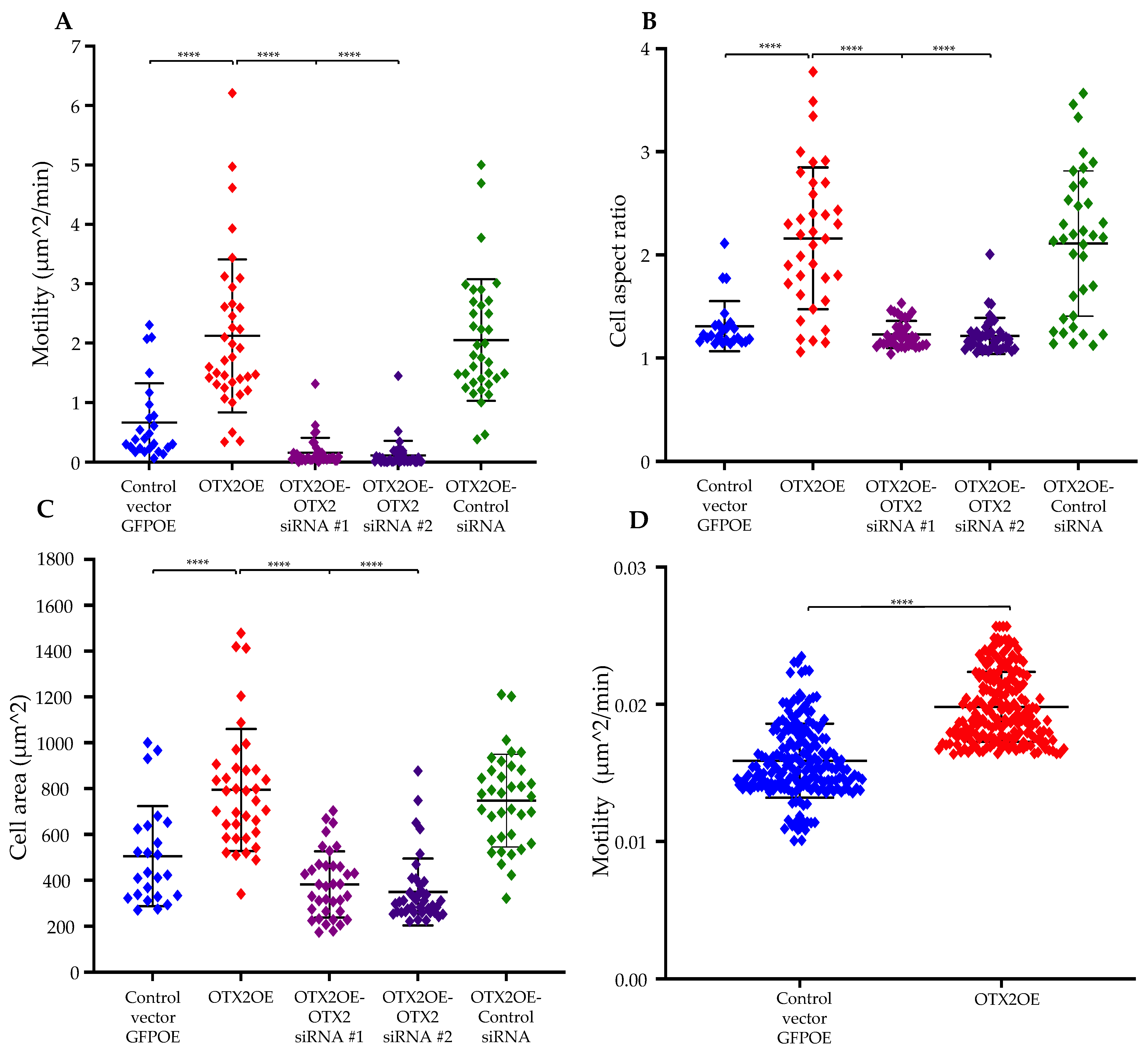
2.1. Overexpression of the OTX2 Gene Increases Cell Motility and Cell Growth
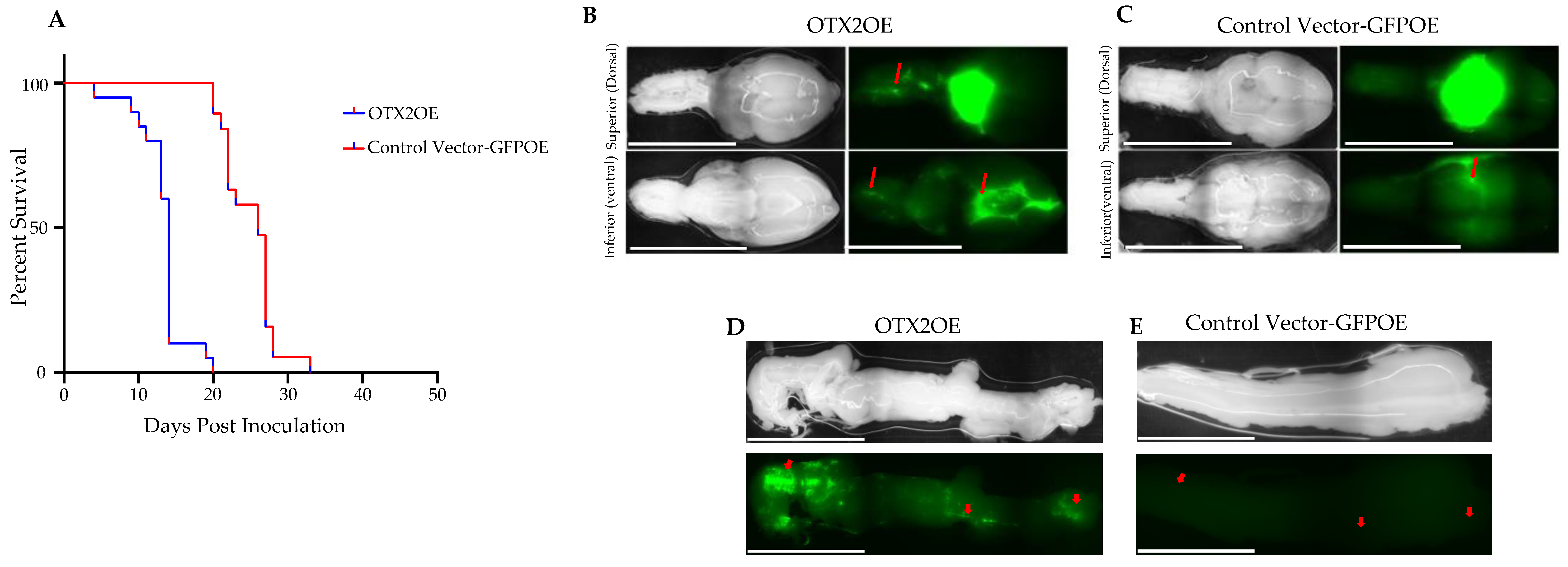
2.2. The OTX2 Gene Induces MB Tumor Growth and Triggers Spinal Cord/Brain Metastases in Cell-Derived Orthotopic Xenograft (CDOX) Mouse Model
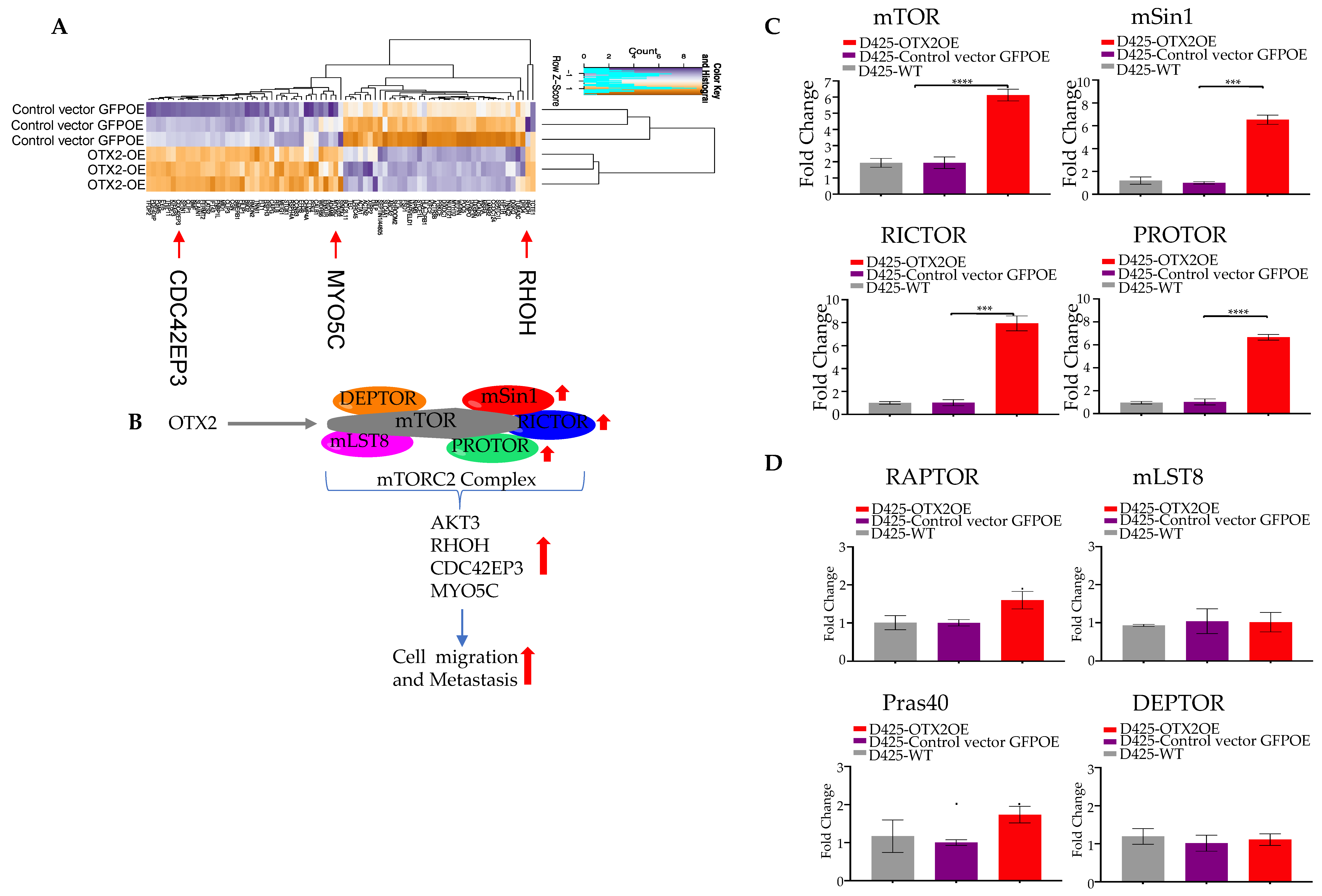
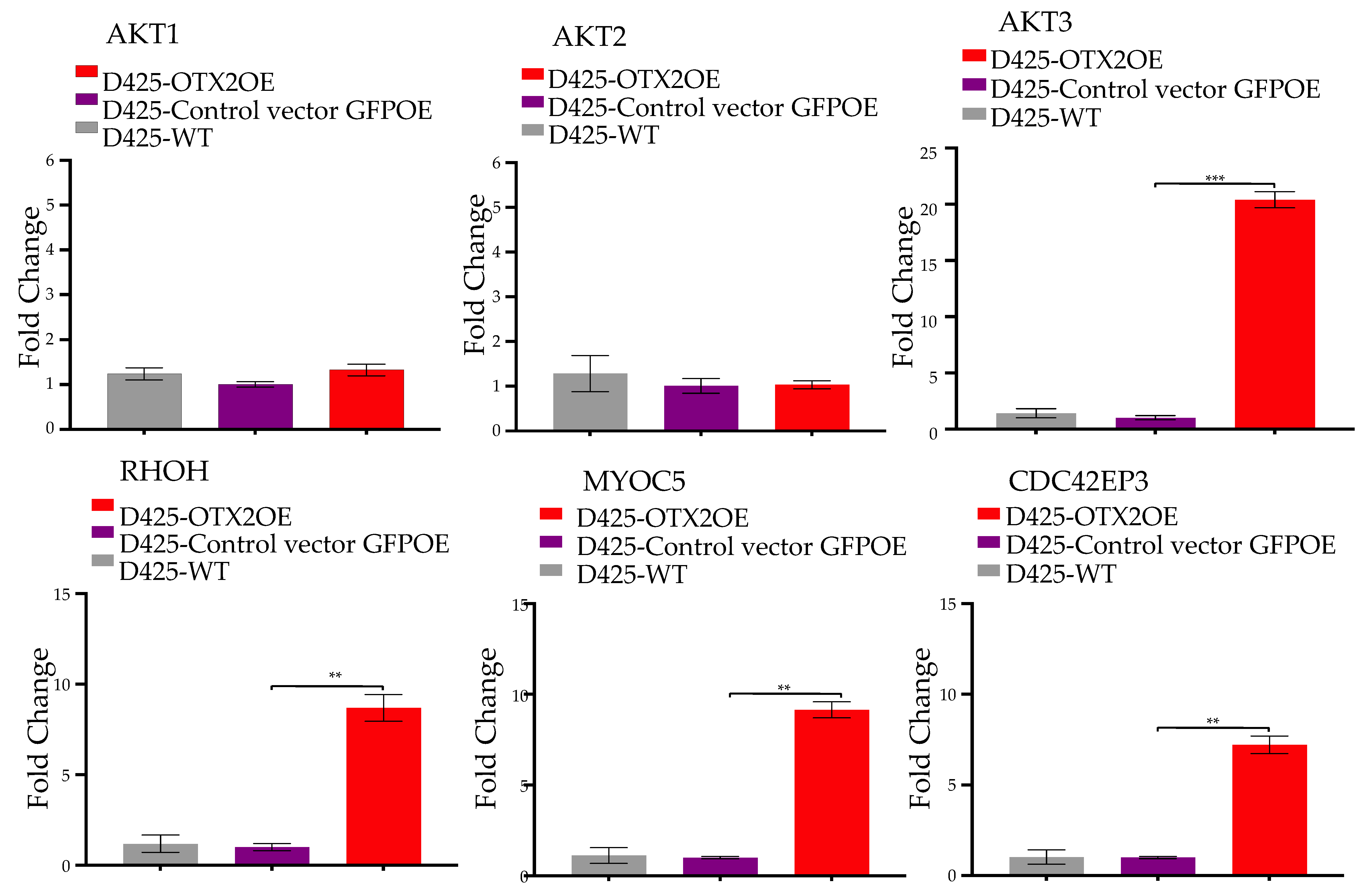
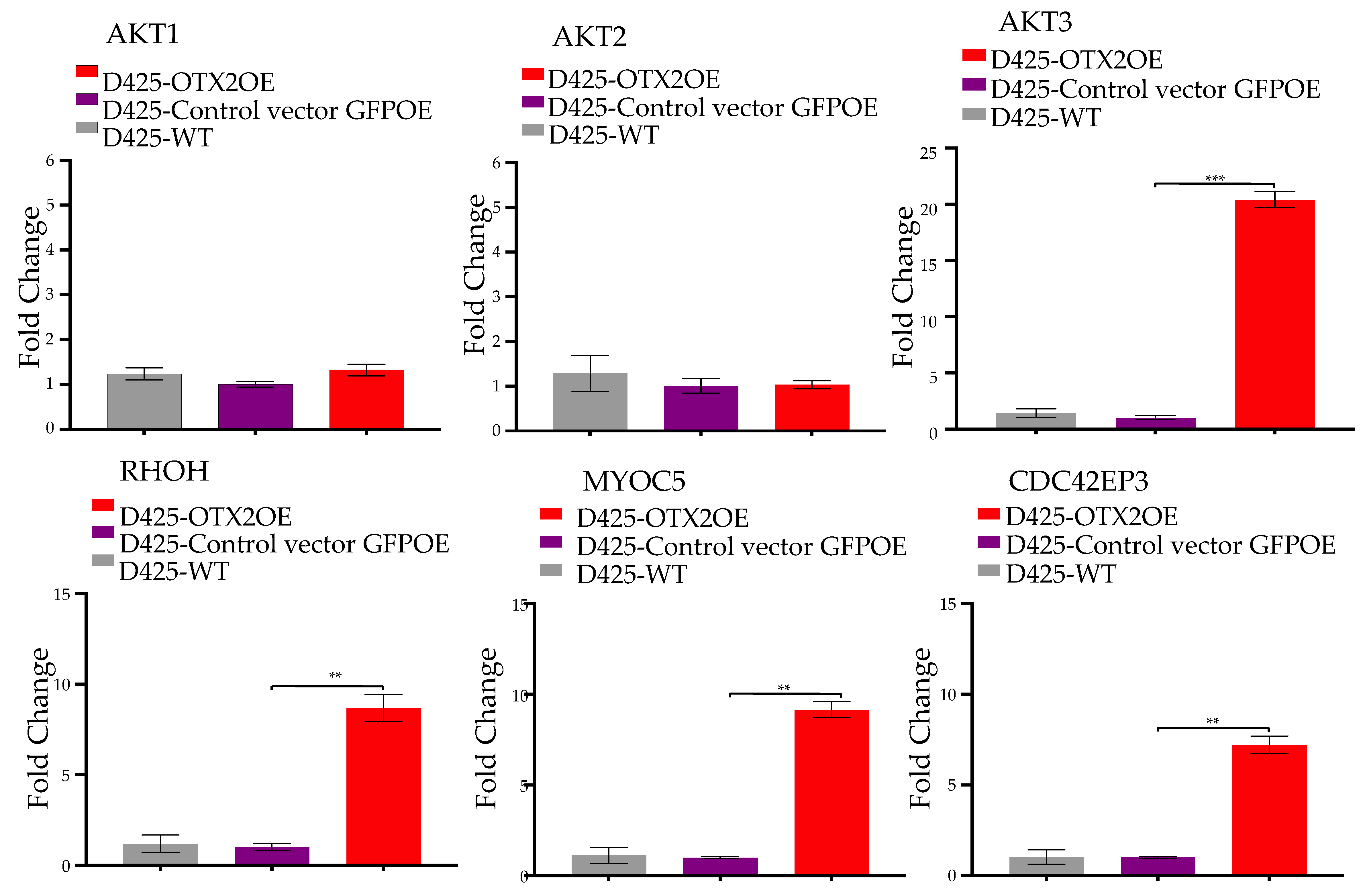
2.3. OTX2 Gene Upregulates the mTORC2 Signaling Pathway
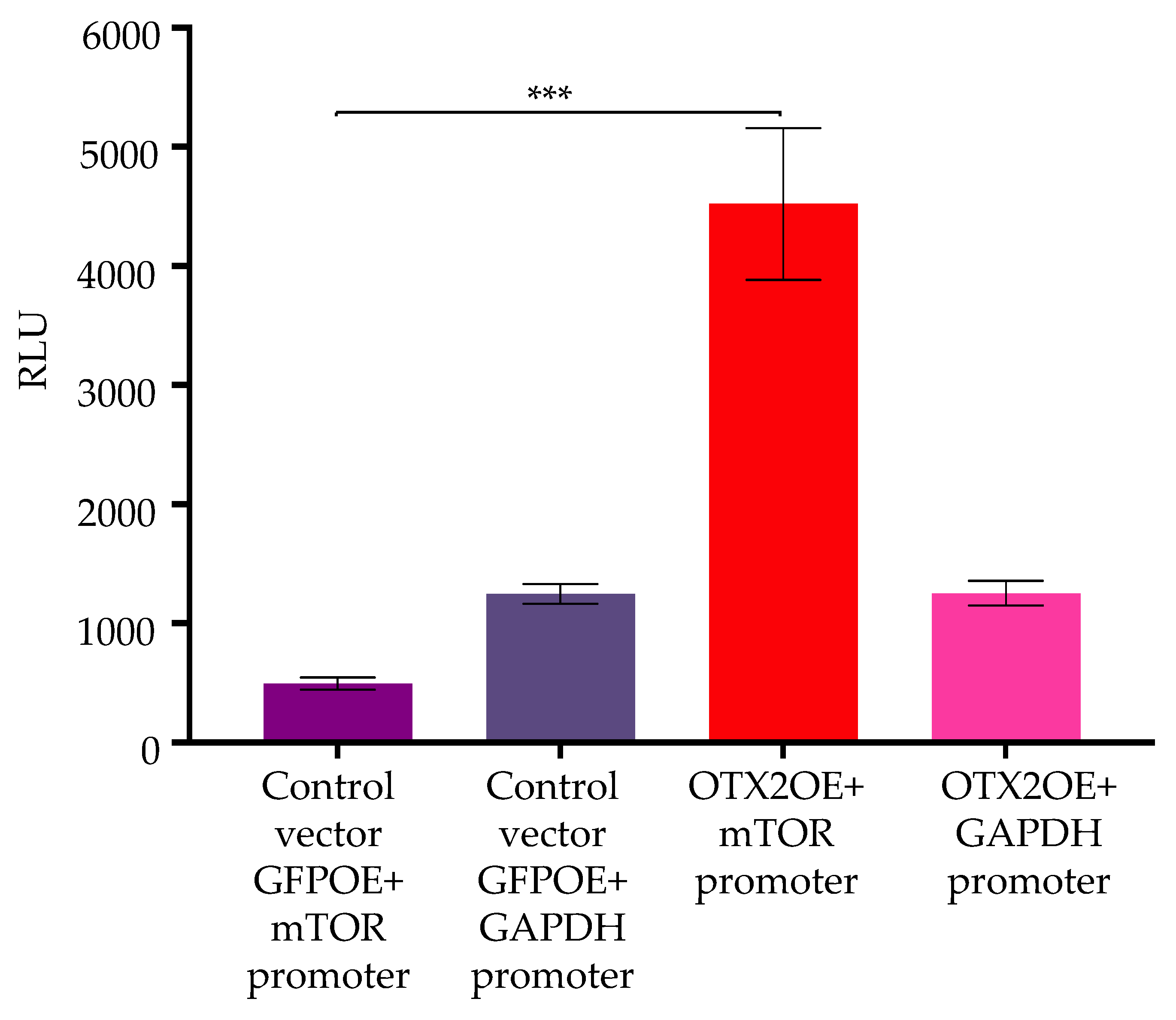
2.4. OTX2 Gene Transcriptionally Activates the mTOR Promoter
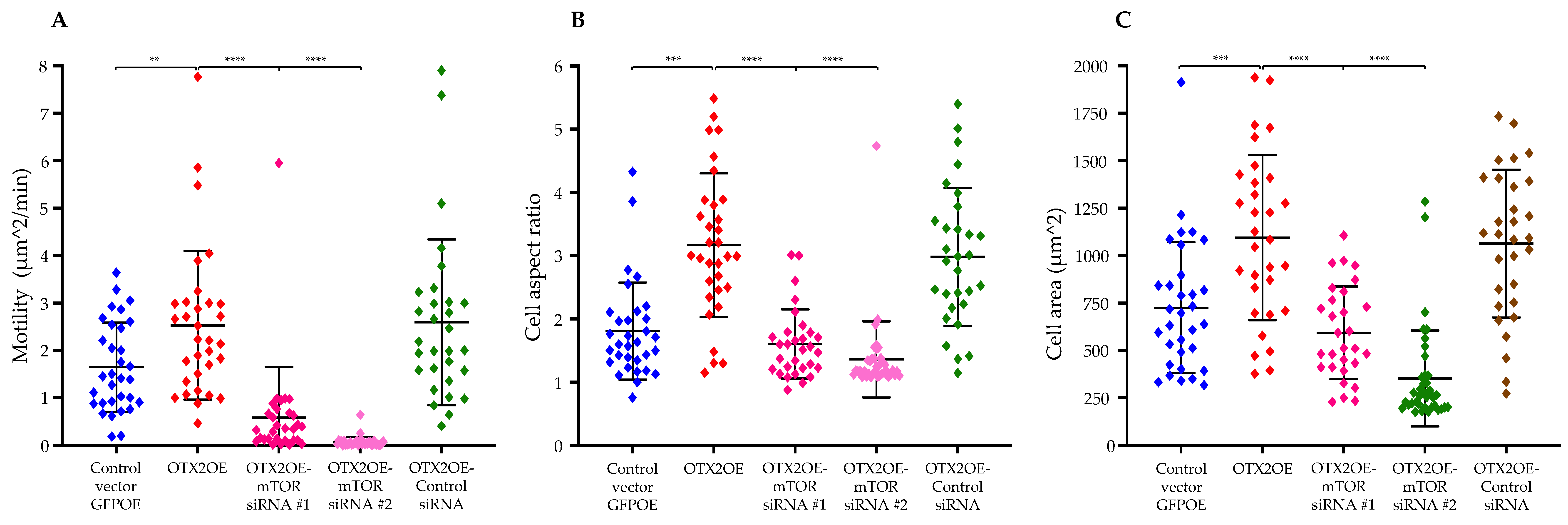
2.5. siRNA-Mediated Knockdown of mTOR mRNA Significantly Reduced OTX2-Mediated Enhancements in Cell Motility and Polarization
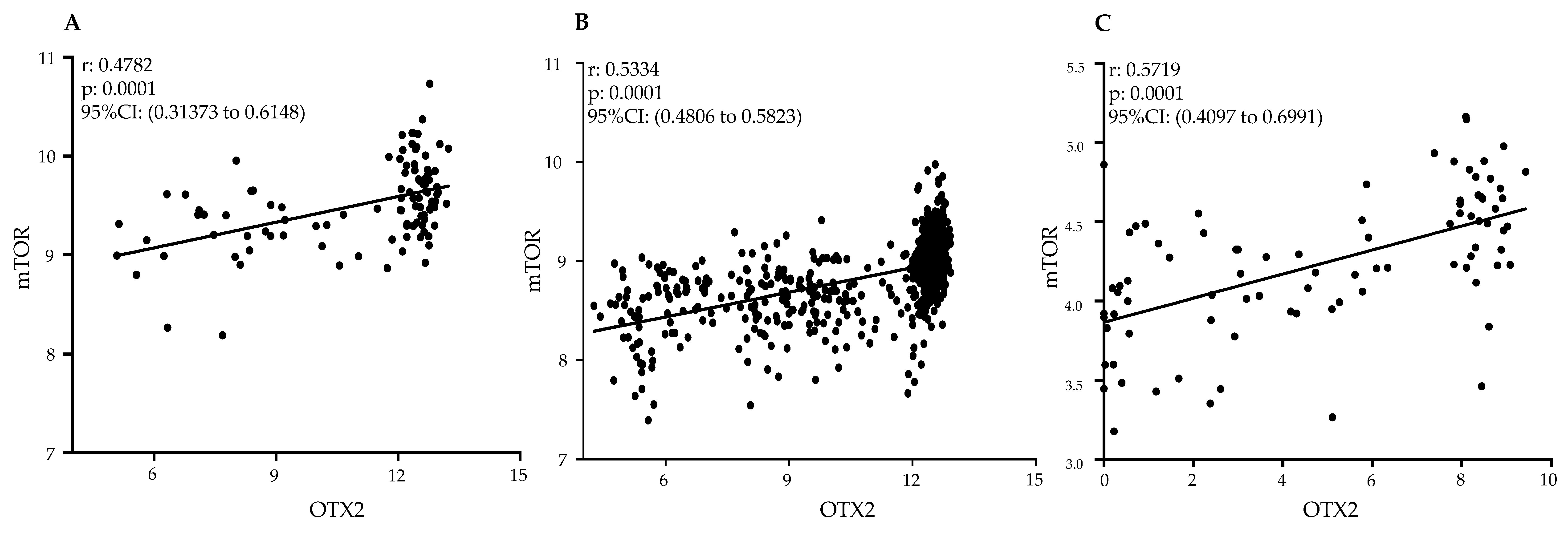
2.6. Clinical Relevance of Gene Expression Analysis and Significant Positive Correlation between OTX2 and mTOR mRNAs in Human MB Tumor Samples
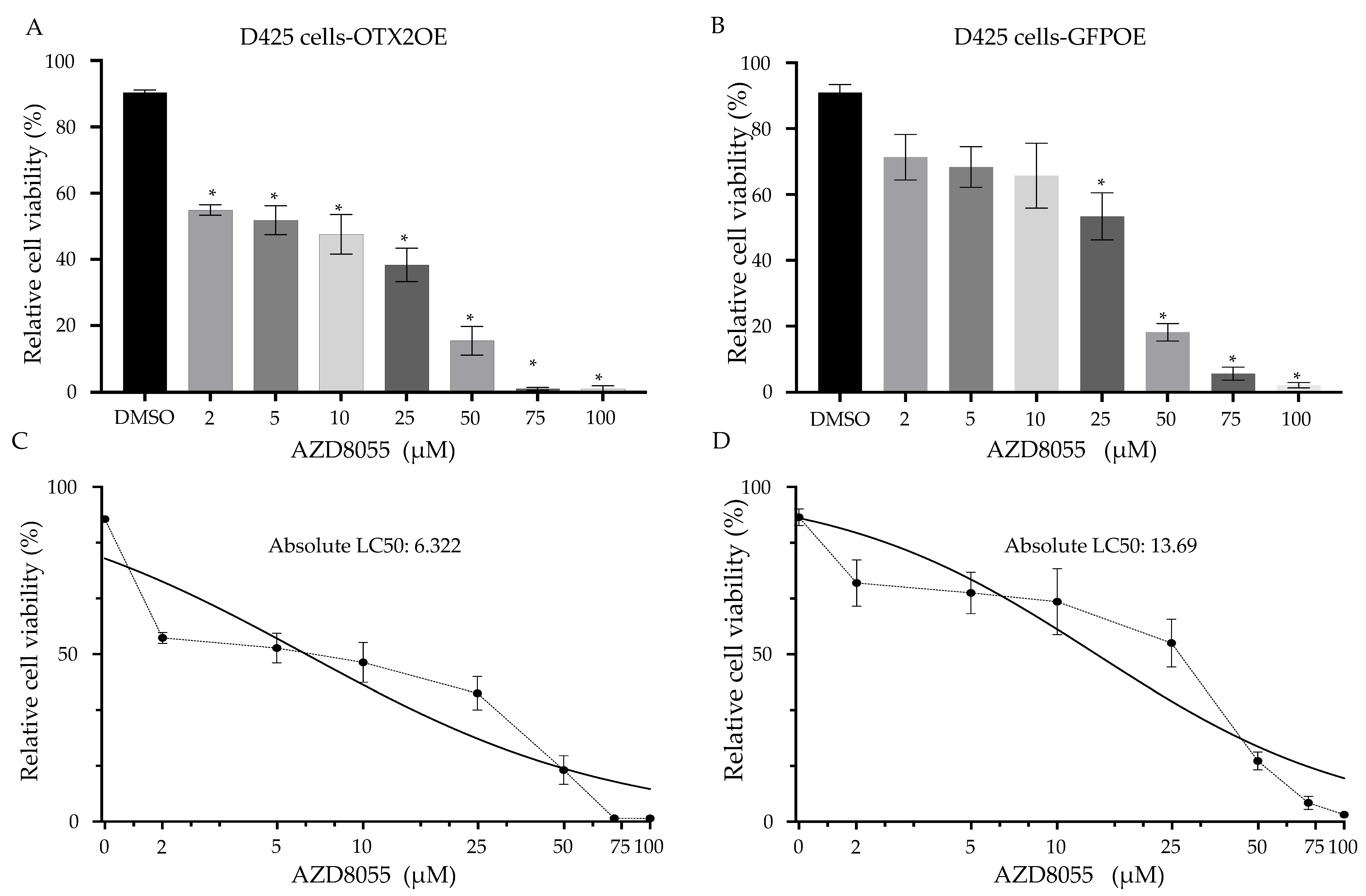
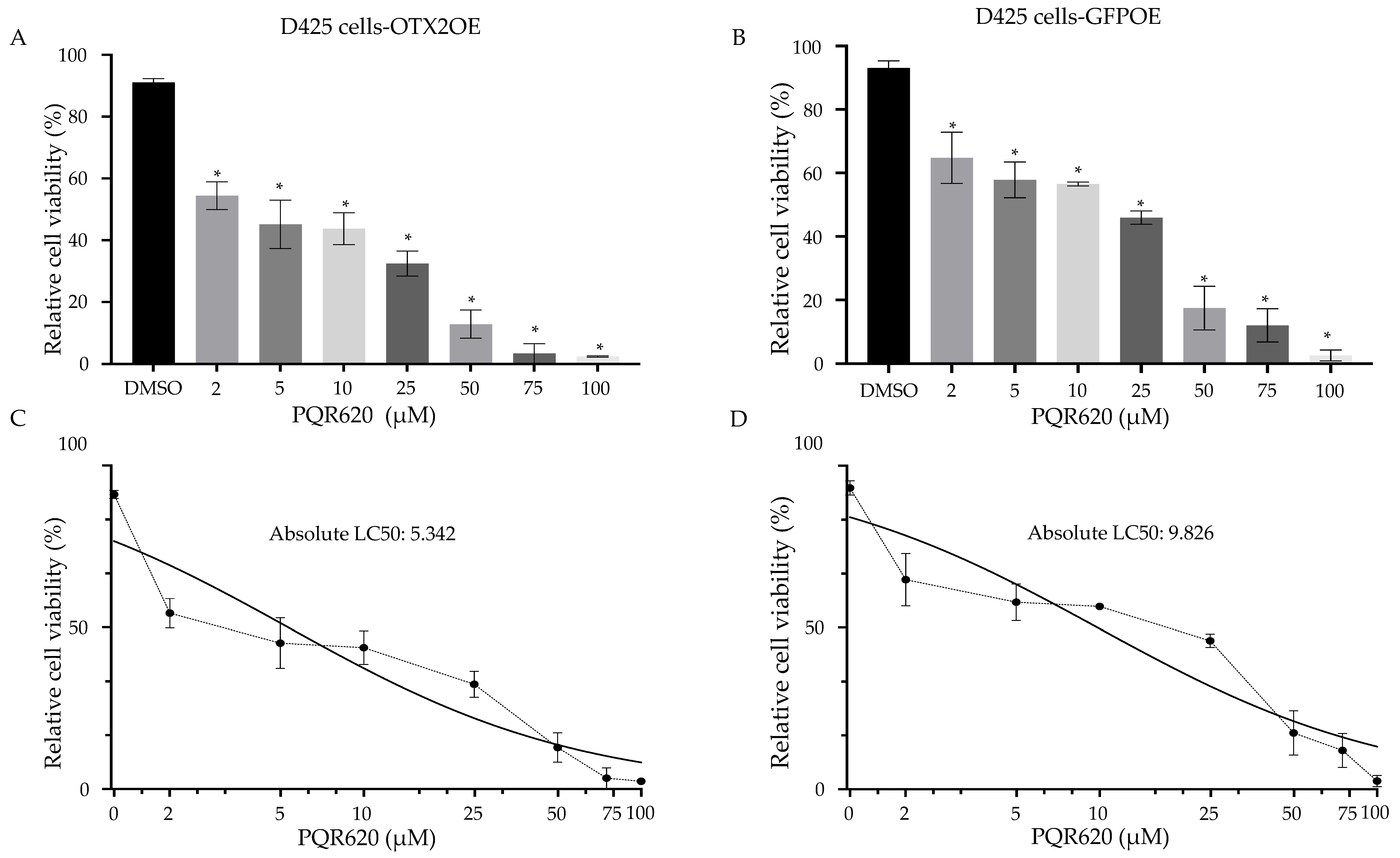
2.7. The Treatment of MB Cells with mTOR Inhibitors Resulted in a Significant Induction of Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cerebellum Inoculation
4.3. Metastasis Acquisition Using GFP Fluorescence
4.4. Lentiviral Infections and Transduction
4.5. RT-qPCR Assays
4.6. Western Blot Analysis
4.7. Collagen-Coated Polyacrylamide Substrate Preparation
4.8. Cell Migration Analysis
4.9. Mouse Cerebellum Slices
4.10. Cells Staining
4.11. Cerebellum-Slice Coculture with Medulloblastoma Cells
4.12. Live-Cell Confocal Fluorescence Time-Lapse Imaging and Analysis of Cell Migration
4.13. siRNA Transfection
4.14. Gaussia Luciferase Activity
4.15. Cell Viability Assay
4.16. Bulk RNAseq
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef]
- Kool, M.; Jones, D.T.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Beaugrand, A.; Pizer, B.; Micheli, J.; Aubelle, M.S.; Fourcade, A.; Couanet, D.; Laplanche, A.; Kalifa, C.; Grill, J. Metastatic Medulloblastoma in Childhood: Chang’s Classification Revisited. Int. J. Surg. Oncol. 2012, 2012, 245385. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Li, K.K.; Lau, K.M.; Ng, H.K. Signaling pathway and molecular subgroups of medulloblastoma. Int. J. Clin. Exp. Pathol. 2013, 6, 1211–1222, Erratum in Int. J. Clin. Exp. Pathol. 2015, 8, 11945. [Google Scholar]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.J.; Robinson, G.W. Medulloblastoma-translating discoveries from the bench to the bedside. Nat. Rev. Clin. Oncol. 2014, 11, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, O.; Giangaspero, F.; Győrffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Boulay, G.; Awad, M.E.; Riggi, N.; Archer, T.C.; Iyer, S.; Boonseng, W.E.; Rossetti, N.E.; Naigles, B.; Rengarajan, S.; Volorio, A.; et al. OTX2 Activity at Distal Regulatory Elements Shapes the Chromatin Landscape of Group 3 Medulloblastoma. Cancer Discov. 2017, 7, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Bunt, J.; Hasselt, N.E.; Zwijnenburg, D.A.; Hamdi, M.; Koster, J.; Versteeg, R.; Kool, M. OTX2 directly activates cell cycle genes and inhibits differentiation in medulloblastoma cells. Int. J. Cancer 2012, 131, E21–E32. [Google Scholar] [CrossRef] [PubMed]
- Bunt, J.; Hasselt, N.A.; Zwijnenburg, D.A.; Koster, J.; Versteeg, R.; Kool, M. OTX2 sustains a bivalent-like state of OTX2-bound promoters in medulloblastoma by maintaining their H3K27me3 levels. Acta Neuropathol. 2013, 125, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e736. [Google Scholar] [CrossRef] [PubMed]
- Cook Sangar, M.L.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef]
- Zapotocky, M.; Mata-Mbemba, D.; Sumerauer, D.; Liby, P.; Lassaletta, A.; Zamecnik, J.; Krskova, L.; Kyncl, M.; Stary, J.; Laughlin, S.; et al. Differential patterns of metastatic dissemination across medulloblastoma subgroups. J. Neurosurg. Pediatr. 2018, 21, 145–152. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Northcott, P.A.; Taylor, M.D. FISH and chips: The recipe for improved prognostication and outcomes for children with medulloblastoma. Cancer Genet. 2011, 204, 577–588. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Langdon, J.A.; Hollander, A.; Hernan, R.; Hogg, T.L.; Gajjar, A.; Fuller, C.; Clifford, S.C. Mutational analysis of PDGFR-RAS/MAPK pathway activation in childhood medulloblastoma. Eur. J. Cancer 2006, 42, 646–649. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.J.; Brown, K.M.; LaFleur, B.; Peterson, K.; Lawlor, C.; Chen, Y.; Packer, R.J.; Cogen, P.; Stephan, D.A. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat. Genet. 2001, 29, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Hernan, R.; Fasheh, R.; Calabrese, C.; Frank, A.J.; Maclean, K.H.; Allard, D.; Barraclough, R.; Gilbertson, R.J. ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res. 2003, 63, 140–148. [Google Scholar]
- Pankratova, S.; Klingelhofer, J.; Dmytriyeva, O.; Owczarek, S.; Renziehausen, A.; Syed, N.; Porter, A.E.; Dexter, D.T.; Kiryushko, D. The S100A4 Protein Signals through the ErbB4 Receptor to Promote Neuronal Survival. Theranostics 2018, 8, 3977–3990. [Google Scholar] [CrossRef] [PubMed]
- Dmytriyeva, O.; Pankratova, S.; Owczarek, S.; Sonn, K.; Soroka, V.; Ridley, C.M.; Marsolais, A.; Lopez-Hoyos, M.; Ambartsumian, N.; Lukanidin, E.; et al. The metastasis-promoting S100A4 protein confers neuroprotection in brain injury. Nat. Commun. 2012, 3, 1197. [Google Scholar] [CrossRef]
- Blom, T.; Roselli, A.; Häyry, V.; Tynninen, O.; Wartiovaara, K.; Korja, M.; Nordfors, K.; Haapasalo, H.; Nupponen, N.N. Amplification and overexpression of KIT, PDGFRA, and VEGFR2 in medulloblastomas and primitive neuroectodermal tumors. J. Neurooncol 2010, 97, 217–224. [Google Scholar] [CrossRef]
- Hartmann, W.; Digon-Söntgerath, B.; Koch, A.; Waha, A.; Endl, E.; Dani, I.; Denkhaus, D.; Goodyer, C.G.; Sörensen, N.; Wiestler, O.D.; et al. Phosphatidylinositol 3′-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin. Cancer Res. 2006, 12, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Rankin, S.L.; Larson, J.D.; Zhu, X.; Chow, L.M.; Qu, C.; Zhang, J.; Ellison, D.W.; Baker, S.J. PTEN Signaling in the Postnatal Perivascular Progenitor Niche Drives Medulloblastoma Formation. Cancer Res. 2017, 77, 123–133. [Google Scholar] [CrossRef]
- Yin, X.L.; Pang, J.C.; Liu, Y.H.; Chong, E.Y.; Cheng, Y.; Poon, W.S.; Ng, H.K. Analysis of loss of heterozygosity on chromosomes 10q, 11, and 16 in medulloblastomas. J. Neurosurg. 2001, 94, 799–805. [Google Scholar] [CrossRef]
- Zakrzewska, M.; Rieske, P.; Debiec-Rychter, M.; Zakrzewski, K.; Polis, L.; Fiks, T.; Liberski, P.P. Molecular abnormalities in pediatric embryonal brain tumors—Analysis of loss of heterozygosity on chromosomes 1, 5, 9, 10, 11, 16, 17 and 22. Clin. Neuropathol. 2004, 23, 209–217. [Google Scholar] [PubMed]
- Weeraratne, S.D.; Amani, V.; Teider, N.; Pierre-Francois, J.; Winter, D.; Kye, M.J.; Sengupta, S.; Archer, T.; Remke, M.; Bai, A.H.; et al. Pleiotropic effects of miR-183~96~182 converge to regulate cell survival, proliferation and migration in medulloblastoma. Acta Neuropathol. 2012, 123, 539–552. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Beckmann, P.J.; Larson, J.D.; Larsson, A.T.; Ostergaard, J.P.; Wagner, S.; Rahrmann, E.P.; Shamsan, G.A.; Otto, G.M.; Williams, R.L.; Wang, J.; et al. Insertional Mutagenesis Reveals Important Genetic Drivers of Central Nervous System Embryonal Tumors. Cancer Res. 2019, 79, 905–917. [Google Scholar] [CrossRef]
- Kahn, S.A.; Wang, X.; Nitta, R.T.; Gholamin, S.; Theruvath, J.; Hutter, G.; Azad, T.D.; Wadi, L.; Bolin, S.; Ramaswamy, V.; et al. Notch1 regulates the initiation of metastasis and self-renewal of Group 3 medulloblastoma. Nat. Commun. 2018, 9, 4121. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Kalra, R.R.; Dubuc, A.; Sivakumar, W.; Pedone, C.A.; Wu, X.; Taylor, M.D.; Fults, D.W. Genetic drivers of metastatic dissemination in sonic hedgehog medulloblastoma. Acta Neuropathol. Commun. 2014, 2, 85. [Google Scholar] [CrossRef]
- Li, M.; Deng, Y.; Zhang, W. Molecular Determinants of Medulloblastoma Metastasis and Leptomeningeal Dissemination. Mol. Cancer Res. 2021, 19, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Van Ommeren, R.; Garzia, L.; Holgado, B.L.; Ramaswamy, V.; Taylor, M.D. The molecular biology of medulloblastoma metastasis. Brain Pathol. 2020, 30, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef]
- Adamson, D.C.; Shi, Q.; Wortham, M.; Northcott, P.A.; Di, C.; Duncan, C.G.; Li, J.; McLendon, R.E.; Bigner, D.D.; Taylor, M.D.; et al. OTX2 is critical for the maintenance and progression of Shh-independent medulloblastomas. Cancer Res. 2010, 70, 181–191. [Google Scholar] [CrossRef]
- Charrier, E.E.; Pogoda, K.; Li, R.; Wells, R.G.; Janmey, P.A. Elasticity-dependent response of malignant cells to viscous dissipation. Biomech. Model. Mechanobiol. 2021, 20, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.C.; Rubiano, A.; Dyson, K.; Simmons, C.S. Mechanical characterization of human brain tumors from patients and comparison to potential surgical phantoms. PLoS ONE 2017, 12, e0177561. [Google Scholar] [CrossRef] [PubMed]
- Bangasser, B.L.; Shamsan, G.A.; Chan, C.E.; Opoku, K.N.; Tüzel, E.; Schlichtmann, B.W.; Kasim, J.A.; Fuller, B.J.; McCullough, B.R.; Rosenfeld, S.S.; et al. Shifting the optimal stiffness for cell migration. Nat. Commun. 2017, 8, 15313. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Federico, A.; Schrimpf, D.; Sievers, P.; Sahm, F.; Koster, J.; Jones, D.T.W.; von Deimling, A.; Pfister, S.M.; Kool, M.; et al. Comparison of transcriptome profiles between medulloblastoma primary and recurrent tumors uncovers novel variance effects in relapses. Acta Neuropathol. Commun. 2023, 11, 7. [Google Scholar] [CrossRef]
- Brandt, C.; Hillmann, P.; Noack, A.; Römermann, K.; Öhler, L.A.; Rageot, D.; Beaufils, F.; Melone, A.; Sele, A.M.; Wymann, M.P.; et al. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology 2018, 140, 107–120. [Google Scholar] [CrossRef]
- Rageot, D.; Bohnacker, T.; Melone, A.; Langlois, J.B.; Borsari, C.; Hillmann, P.; Sele, A.M.; Beaufils, F.; Zvelebil, M.; Hebeisen, P.; et al. Discovery and Preclinical Characterization of 5-[4,6-Bis({3-oxa-8-azabicyclo[3.2.1]octan-8-yl})-1,3,5-triazin-2-yl]-4-(difluoromethyl)pyridin-2-amine (PQR620), a Highly Potent and Selective mTORC1/2 Inhibitor for Cancer and Neurological Disorders. J. Med. Chem. 2018, 61, 10084–10105. [Google Scholar] [CrossRef]
- Luchman, H.A.; Stechishin, O.D.; Nguyen, S.A.; Lun, X.Q.; Cairncross, J.G.; Weiss, S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin. Cancer Res. 2014, 20, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Aiken, C.; Morrison, L.C.; Rao, R.; Del Bigio, M.R.; Rampalli, S.; Werbowetski-Ogilvie, T. OTX2 exhibits cell-context-dependent effects on cellular and molecular properties of human embryonic neural precursors and medulloblastoma cells. Dis. Model. Mech. 2015, 8, 1295–1309. [Google Scholar] [CrossRef]
- Zagozewski, J.; Shahriary, G.M.; Morrison, L.C.; Saulnier, O.; Stromecki, M.; Fresnoza, A.; Palidwor, G.; Porter, C.J.; Forget, A.; Ayrault, O.; et al. An OTX2-PAX3 signaling axis regulates Group 3 medulloblastoma cell fate. Nat. Commun. 2020, 11, 3627. [Google Scholar] [CrossRef]
- Stromecki, M.; Tatari, N.; Morrison, L.C.; Kaur, R.; Zagozewski, J.; Palidwor, G.; Ramaswamy, V.; Skowron, P.; Wölfl, M.; Milde, T.; et al. Characterization of a novel OTX2-driven stem cell program in Group 3 and Group 4 medulloblastoma. Mol. Oncol. 2018, 12, 495–513. [Google Scholar] [CrossRef]
- Ferrucci, V.; de Antonellis, P.; Pennino, F.P.; Asadzadeh, F.; Virgilio, A.; Montanaro, D.; Galeone, A.; Boffa, I.; Pisano, I.; Scognamiglio, I.; et al. Metastatic group 3 medulloblastoma is driven by PRUNE1 targeting NME1-TGF-β-OTX2-SNAIL via PTEN inhibition. Brain 2018, 141, 1300–1319. [Google Scholar] [CrossRef] [PubMed]
- Bakhshinyan, D.; Venugopal, C.; Adile, A.A.; Garg, N.; Manoranjan, B.; Hallett, R.; Wang, X.; Mahendram, S.; Vora, P.; Vijayakumar, T.; et al. BMI1 is a therapeutic target in recurrent medulloblastoma. Oncogene 2019, 38, 1702–1716. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Burger, P.C.; Bigner, S.H.; Trojanowski, J.Q.; Wikstrand, C.J.; Halperin, E.C.; Bigner, D.D. Establishment and characterization of the human medulloblastoma cell line and transplantable xenograft D283 Med. J. Neuropathol. Exp. Neurol. 1985, 44, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, P.F.; Jenkyn, D.J.; Papadimitriou, J.M. Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J. Neuropathol. Exp. Neurol. 1985, 44, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Burger, P.C.; Bigner, S.H.; Trojanowski, J.Q.; Brodeur, G.M.; He, X.M.; Wikstrand, C.J.; Kurtzberg, J.; Berens, M.E.; Halperin, E.C. Phenotypic and genotypic analysis of a human medulloblastoma cell line and transplantable xenograft (D341 Med) demonstrating amplification of c-myc. Am. J. Pathol. 1988, 130, 472–484. [Google Scholar] [PubMed]
- Choi, S.A.; Kwak, P.A.; Kim, S.K.; Park, S.H.; Lee, J.Y.; Wang, K.C.; Oh, H.J.; Kim, K.; Lee, D.S.; Hwang, D.W.; et al. In vivo bioluminescence imaging for leptomeningeal dissemination of medulloblastoma in mouse models. BMC Cancer 2016, 16, 723. [Google Scholar] [CrossRef] [PubMed]
- Shackleford, G.M.; Mahdi, M.Y.; Moats, R.A.; Hawes, D.; Tran, H.C.; Finlay, J.L.; Hoang, T.Q.; Meng, E.F.; Erdreich-Epstein, A. Continuous and bolus intraventricular topotecan prolong survival in a mouse model of leptomeningeal medulloblastoma. PLoS ONE 2019, 14, e0206394. [Google Scholar] [CrossRef]
- Shu, Q.; Antalffy, B.; Su, J.M.; Adesina, A.; Ou, C.N.; Pietsch, T.; Blaney, S.M.; Lau, C.C.; Li, X.N. Valproic Acid prolongs survival time of severe combined immunodeficient mice bearing intracerebellar orthotopic medulloblastoma xenografts. Clin. Cancer Res. 2006, 12, 4687–4694. [Google Scholar] [CrossRef]
- Navarro, K.L.; Huss, M.; Smith, J.C.; Sharp, P.; Marx, J.O.; Pacharinsak, C. Mouse Anesthesia: The Art and Science. ILAR J. 2021, 62, 238–273. [Google Scholar] [CrossRef]
- Yang, M.; Baranov, E.; Jiang, P.; Sun, F.X.; Li, X.M.; Li, L.; Hasegawa, S.; Bouvet, M.; Al-Tuwaijri, M.; Chishima, T.; et al. Whole-body optical imaging of green fluorescent protein-expressing tumors and metastases. Proc. Natl. Acad. Sci. USA 2000, 97, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Benabdellah, K.; Cobo, M.; Muñoz, P.; Toscano, M.G.; Martin, F. Development of an all-in-one lentiviral vector system based on the original TetR for the easy generation of Tet-ON cell lines. PLoS ONE 2011, 6, e23734. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Pak, J.E.; Gruswitz, F.; Sharma, V.; Stroud, R.M. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat. Protoc. 2012, 7, 453–466. [Google Scholar] [CrossRef]
- Saydam, O.; Saydam, N. Deficiency of Ku Induces Host Cell Exploitation in Human Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 651818. [Google Scholar] [CrossRef]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Ströbel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Senol, O.; Schaaij-Visser, T.B.; Erkan, E.P.; Dorfer, C.; Lewandrowski, G.; Pham, T.V.; Piersma, S.R.; Peerdeman, S.M.; Ströbel, T.; Tannous, B.; et al. miR-200a-mediated suppression of non-muscle heavy chain IIb inhibits meningioma cell migration and tumor growth in vivo. Oncogene 2015, 34, 1790–1798. [Google Scholar] [CrossRef]
- Senfter, D.; Erkan, E.P.; Özer, E.; Jungwirth, G.; Madlener, S.; Kool, M.; Ströbel, T.; Saydam, N.; Saydam, O. Overexpression of minichromosome maintenance protein 10 in medulloblastoma and its clinical implications. Pediatr. Blood Cancer 2017, 64, e26670. [Google Scholar] [CrossRef]
- Ströbel, T.; Madlener, S.; Tuna, S.; Vose, S.; Lagerweij, T.; Wurdinger, T.; Vierlinger, K.; Wöhrer, A.; Price, B.D.; Demple, B.; et al. Ape1 guides DNA repair pathway choice that is associated with drug tolerance in glioblastoma. Sci. Rep. 2017, 7, 9674. [Google Scholar] [CrossRef]
- Erkan, E.P.; Ströbel, T.; Lewandrowski, G.; Tannous, B.; Madlener, S.; Czech, T.; Saydam, N.; Saydam, O. Depletion of minichromosome maintenance protein 7 inhibits glioblastoma multiforme tumor growth in vivo. Oncogene 2014, 33, 4778–4785. [Google Scholar] [CrossRef]
- Erkan, E.P.; Ströbel, T.; Dorfer, C.; Sonntagbauer, M.; Weinhäusel, A.; Saydam, N.; Saydam, O. Circulating Tumor Biomarkers in Meningiomas Reveal a Signature of Equilibrium Between Tumor Growth and Immune Modulation. Front. Oncol. 2019, 9, 1031. [Google Scholar] [CrossRef]
- Wang, Y.L.; Pelham, R.J. Preparation of a flexible, porous polyacrylamide substrate for mechanical studies of cultured cells. Methods Enzymol. 1998, 298, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Shamsan, G.A.; Akkin, T.; Odde, D.J. Glioma Cell Migration Dynamics in Brain Tissue Assessed by Multimodal Optical Imaging. Biophys. J. 2019, 117, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Christie, B.R. Isolectin-IB 4 as a vascular stain for the study of adult neurogenesis. J. Neurosci. Methods 2006, 150, 138–142. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ampudia-Mesias, E.; Cameron, C.S.; Yoo, E.; Kelly, M.; Anderson, S.M.; Manning, R.; Abrahante Lloréns, J.E.; Moertel, C.L.; Yim, H.; Odde, D.J.; et al. The OTX2 Gene Induces Tumor Growth and Triggers Leptomeningeal Metastasis by Regulating the mTORC2 Signaling Pathway in Group 3 Medulloblastomas. Int. J. Mol. Sci. 2024, 25, 4416. https://doi.org/10.3390/ijms25084416
Ampudia-Mesias E, Cameron CS, Yoo E, Kelly M, Anderson SM, Manning R, Abrahante Lloréns JE, Moertel CL, Yim H, Odde DJ, et al. The OTX2 Gene Induces Tumor Growth and Triggers Leptomeningeal Metastasis by Regulating the mTORC2 Signaling Pathway in Group 3 Medulloblastomas. International Journal of Molecular Sciences. 2024; 25(8):4416. https://doi.org/10.3390/ijms25084416
Chicago/Turabian StyleAmpudia-Mesias, Elisabet, Charles S. Cameron, Eunjae Yoo, Marcus Kelly, Sarah M. Anderson, Riley Manning, Juan E. Abrahante Lloréns, Christopher L. Moertel, Hyungshin Yim, David J. Odde, and et al. 2024. "The OTX2 Gene Induces Tumor Growth and Triggers Leptomeningeal Metastasis by Regulating the mTORC2 Signaling Pathway in Group 3 Medulloblastomas" International Journal of Molecular Sciences 25, no. 8: 4416. https://doi.org/10.3390/ijms25084416
APA StyleAmpudia-Mesias, E., Cameron, C. S., Yoo, E., Kelly, M., Anderson, S. M., Manning, R., Abrahante Lloréns, J. E., Moertel, C. L., Yim, H., Odde, D. J., Saydam, N., & Saydam, O. (2024). The OTX2 Gene Induces Tumor Growth and Triggers Leptomeningeal Metastasis by Regulating the mTORC2 Signaling Pathway in Group 3 Medulloblastomas. International Journal of Molecular Sciences, 25(8), 4416. https://doi.org/10.3390/ijms25084416