
Moving toward the Inclusion of Epigenomics in Bacterial Genome Evolution: Perspectives and Challenges



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Classification and Mode of Action of DNA Methyltransferases
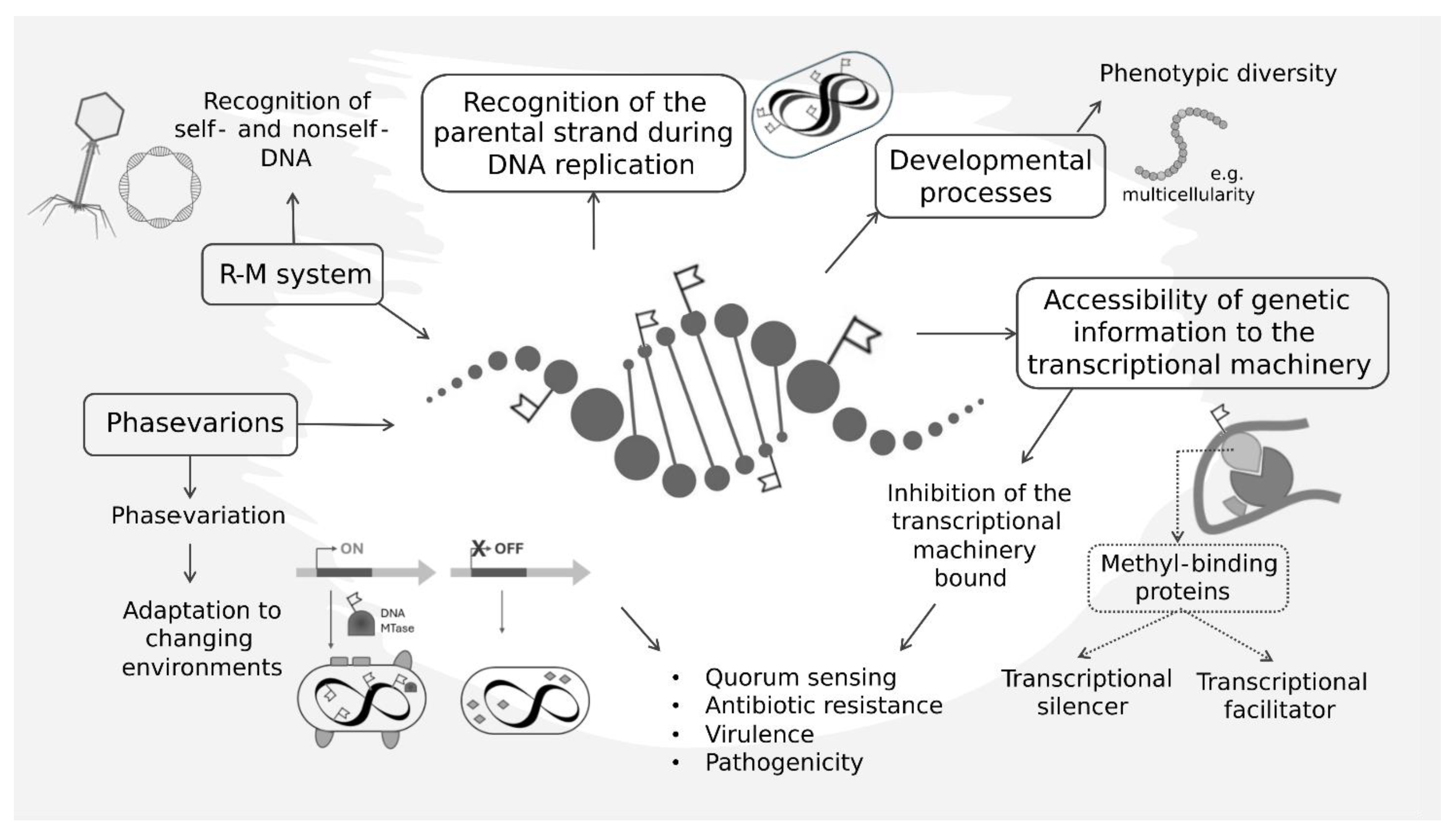
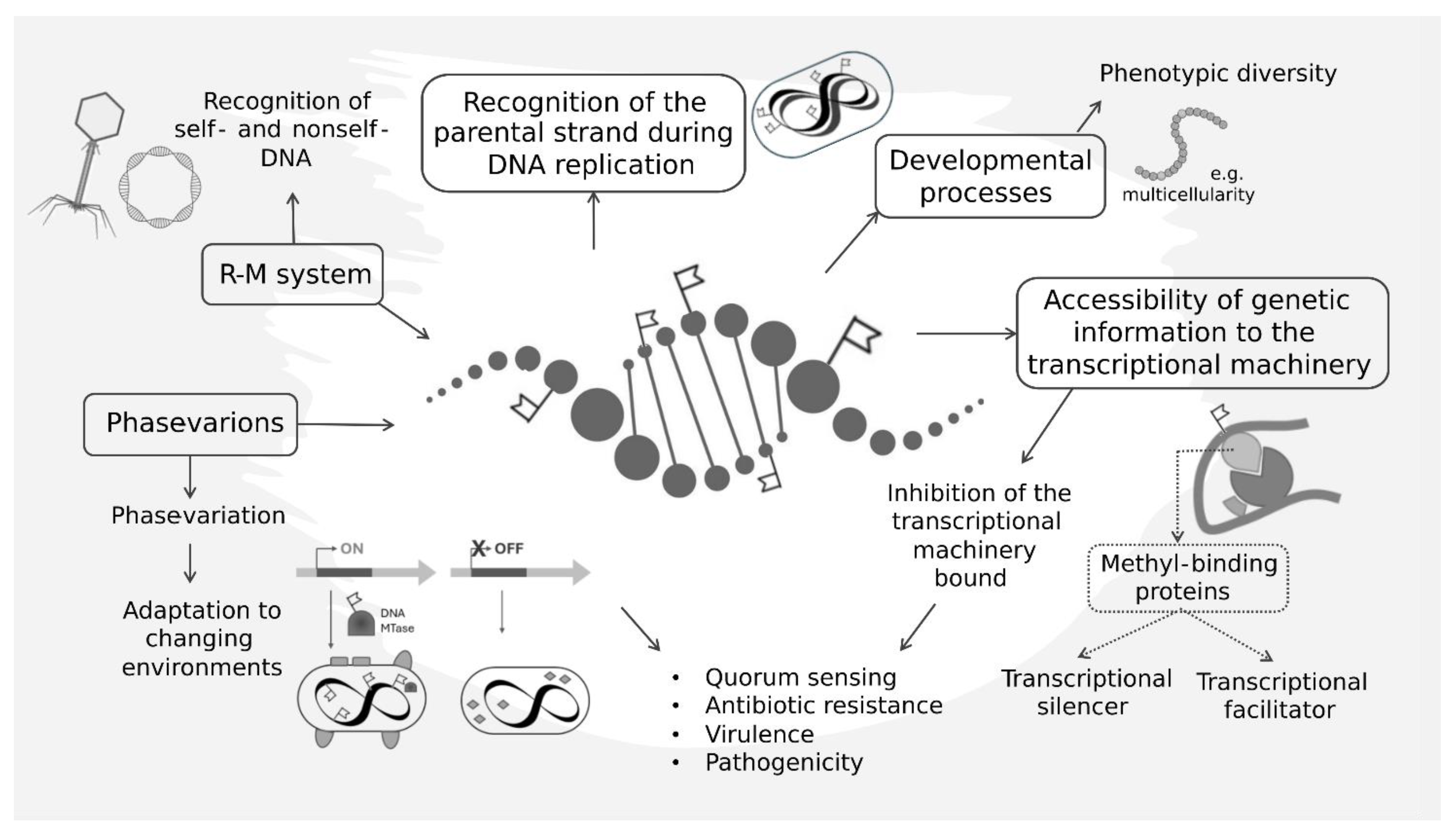
3. DNA Methylation Pattern Is Involved in Core Cellular Processes
4. Impact of DNA Methylation Patterns on Gene Expression
5. Epigenomics and Genome Evolution
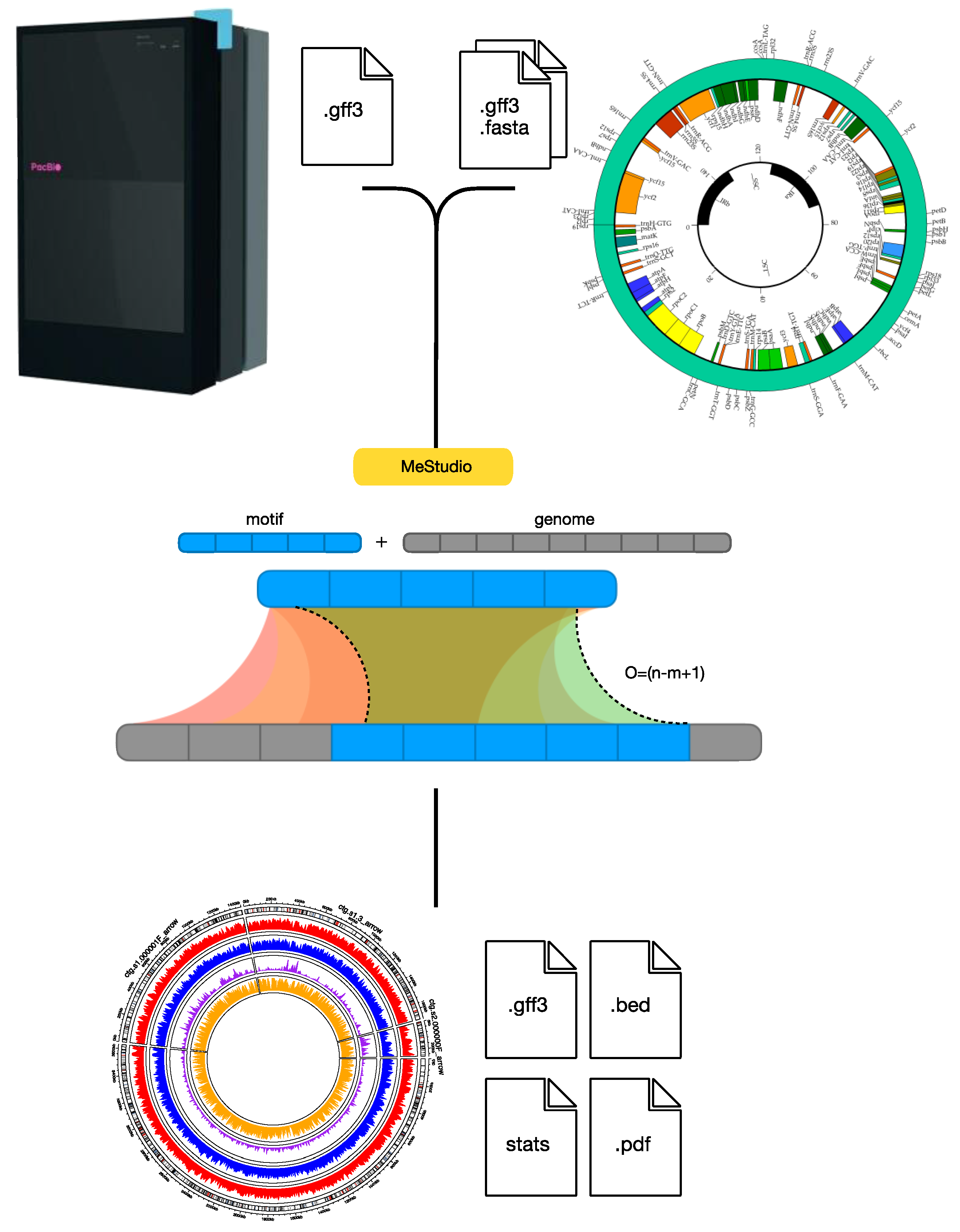
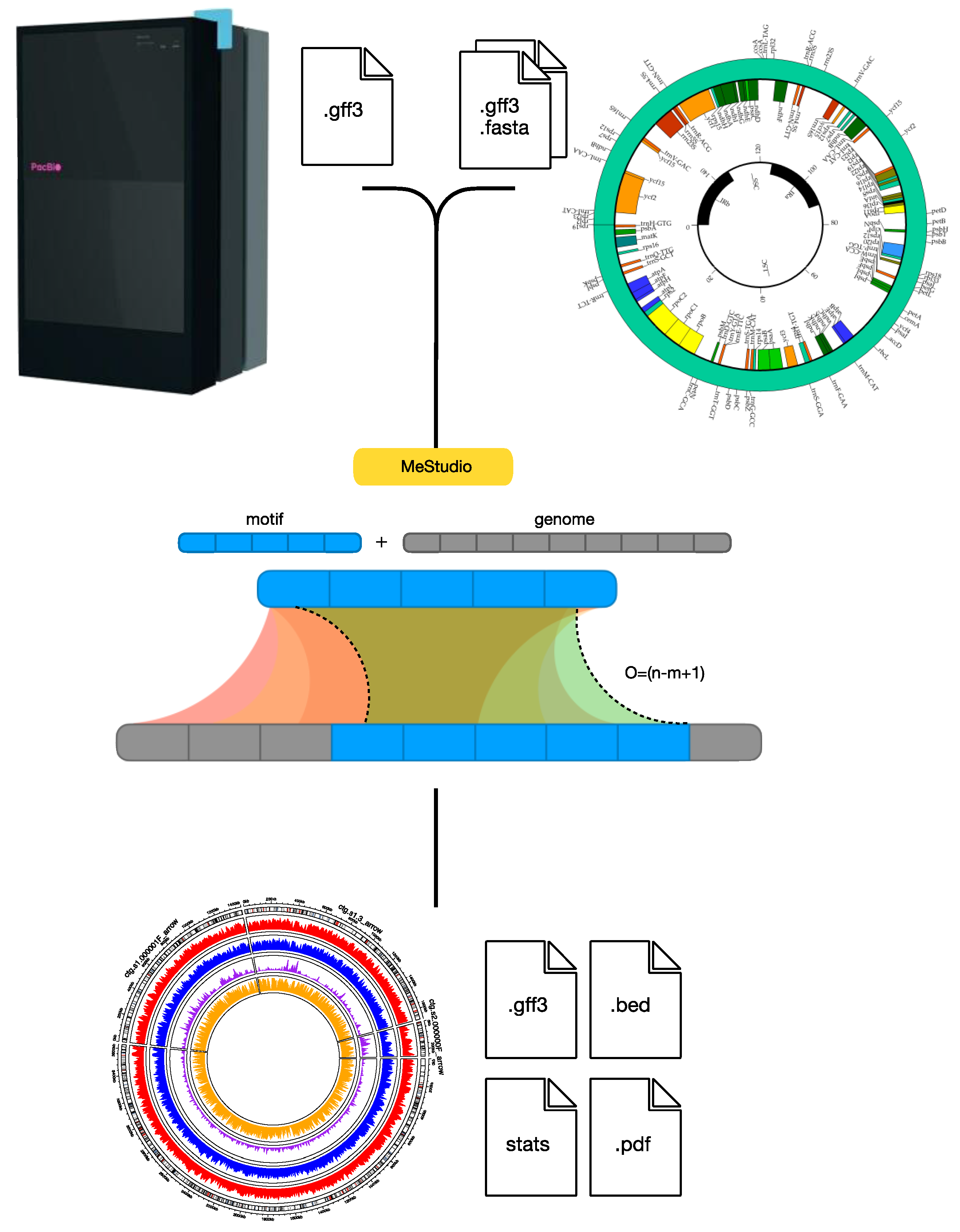
6. Computational Tools for Bacterial Epigenomic Studies
7. Conclusions and Future Directions
The Need for Experimental Validation of Predicted Effects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sánchez-Romero, M.A.; Casadesús, J. The Bacterial Epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Fang, G. Conserved DNA Methyltransferases: A Window into Fundamental Mechanisms of Epigenetic Regulation in Bacteria. Trends Microbiol. 2021, 29, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Curtis, P.D. DNA Methyltransferases and Epigenetic Regulation in Bacteria. FEMS Microbiol. Rev. 2016, 40, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural History of Eukaryotic DNA Methylation Systems. Prog. Mol. Biol. Transl. Sci. 2011, 101, 25–104. [Google Scholar] [CrossRef]
- Gao, Q.; Lu, S.; Wang, Y.; He, L.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Zhao, X.; et al. Bacterial DNA Methyltransferase: A Key to the Epigenetic World with Lessons Learned from Proteobacteria. Front. Microbiol. 2023, 14, 1129437. [Google Scholar] [CrossRef] [PubMed]
- Janulaitis, A.; Petrusyte, M.; Maneliene, Z.; Klimasauskas, S.; Butkus, V. Purification and Properties of the Eco57l Restriction Endonuclease and Methylas—Prototypes of a New Class (Type IV). Nucleic Acids. Res. 1992, 20, 6043–6049. [Google Scholar] [CrossRef]
- Lepikhov, K.; Tchernov, A.; Zheleznaja, L.; Matvienko, N.; Walter, J.; Trautner, T.A. Characterization of the Type IV Restriction Modification System BspLU11III from Bacillus Sp. LU11. Nucleic Acids Res. 2001, 29, 4691–4698. [Google Scholar] [CrossRef]
- Anton, B.P.; Roberts, R.J. Beyond Restriction Modification: Epigenomic Roles of DNA Methylation in Prokaryotes. Annu Rev. Microbiol. 2021, 75, 129–149. [Google Scholar] [CrossRef]
- Militello, K.T.; Mandarano, A.H.; Varechtchouk, O.; Simon, R.D. Cytosine DNA Methylation Influences Drug Resistance in Escherichia coli through Increased SugE Expression. FEMS Microbiol. Lett. 2014, 350, 100–106. [Google Scholar] [CrossRef]
- Mehershahi, K.S.; Chen, S.L. DNA Methylation by Three Type I Restriction Modification Systems of Escherichia Coli Does Not Influence Gene Regulation of the Host Bacterium. Nucleic Acids Res. 2021, 49, 7375–7388. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Romero, M.A.; Cota, I.; Casadesús, J. DNA Methylation in Bacteria: From the Methyl Group to the Methylome. Curr. Opin. Microbiol. 2015, 25, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Casadesús, J. Bacterial DNA Methylation and Methylomes. Adv. Exp. Med. Biol. 2016, 945, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Løbner-Olesen, A.; Skovgaard, O.; Marinus, M.G. Dam Methylation: Coordinating Cellular Processes. Curr. Opin. Microbiol. 2005, 8, 154–160. [Google Scholar] [CrossRef] [PubMed]
- diCenzo, G.C.; Cangioli, L.; Nicoud, Q.; Cheng, J.H.T.; Blow, M.J.; Shapiro, N.; Woyke, T.; Biondi, E.G.; Alunni, B.; Mengoni, A.; et al. DNA Methylation in Ensifer Species during Free-Living Growth and during Nitrogen-Fixing Symbiosis with Medicago Spp. mSystems 2022, 7, e0109221. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Biondi, E.G. Coordination of Symbiosis and Cell Cycle Functions in Sinorhizobium meliloti. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Kozdon, J.B.; McAdams, H.H.; Shapiro, L.; Collier, J. The Functions of DNA Methylation by CcrM in Caulobacter Crescentus: A Global Approach. Nucleic Acids Res. 2014, 42, 3720–3735. [Google Scholar] [CrossRef] [PubMed]
- Collier, J. Epigenetic Regulation of the Bacterial Cell Cycle. Curr. Opin. Microbiol. 2009, 12, 722–729. [Google Scholar] [CrossRef]
- Mohapatra, S.S.; Fioravanti, A.; Biondi, E.G. DNA Methylation in Caulobacter and Other Alphaproteobacteria during Cell Cycle Progression. Trends Microbiol. 2014, 22, 528–535. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Casadesús, J.; Low, D. Epigenetic Gene Regulation in the Bacterial World. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef] [PubMed]
- Bošković, A.; Rando, O.J. Transgenerational Epigenetic Inheritance. Annu Rev. Genet. 2018, 52, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Casadesús, J.; Low, D.A. Programmed Heterogeneity: Epigenetic Mechanisms in Bacteria. J. Biol. Chem. 2013, 288, 13929–13935. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA Methylation: The Mark and Its Mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Boulias, K.; Greer, E.L. Means, Mechanisms and Consequences of Adenine Methylation in DNA. Nat. Rev. Genet. 2022, 23, 411–428. [Google Scholar] [CrossRef]
- Kahramanoglou, C.; Prieto, A.I.; Khedkar, S.; Haase, B.; Gupta, A.; Benes, V.; Fraser, G.M.; Luscombe, N.M.; Seshasayee, A.S.N. Genomics of DNA Cytosine Methylation in Escherichia coli Reveals Its Role in Stationary Phase Transcription. Nat. Commun. 2012, 3, 886. [Google Scholar] [CrossRef] [PubMed]
- Bruneaux, M.; Kronholm, I.; Ashrafi, R.; Ketola, T. Roles of Adenine Methylation and Genetic Mutations in Adaptation to Different Temperatures in Serratia marcescens. Epigenetics 2022, 17, 861–881. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xiao, P.; Jiang, Y.; Dong, M.; Chen, Z.; Li, H.; Hu, Z.; Lei, A.; Wang, J. Transgenerational Epigenetic Inheritance under Environmental Stress by Genome-Wide DNA Methylation Profiling in Cyanobacterium. Front. Microbiol. 2018, 9, 1479. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, W.; Yin, M.; Huang, W.; Ye, B.; Li, P.; Shi, S.; Bai, G.; Guo, X.; Jin, Y.; et al. Changes in Higher-Order Chromosomal Structure of Klebsiella pneumoniae Under Simulated Microgravity. Front. Microbiol. 2022, 13, 879321. [Google Scholar] [CrossRef]
- Shell, S.S.; Prestwich, E.G.; Baek, S.H.; Shah, R.R.; Sassetti, C.M.; Dedon, P.C.; Fortune, S.M. DNA Methylation Impacts Gene Expression and Ensures Hypoxic Survival of Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003419. [Google Scholar] [CrossRef]
- Moler, E.R.V.; Abakir, A.; Eleftheriou, M.; Johnson, J.S.; Krutovsky, K.V.; Lewis, L.C.; Ruzov, A.; Whipple, A.V.; Rajora, O.P. Population Epigenomics: Advancing Understanding of Phenotypic Plasticity, Acclimation, Adaptation and Diseases. In Population Genomics; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Ribis, J.W.; Garrett, E.M.; Trzilova, D.; Kim, A.; Sekulovic, O.; Mead, E.A.; Pak, T.; Zhu, S.; Deikus, G.; et al. Epigenomic Characterization of Clostridioides difficile Finds a Conserved DNA Methyltransferase That Mediates Sporulation and Pathogenesis. Nat. Microbiol. 2020, 5, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Martín-Rodríguez, A.J. Respiration-Induced Biofilm Formation as a Driver for Bacterial Niche Colonization. Trends Microbiol. 2023, 31, 120–134. [Google Scholar] [CrossRef]
- Atack, J.M.; Yang, Y.; Seib, K.L.; Zhou, Y.; Jennings, M.P. A Survey of Type III Restriction-Modification Systems Reveals Numerous, Novel Epigenetic Regulators Controlling Phase-Variable Regulons; Phasevarions. Nucleic Acids Res. 2018, 46, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.R.; Owen, P.; Nataro, J.P. Molecular Switches—The ON and OFF of Bacterial Phase Variation. Mol. Microbiol. 1999, 33, 919–932. [Google Scholar] [CrossRef]
- Van Der Woude, M.W.; Bäumler, A.J. Phase and Antigenic Variation in Bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.S.; Chai, M.H.; Atack, J.M.; Furi, L.; De Ste Croix, M.; Haigh, R.; Trappetti, C.; Ogunniyi, A.D.; Shewell, L.K.; Boitano, M.; et al. A Random Six-Phase Switch Regulates Pneumococcal Virulence via Global Epigenetic Changes. Nat. Commun. 2014, 5, 5055. [Google Scholar] [CrossRef]
- Nye, T.M.; Fernandez, N.L.; Simmons, L.A. A Positive Perspective on DNA Methylation: Regulatory Functions of DNA Methylation Outside of Host Defense in Gram-Positive Bacteria. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 576–591. [Google Scholar] [CrossRef]
- Oliveira, F.A.; Paludo, K.S.; Arend, L.N.V.S.; Farah, S.M.S.S.; Pedrosa, F.O.; Souza, E.M.; Surek, M.; Picheth, G.; Fadel-Picheth, C.M.T. Virulence Characteristics and Antimicrobial Susceptibility of Uropathogenic Escherichia coli Strains. Genet. Mol. Res. 2011, 10, 4114–4125. [Google Scholar] [CrossRef]
- van der Woude, M.W.; Braaten, B.A.; Low, D.A. Evidence for Global Regulatory Control of Pilus Expression in Escherichia Coli by Lrp and DNA Methylation: Model Building Based on Analysis of Pap. Mol. Microbiol. 1992, 6, 2429–2435. [Google Scholar] [CrossRef]
- Vandenbussche, I.; Sass, A.; Pinto-Carbó, M.; Mannweiler, O.; Eberl, L.; Coenye, T. DNA Methylation Epigenetically Regulates Gene Expression in Burkholderia cenocepacia and Controls Biofilm Formation, Cell Aggregation, and Motility. mSphere 2020, 5, e00455-20. [Google Scholar] [CrossRef] [PubMed]
- Chhotaray, C.; Wang, S.; Tan, Y.; Ali, A.; Shehroz, M.; Fang, C.; Liu, Y.; Lu, Z.; Cai, X.; Adnan Hameed, H.M.; et al. Comparative Analysis of Whole-Genome and Methylome Profiles of a Smooth and a Rough Mycobacterium abscessus Clinical Strain. G3 Genes Genomes Genet. 2020, 10, 13–22. [Google Scholar] [CrossRef]
- García-Del Portillo, F.; Pucciarelli, M.G.; Casadesús, J. DNA Adenine Methylase Mutants of Salmonella typhimurium Show Defects in Protein Secretion, Cell Invasion, and M Cell Cytotoxicity. Proc. Natl. Acad. Sci. USA 1999, 96, 11578–11583. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.E.; Jarisch, J.; Smith, A.L. Inactivation of Deoxyadenosine Methyltransferase (Dam) Attenuates Haemophilus influenzae Virulence. Mol. Microbiol. 2004, 53, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Julio, S.M.; Heithoff, D.M.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. DNA Adenine Methylase Overproduction in Yersinia pseudotuberculosis Alters YopE Expression and Secretion and Host Immune Responses to Infection. Infect. Immun. 2002, 70, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Balbontín, R.; Rowley, G.; Pucciarelli, M.G.; López-Garrido, J.; Wormstone, Y.; Lucchini, S.; García-Del Portillo, F.; Hinton, J.C.D.; Casadesús, J. DNA Adenine Methylation Regulates Virulence Gene Expression in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2006, 188, 8160–8168. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Di Silvestre, C.; Gomez-Valero, L.; Buchrieser, C. Bacterial Methyltransferases: From Targeting Bacterial Genomes to Host Epigenetics. MicroLife 2022, 3, uqac014. [Google Scholar] [CrossRef] [PubMed]
- Douvlataniotis, K.; Bensberg, M.; Lentini, A.; Gylemo, B.; Nestor, C.E. No Evidence for DNA N6-Methyladenine in Mammals. Sci. Adv. 2020, 6, eaay3335. [Google Scholar] [CrossRef] [PubMed]
- Stolzenburg, S.; Goubert, D.; Rots, M.G. DNA Methyltransferases—Role and Function; Springer International: Berlin/Heidelberg, Germany, 2016; p. 945. [Google Scholar]
- Cohen, N.R.; Ross, C.A.; Jain, S.; Shapiro, R.S.; Gutierrez, A.; Belenky, P.; Li, H.; Collins, J.J. A Role for the Bacterial GATC Methylome in Antibiotic Stress Survival. Nat. Genet. 2016, 48, 581–586. [Google Scholar] [CrossRef]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage Orphan DNA Methyltransferases: Insights from Their Bacterial Origin, Function, and Occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef]
- Poole, A.M.; Phillips, M.J.; Penny, D. Prokaryote and Eukaryote Evolvability. In Proceedings of the BioSystems; Elsevier: Amsterdam, The Netherlands, 2003; Volume 69. [Google Scholar]
- Johnston, C.D.; Cotton, S.L.; Rittling, S.R.; Starr, J.R.; Borisy, G.G.; Dewhirst, F.E.; Lemon, K.P. Systematic Evasion of the Restriction-Modification Barrier in Bacteria. Proc. Natl. Acad. Sci. USA 2019, 166, 11454–11459. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.J.; Tricker, P.J. Epigenomic Plasticity within Populations: Its Evolutionary Significance and Potential. Heredity 2010, 105, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, S.; Okazaki, Y.; Anda, M.; Toyoda, A.; Nakano, S.-I.; Iwasaki, W. Metaepigenomic Analysis Reveals the Unexplored Diversity of DNA Methylation in an Environmental Prokaryotic Community. Nat. Commun. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.J.; Han, S.W.; Sul, W.J. Prokaryotic DNA Methylation and Its Functional Roles. J. Microbiol. 2021, 59, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P.C. Regulation of Genetic Flux between Bacteria by Restriction-Modification Systems. Proc. Natl. Acad. Sci. USA 2016, 113, 5658–5663. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P.C. The Interplay of Restriction-Modification Systems with Mobile Genetic Elements and Their Prokaryotic Hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef]
- Chen, Z.; Shen, M.; Mao, C.; Wang, C.; Yuan, P.; Wang, T.; Sun, D. A Type I Restriction Modification System Influences Genomic Evolution Driven by Horizontal Gene Transfer in Paenibacillus polymyxa. Front. Microbiol. 2021, 12, 709571. [Google Scholar] [CrossRef] [PubMed]
- Reva, O.N.; Swanevelder, D.Z.H.; Mwita, L.A.; Mwakilili, A.D.; Muzondiwa, D.; Joubert, M.; Chan, W.Y.; Lutz, S.; Ahrens, C.H.; Avdeeva, L.V.; et al. Genetic, Epigenetic and Phenotypic Diversity of Four Bacillus velezensis Strains Used for Plant Protection or as Probiotics. Front. Microbiol. 2019, 10, 2610. [Google Scholar] [CrossRef] [PubMed]
- Dordet-Frisoni, E.; Vandecasteele, C.; Contarin, R.; Sagné, E.; Baranowski, E.; Klopp, C.; Nouvel, L.X.; Citti, C. Impacts of Mycoplasma Agalactiae Restriction-Modification Systems on Pan-Epigenome Dynamics and Genome Plasticity. Microb. Genom. 2022, 8, mgen000829. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct Detection of DNA Methylation during Single-Molecule, Real-Time Sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA Cytosine Methylation Using Nanopore Sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.; Otto, C.; Stadler, P.F.; Langenberger, D. Comprehensive Benchmarking of Software for Mapping Whole Genome Bisulfite Data: From Read Alignment to DNA Methylation Analysis. Brief. Bioinform. 2021, 22, bbab021. [Google Scholar] [CrossRef]
- Su, S.; Gouil, Q.; Blewitt, M.E.; Cook, D.; Hickey, P.F.; Ritchie, M.E. NanoMethViz: An R/Bioconductor Package for Visualizing Long-Read Methylation Data. PLoS Comput. Biol. 2021, 17, e1009524. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; Stovner, E.B.; Strazisar, M. Methplotlib: Analysis of Modified Nucleotides from Nanopore Sequencing. Bioinformatics 2020, 36, 3236–3238. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Passeri, I.; Cangioli, L.; Fagorzi, C.; Mengoni, A.; Fondi, M. MeStudio: Crossing Methylation and Genomic Features for Comparative Epigenomic Analyses. Int. J. Mol. Sci. 2023, 24, 159. [Google Scholar] [CrossRef]
- Mouammine, A.; Collier, J. The Impact of DNA Methylation in Alphaproteobacteria. Mol. Microbiol. 2018, 110, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chahar, S.; Elsawy, H.; Ragozin, S.; Jeltsch, A. Changing the DNA Recognition Specificity of the EcoDam DNA-(Adenine-N6)-Methyltransferase by Directed Evolution. J. Mol. Biol. 2010, 395, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, L.; Podger, D.M.; Hall, R.M. A Mutation in the DNA Adenine Methylase Gene (Dam) of Salmonella typhimurium Decrease Susceptibility to 9-Aminoacridine-Induced Frameshift Mutagenesis. Mutat. Res. DNA Repair Rep. 1988, 194, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating Transcriptome and Proteome Profiling: Strategies and Applications. Proteomics 2016, 16, 2533–2544. [Google Scholar] [CrossRef]
- Bottacini, F.; Morrissey, R.; Roberts, R.J.; James, K.; Van Breen, J.; Egan, M.; Lambert, J.; Van Limpt, K.; Knol, J.; Motherway, M.O.C.; et al. Comparative Genome and Methylome Analysis Reveals Restriction/Modification System Diversity in the Gut Commensal Bifidobacterium breve. Nucleic Acids Res. 2018, 46, 1860–1877. [Google Scholar] [CrossRef]
- Gonzalez, D.; Collier, J. Genomic Adaptations to the Loss of a Conserved Bacterial DNA Methyltransferase. mBio 2015, 6, e00952-15. [Google Scholar] [CrossRef] [PubMed]
- Catania, S.; Dumesic, P.A.; Pimentel, H.; Nasif, A.; Stoddard, C.I.; Burke, J.E.; Diedrich, J.K.; Cook, S.; Shea, T.; Geinger, E.; et al. Evolutionary Persistence of DNA Methylation for Millions of Years after Ancient Loss of a De Novo Methyltransferase. Cell 2020, 180, 263–277.e20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, J.L.; Huang, L.N.; Mengoni, A.; Shu, W.S. Metagenomic Assembly: Reconstructing Genomes from Metagenomes. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2021; Volume 2242. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passeri, I.; Vaccaro, F.; Mengoni, A.; Fagorzi, C. Moving toward the Inclusion of Epigenomics in Bacterial Genome Evolution: Perspectives and Challenges. Int. J. Mol. Sci. 2024, 25, 4425. https://doi.org/10.3390/ijms25084425
Passeri I, Vaccaro F, Mengoni A, Fagorzi C. Moving toward the Inclusion of Epigenomics in Bacterial Genome Evolution: Perspectives and Challenges. International Journal of Molecular Sciences. 2024; 25(8):4425. https://doi.org/10.3390/ijms25084425
Chicago/Turabian StylePasseri, Iacopo, Francesca Vaccaro, Alessio Mengoni, and Camilla Fagorzi. 2024. "Moving toward the Inclusion of Epigenomics in Bacterial Genome Evolution: Perspectives and Challenges" International Journal of Molecular Sciences 25, no. 8: 4425. https://doi.org/10.3390/ijms25084425
APA StylePasseri, I., Vaccaro, F., Mengoni, A., & Fagorzi, C. (2024). Moving toward the Inclusion of Epigenomics in Bacterial Genome Evolution: Perspectives and Challenges. International Journal of Molecular Sciences, 25(8), 4425. https://doi.org/10.3390/ijms25084425