

Gut Bacteria Provide Genetic and Molecular Reporter Systems to Identify Specific Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
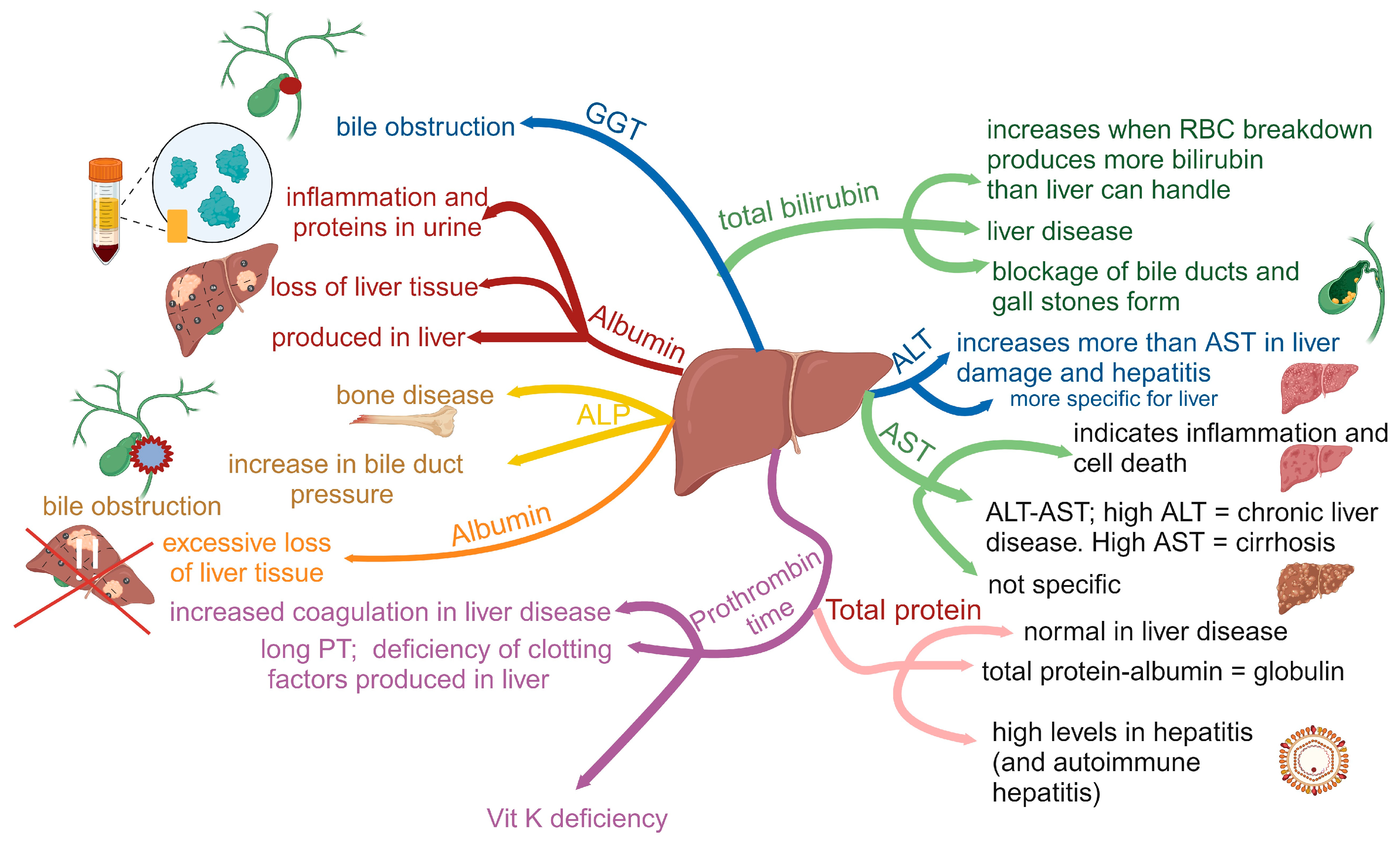
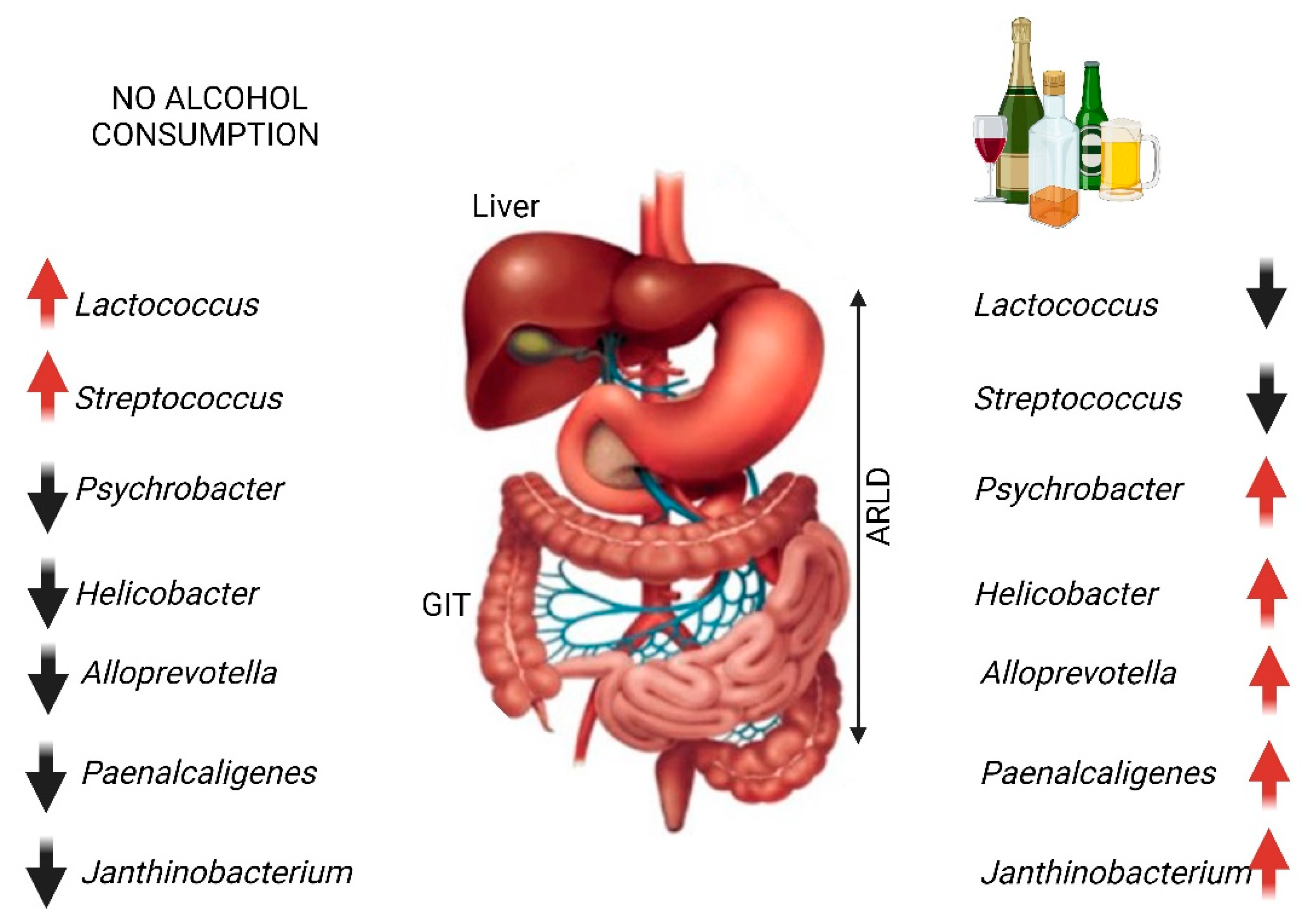
2. Alcohol-Related Liver Disease (ARLD)
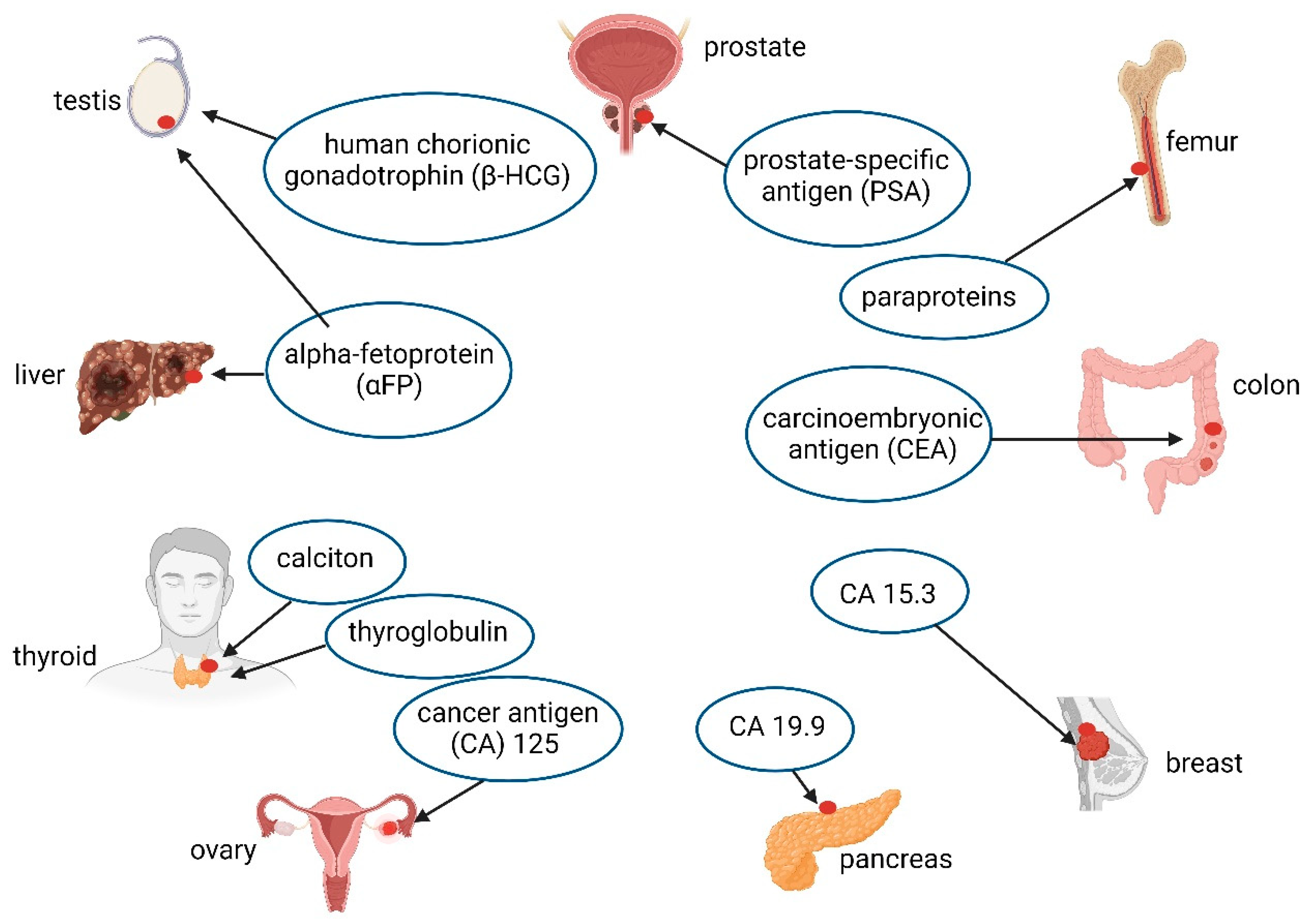
3. Cancer
4. Cognitive Impairment
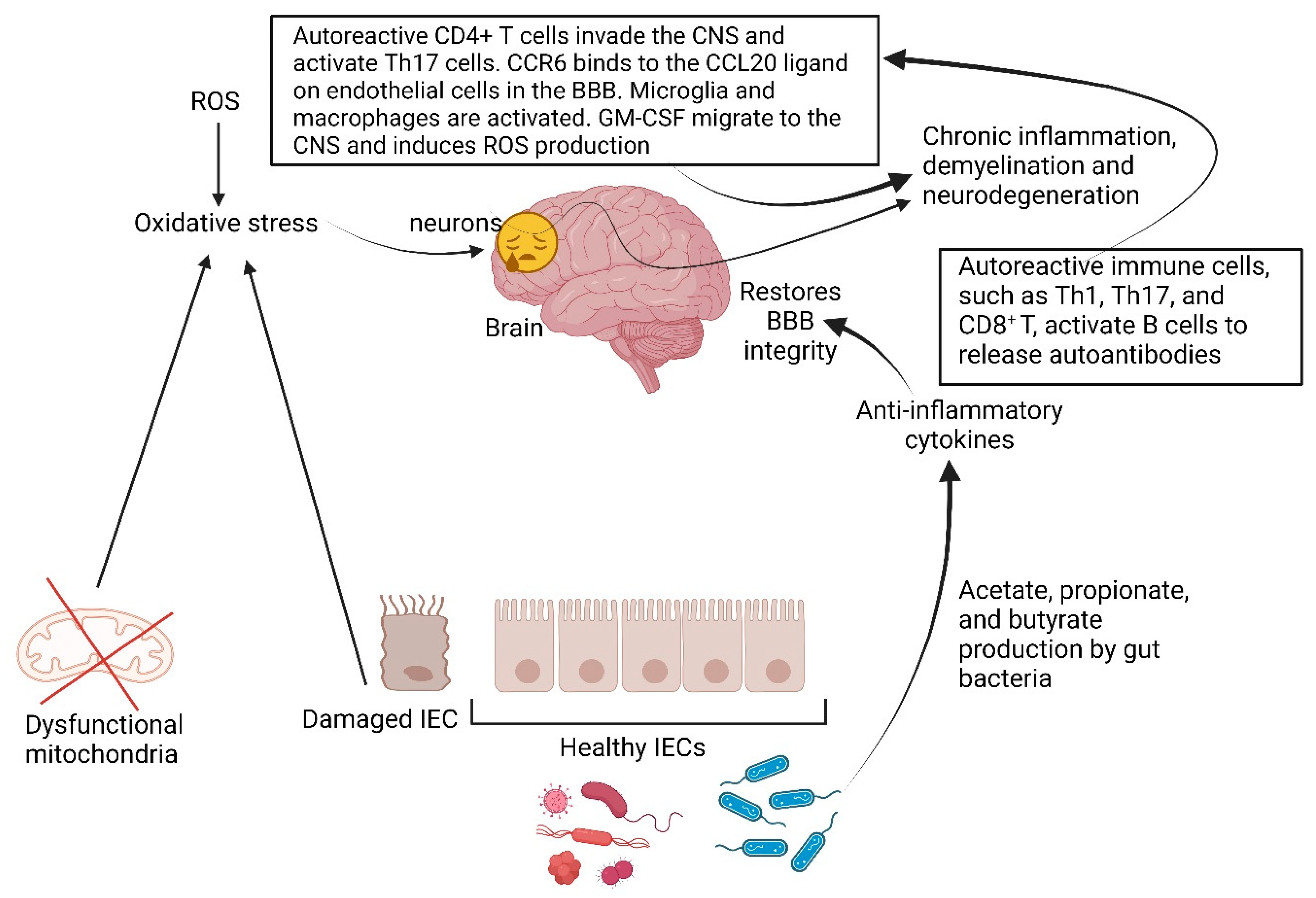
5. Multiple Sclerosis
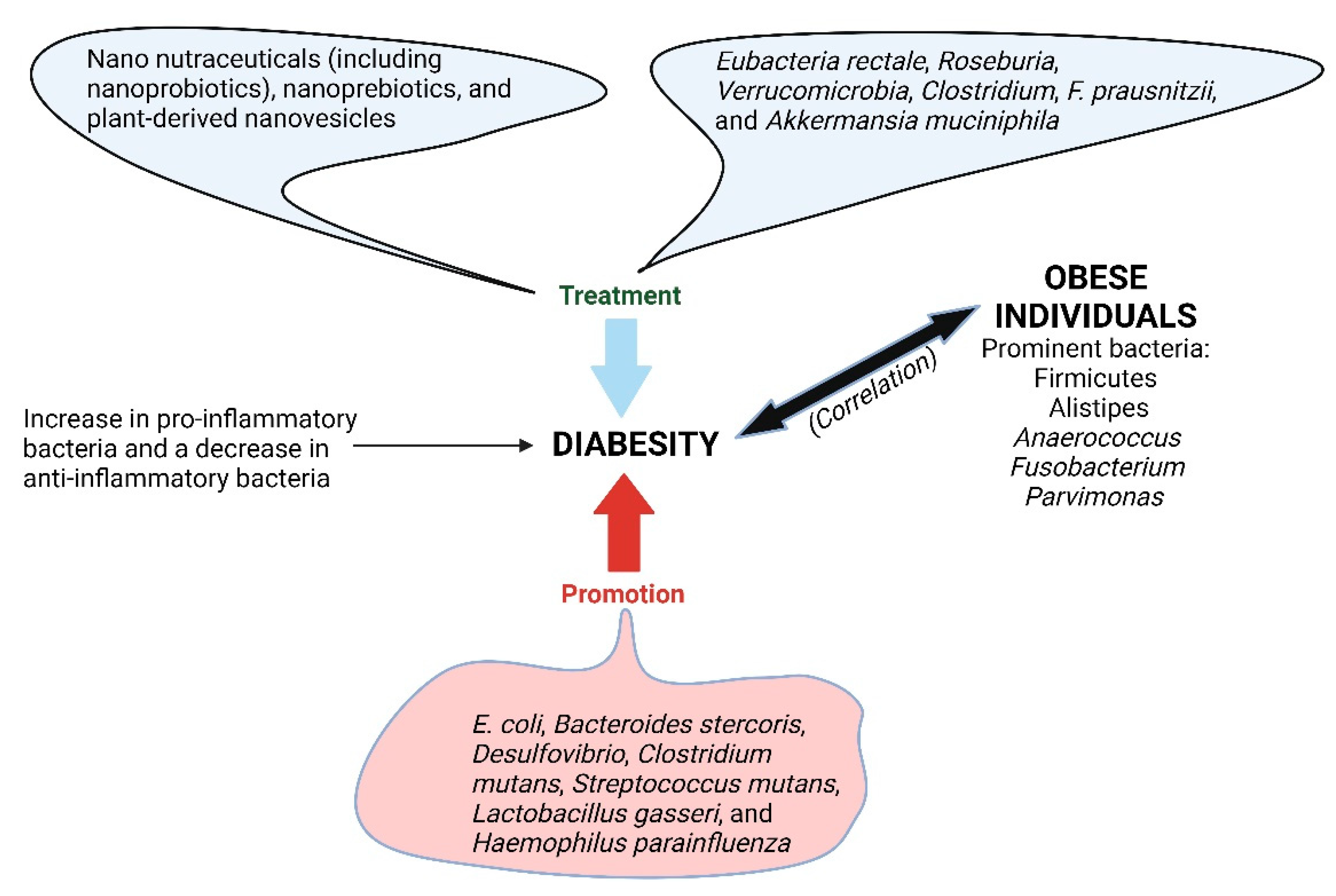
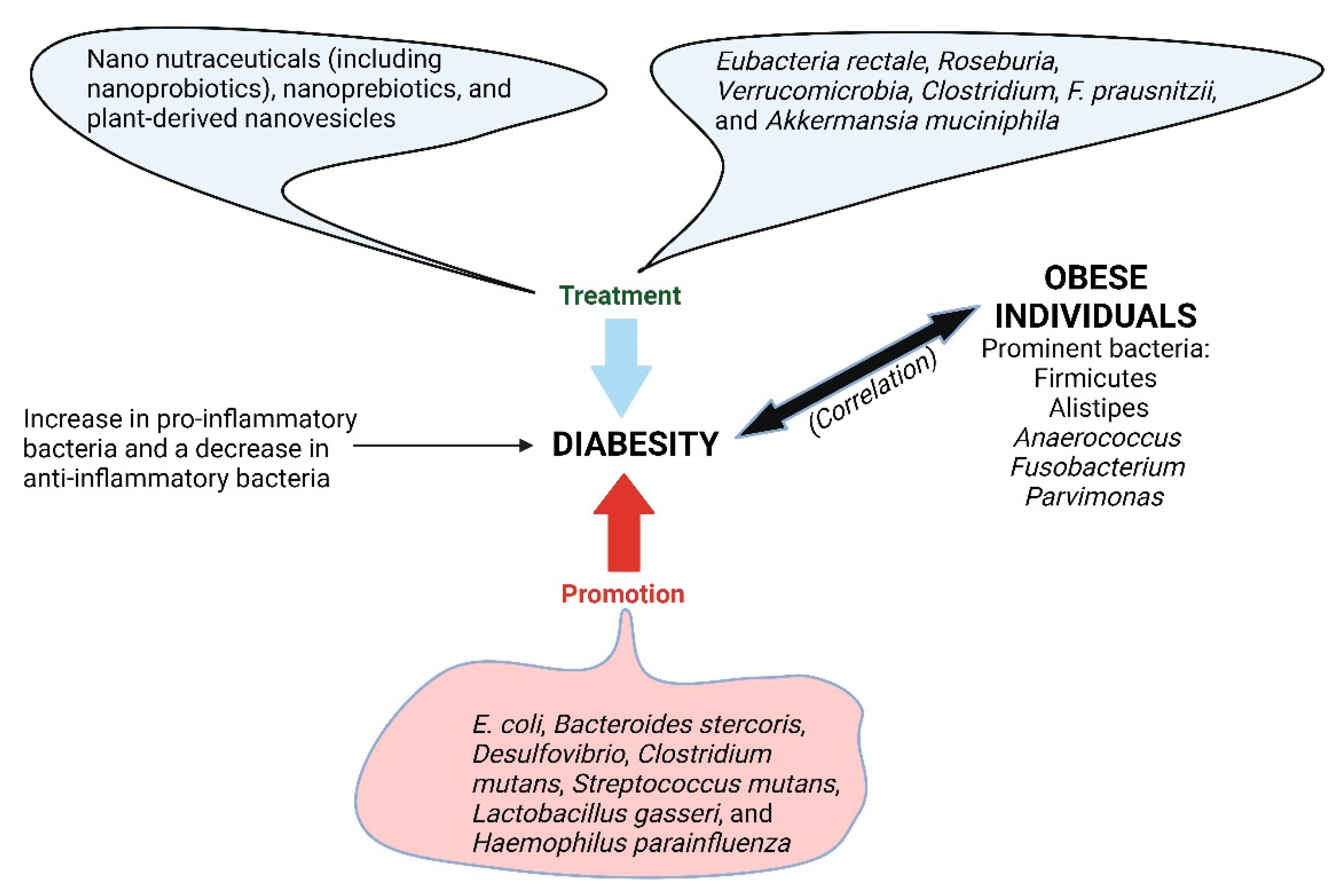
6. Diabesity
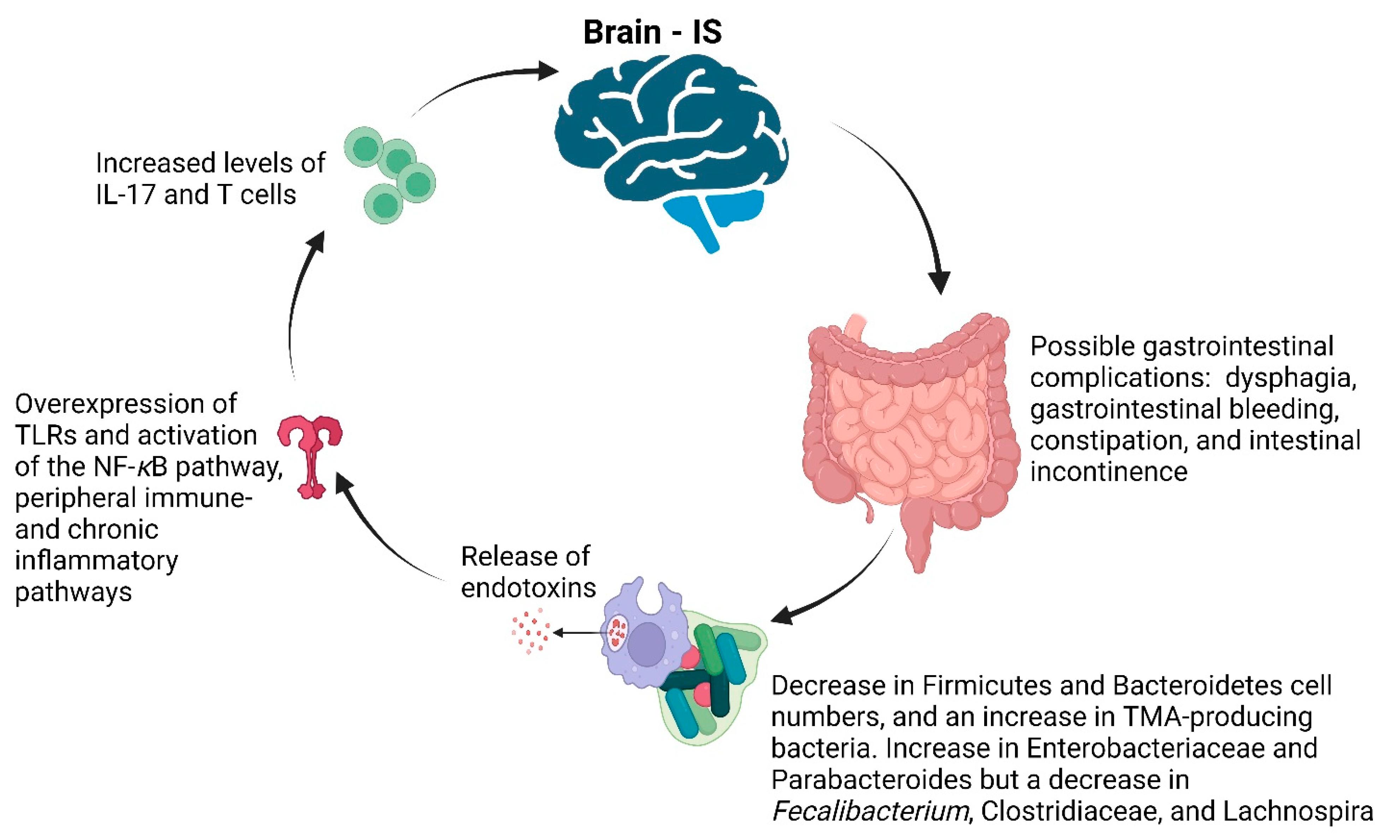
7. Stroke
8. Conclusions
Funding
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Dicks, L.; Geldenhuys, J.; Mikkelsen, L.; Brandsborg, E.; Marcotte, H. Our gut microbiota: A long walk to homeostasis. Benef. Microbes 2018, 9, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10, 1838. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Kist, M.; Blum, H.E. Diagnosis of small intestinal bacterial overgrowth in patients with cirrhosis of the liver: Poor performance of the glucose breath hydrogen test. J. Hepatol. 2000, 33, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Simren, M.; Stotzer, P.-O. Use and abuse of hydrogen breath tests. Gut 2006, 55, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Canakis, A.; Haroon, M.; Weber, H.C. Irritable bowel syndrome and gut microbiota. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 28–35. [Google Scholar] [CrossRef]
- Hillestad, E.M.R.; van der Meeren, A.; Nagaraja, B.H.; Bjørsvik, B.R.; Haleem, N.; Benitez-Paez, A.; Sanz, Y.; Hausken, T.; Lied, G.A.; Lundervold, A.; et al. Gut bless you: The microbiota-gut-brain axis in irritable bowel syndrome. World J. Gastroenterol. 2022, 28, 412–431. [Google Scholar] [CrossRef]
- Mandarino, F.V.; Sinagra, E.; Barchi, A.; Verga, M.C.; Brinch, D.; Raimondo, D.; Danese, S. Gastroparesis: The complex interplay with microbiota and the role of exogenous infections in the pathogenesis of the disease. Microorganisms 2023, 11, 1122. [Google Scholar] [CrossRef]
- Salmeri, N.; Sinagra, E.; Dolci, C.; Buzzaccarini, G.; Sozzi, G.; Sutera, M.; Candiani, M.; Ungaro, F.; Massimino, L.; Danese, S.; et al. Microbiota in irritable bowel syndrome and endometriosis: Birds of a feather flock together—A review. Microorganisms 2023, 11, 2089. [Google Scholar] [CrossRef]
- Duan, R.; Zhu, S.; Wang, B.; Duan, L. Alterations of gut microbiota in patients with irritable bowel syndrome based on 16S rRNA-targeted sequencing: A systematic review. Clin. Transl. Gastroenterol. 2019, 10, e00012. [Google Scholar] [CrossRef]
- Wang, L.; Alammar, N.; Singh, R.; Nanavati, J.; Song, Y.; Chaudhary, R.; Mullin, G.E. Gut microbial dysbiosis in the irritable bowel syndrome: A systematic review and meta-analysis of case-control studies. J. Acad. Nutr. Diet. 2020, 120, 565–586. [Google Scholar] [CrossRef]
- Saadi, M.; McCallum, R.W. Rifaximin in irritable bowel syndrome: Rationale, evidence and clinical use. Ther. Adv. Chronic Dis. 2013, 4, 71–75. [Google Scholar] [CrossRef]
- Lacy, B.E.; Chang, L.; Rao, S.S.; Heimanson, Z.; Sayuk, G.S. Rifaximin treatment for individual and multiple symptoms of irritable bowel syndrome with diarrhea: An analysis using new end points. Clin. Ther. 2023, 45, 198–209. [Google Scholar] [CrossRef]
- Fodor, A.A.; Pimentel, M.; Chey, W.D.; Lembo, A.; Golden, P.L.; Israel, R.J.; Carroll, I.M. Rifaximin is associated with modest, transient decreases in multiple taxa in the gut microbiota of patients with diarrhoea-predominant irritable bowel syndrome. Gut Microbes 2019, 10, 22–33. [Google Scholar] [CrossRef]
- Jung, H.K.; Locke, G.R., III; Schleck, C.D.; Zinsmeister, A.R.; Szarka, L.A.; Mullan, B.; Talley, N.J. The incidence, prevalence, and outcomes of patients with gastroparesis in Olmsted County, Minnesota, from 1996 to 2006. Gastroenterology 2009, 136, 1225–1233. [Google Scholar] [CrossRef]
- Ye, Y.; Jiang, B.; Manne, S.; Moses, P.L.; Almansa, C.; Bennett, D.; Dolin, P.; Ford, A.C. Epidemiology and outcomes of gastroparesis, as documented in general practice records, in the United Kingdom. Gut 2021, 70, 644–653. [Google Scholar] [CrossRef]
- Zhong, L.; Shanahan, E.R.; Raj, A.; Koloski, N.A.; Fletcher, L.; Morrison, M.; Walker, M.M.; Talley, N.J.; Holtmann, G. Dyspepsia and the microbiome: Time to focus on the small intestine. Gut 2017, 66, 1168–1169. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Viganò, P. Endometriosis. Nat. Rev. Dis. Prim. 2018, 4, 9. [Google Scholar] [CrossRef]
- Leonardi, M.; Hicks, C.; El-Assaad, F.; El-Omar, E.; Condous, G. Endometriosis and the microbiome: A systematic review. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 239–249. [Google Scholar] [CrossRef]
- Yuan, M.; Li, D.; Zhang, Z.; Sun, H.; An, M.; Wang, G. Endometriosis induces gut microbiota alterations in mice. Hum. Reprod. 2018, 33, 607–616. [Google Scholar] [CrossRef]
- Ni, Z.; Sun, S.; Bi, Y.; Ding, J.; Cheng, W.; Yu, J.; Zhou, L.; Li, M.; Yu, C. Correlation of fecal metabolomics and gut microbiota in mice with endometriosis. Am. J. Reprod. Immunol. 2020, 84, e13307. [Google Scholar] [CrossRef]
- Shan, J.; Ni, Z.; Cheng, W.; Zhou, L.; Zhai, D.; Sun, S.; Yu, C. Gut microbiota imbalance and its correlations with hormone and inflammatory factors in patients with stage 3/4 endometriosis. Arch. Gynecol. Obstet. 2021, 304, 1363–1373. [Google Scholar] [CrossRef]
- Ata, B.; Yildiz, S.; Turkgeldi, E.; Brocal, V.P.; Dinleyici, E.C.; Moya, A.; Urman, B. The endobiota study: Comparison of vaginal, cervical and gut microbiota between women with stage 3/4 endometriosis and healthy controls. Sci. Rep. 2019, 9, 2204. [Google Scholar] [CrossRef]
- Khan, K.N.; Kitajima, M.; Hiraki, K.; Yamaguchi, N.; Katamine, S.; Matsuyama, T.; Nakashima, M.; Fujishita, A.; Ishimaru, T.; Masuzaki, H. Escherichia coli contamination of menstrual blood and effect of bacterial endotoxin on endometriosis. Fertil. Steril. 2010, 94, 2860–2863.e3. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Hurn, D.; Hermanus, D. Gut bacteria and neuropsychiatric disorders. Microorganisms 2021, 9, 2583. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Van de Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef]
- Goldstein, A.; Hofstra, R.; Burns, A. Building a brain in the gut: Development of the enteric nervous system. Clin. Genet. 2013, 83, 307–316. [Google Scholar] [CrossRef]
- Rao, M.; Gershon, M.D. Neurogastroenterology: The dynamic cycle of life in the enteric nervous system. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 453–454. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Saberi, B.; Dadabhai, A.S.; Jang, Y.-Y.; Gurakar, A.; Mezey, E. Current management of alcoholic hepatitis and future therapies. J. Clin. Transl. Hepatol. 2016, 4, 113–122. [Google Scholar] [CrossRef]
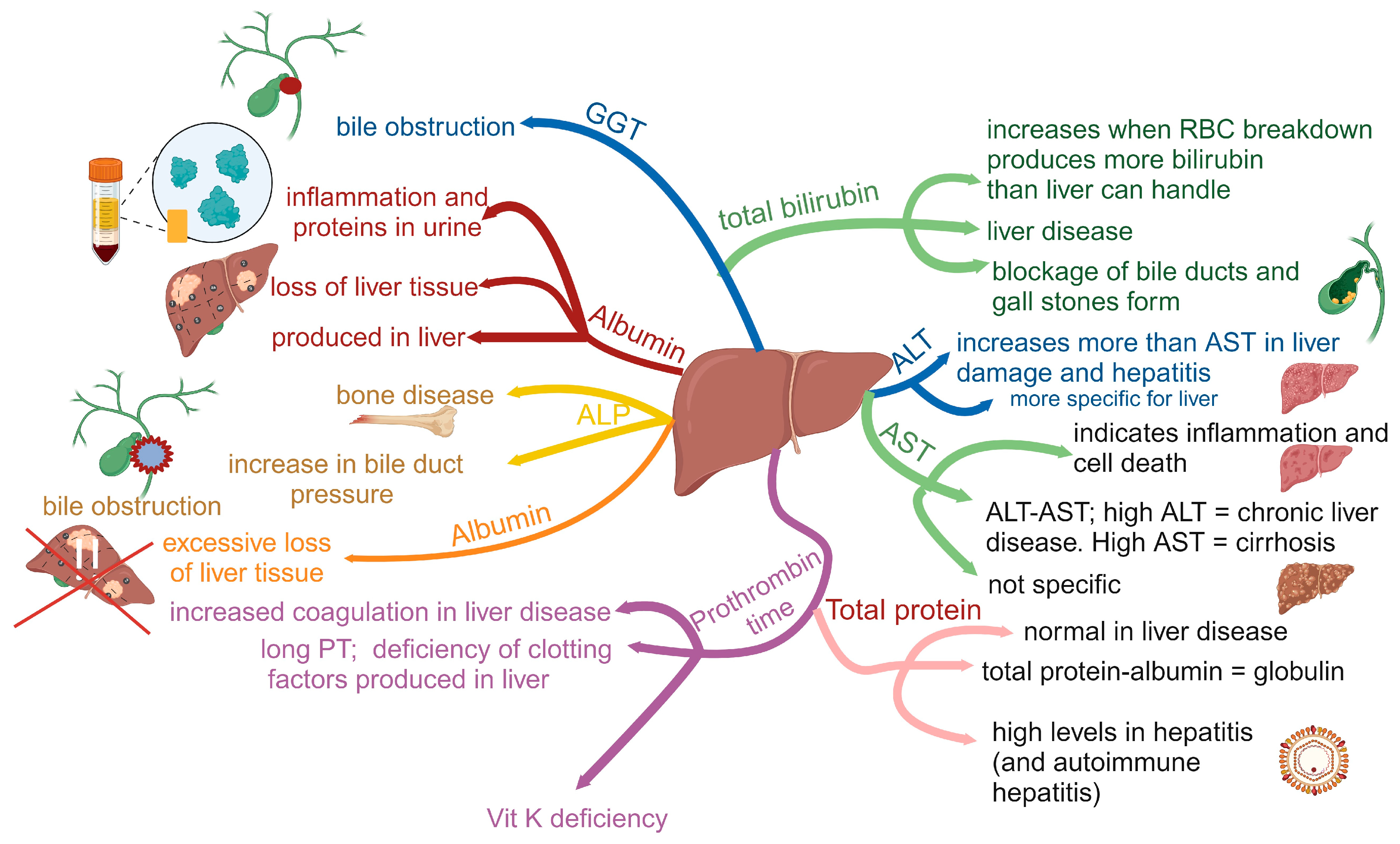
- Torruellas, C.; French, S.W.; Medici, V. Diagnosis of alcoholic liver disease. World J. Gastroenterol. 2014, 20, 11684–11699. [Google Scholar] [CrossRef]
- Conigrave, K.M.; Degenhardt, L.J.; Whitfield, J.B.; Saunders, J.B.; Helander, A.; Tabakoff, B. CDT, GGT, and AST as markers of alcohol use: The WHO/ISBRA collaborative project. Alcohol. Clin. Exp. Res. 2002, 26, 332–339. [Google Scholar] [CrossRef]
- Bortolotti, F.; De Paoli, G.; Tagliaro, F. Carbohydrate-deficient transferrin (CDT) as a marker of alcohol abuse: A critical review of the literature 2001–2005. J. Chromatogr. B 2006, 841, 96–109. [Google Scholar] [CrossRef]
- Rosman, A.S.; Lieber, C.S. Diagnostic utility of laboratory tests in alcoholic liver disease. Clin. Chem. 1994, 40, 1641–1651. [Google Scholar] [CrossRef]
- Diehl, A.M. Liver disease in alcohol abusers: Clinical perspective. Alcohol 2002, 27, 7–11. [Google Scholar] [CrossRef]
- Hietala, J.; Koivisto, H.; Latvala, J.; Anttila, P.; Niemelä, O. IgAs against acetaldehyde-modified red cell protein as a marker of ethanol consumption in male alcoholic subjects, moderate drinkers, and abstainers. Alcohol. Clin. Exp. Res. 2006, 30, 1693–1698. [Google Scholar] [CrossRef]
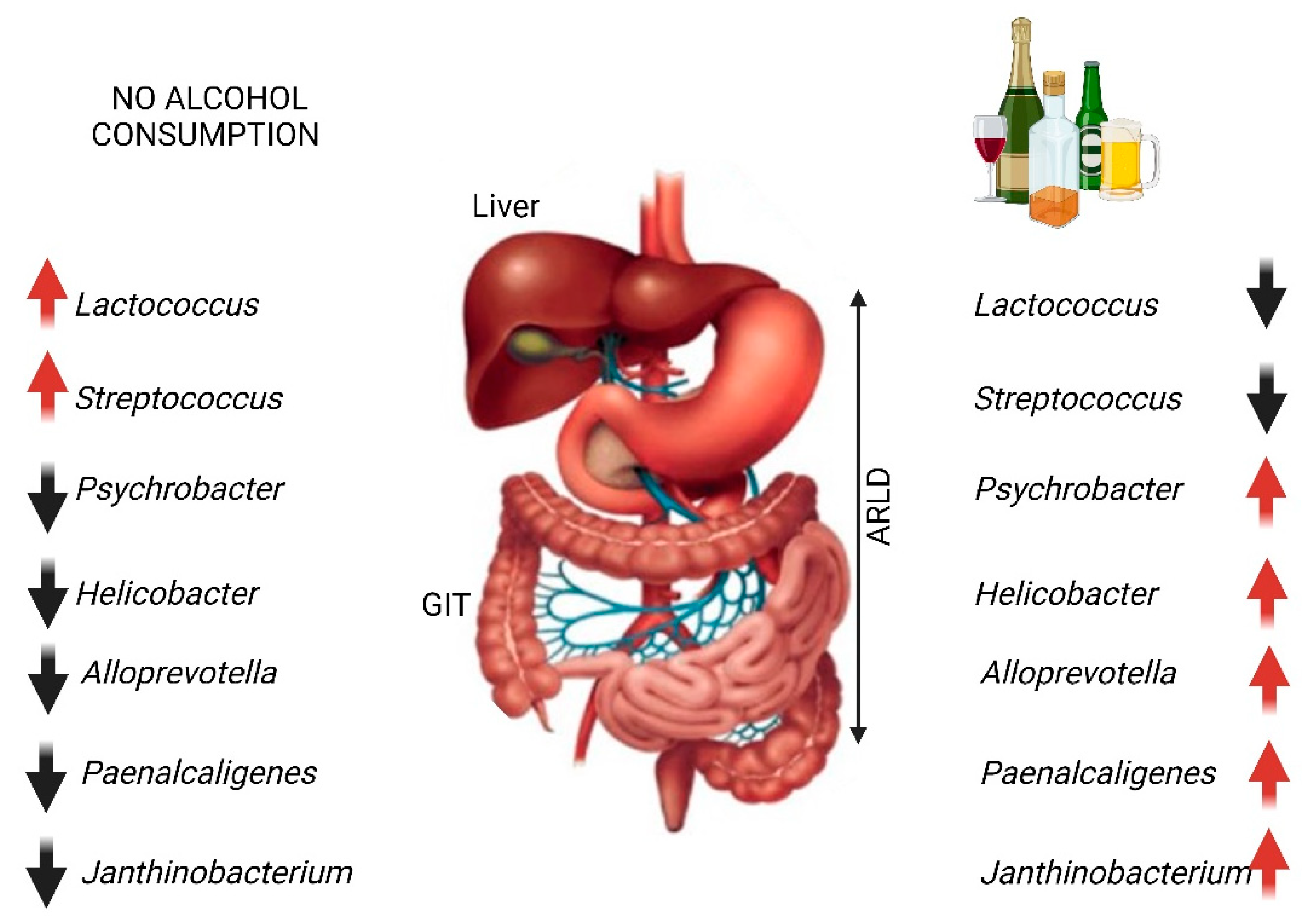
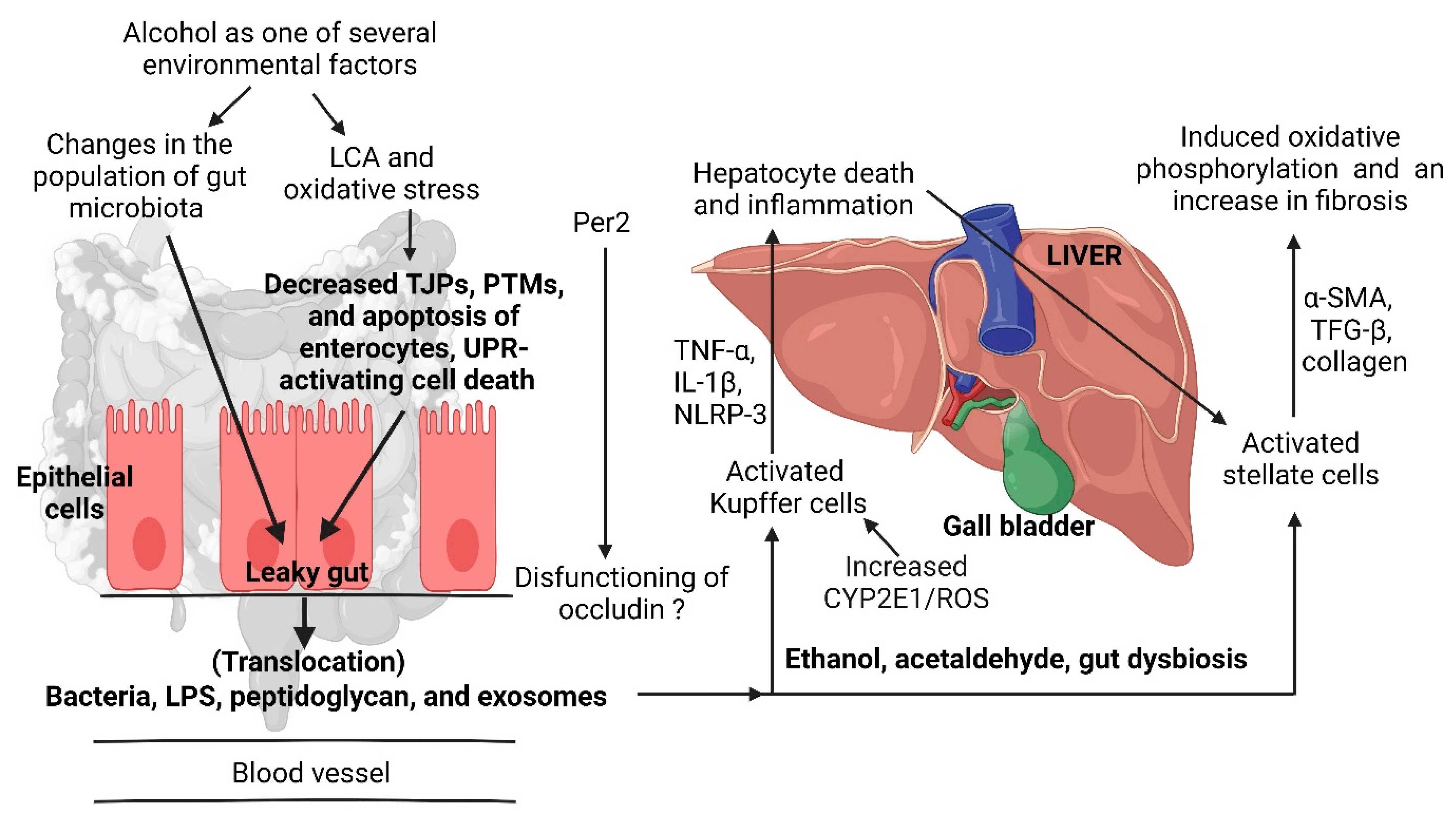
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Pan, C.; Liu, C.; Jia, W.; Zhao, D.; Chen, X.; Zhu, X.; Yang, M.; Wang, L. Alcohol drinking alters oral microbiota to modulate the progression of alcohol-related liver disease. iScience 2023, 26, 107977. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn’s disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol. Clin. Exp. Res. 2014, 38, 1489–1501. [Google Scholar] [CrossRef]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Stärkel, P.; van Pijkeren, J.-P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015, 148, 203–214.e16. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, srep08096. [Google Scholar] [CrossRef]
- Zeng, H.; Liu, J.; Jackson, M.I.; Zhao, F.Q.; Yan, L.; Combs, G.F., Jr. Fatty liver accompanies an increase in lactobacillus species in the hind gut of C57BL/6 mice fed a high-fat diet. J. Nutr. 2013, 143, 627–631. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Vermeulen, W. Do bacteria provide an alternative to cancer treatment and what role does lactic acid bacteria play? Microorganisms 2022, 10, 1733. [Google Scholar] [CrossRef]
- Hartmann, P.; Hochrath, K.; Horvath, A.; Chen, P.; Seebauer, C.T.; Llorente, C.; Wang, L.; Alnouti, Y.; Fouts, D.E.; Stärkel, P.; et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. J. Hepatol. 2017, 67, 2150–2166. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Adachi, T.; Kaminaga, T.; Yasuda, H.; Kamiya, T.; Hara, H. The involvement of endoplasmic reticulum stress in bile acid-induced hepatocellular injury. J. Clin. Biochem. Nutr. 2014, 54, 129–135. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef]
- Brandl, K.; Hartmann, P.; Jih, L.J.; Pizzo, D.P.; Argemi, J.; Ventura-Cots, M.; Coulter, S.; Liddle, C.; Ling, L.; Rossi, S.J.; et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J. Hepatol. 2018, 69, 396–405. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Wang, L.; Fouts, D.E.; Stärkel, P.; Hartmann, P.; Chen, P.; Llorente, C.; DePew, J.; Moncera, K.; Ho, S.B.; Brenner, D.A.; et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe 2016, 19, 227–239. [Google Scholar] [CrossRef]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef]
- Llorente, C.; Jepsen, P.; Inamine, T.; Wang, L.; Bluemel, S.; Wang, H.J. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat. Commun. 2017, 8, 837. [Google Scholar] [CrossRef]
- Bluemel, S.; Wang, L.; Martino, C.; Lee, S.; Wang, Y.; Williams, B.; Horvath, A.; Stadlbauer, V.; Zengler, K.; Schnabl, B. The role of intestinal C-type regenerating islet derived-3 lectins for nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Dicks, L.M.T.; Endo, A. Are fructophilic lactic acid bacteria (FLAB) beneficial to humans? Benef. Microbes 2022, 13, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Freese, K.; Waligora-Dupriet, A.-J.; Nubret, E.; Butel, M.-J.; Bergheim, I.; De Bandt, J.-P. Preventive effects of citrulline on Western diet-induced non-alcoholic fatty liver disease in rats. Br. J. Nutr. 2016, 116, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Rajilic-Stojanovic, M.; Jonkers, D.M.; Salonen, A.; Hanevik, K.; Raes, J.; Jalanka, J.; De Vos, W.M.; Manichanh, C.; Golic, N.; Enck, P.; et al. Intestinal microbiota and diet in IBS: Causes, consequences, or epiphenomena? Am. J. Gastroenterol. 2015, 110, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; He, J.; Gao, N.; Lu, X.; Li, M.; Wu, X.; Liu, Z.; Jin, Y.; Liu, J.; Xu, J.; et al. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci. Rep. 2017, 7, 45176. [Google Scholar] [CrossRef]
- Swanson, G.; Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Turek, F.W.; Keshavarzian, A. Role of intestinal circadian genes in alcohol-induced gut leakiness. Alcohol. Clin. Exp. Res. 2011, 35, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Sarin, S.K.; Pande, A.; Schnabl, B. Microbiome as a therapeutic target in alcohol-related liver disease. J. Hepatol. 2019, 70, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Min, Y.; Pang, K.; Wu, D. Therapeutic approach targeting gut microbiome in gastrointestinal infectious diseases. Int. J. Mol. Sci. 2023, 24, 15654. [Google Scholar] [CrossRef]
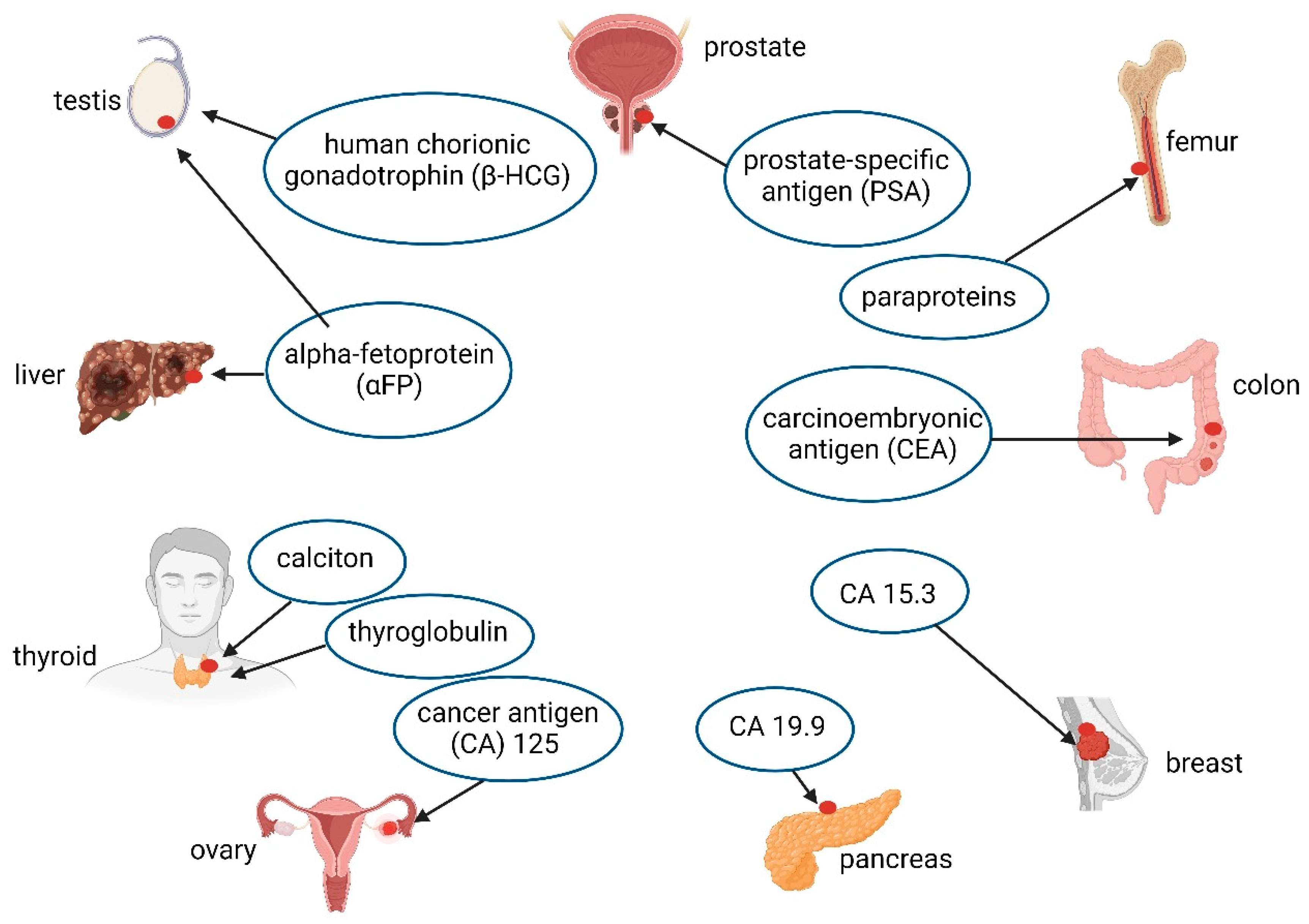
- Chu, T.M. Biochemical markers for human cancer. In Morphological Tumor Markers; Seifer, G., Ed.; Current Topics in Pathology; Springer: Berlin/Heidelberg, Germany, 1987; Volume 77. [Google Scholar] [CrossRef]
- Sayed, I.M.; Sahan, A.Z.; Venkova, T.; Chakraborty, A.; Mukhopadhyay, D.; Bimczok, D.; Beswick, E.J.; Reyes, V.E.; Pinchuk, I.; Sahoo, D.; et al. Helicobacter pylori infection downregulates the DNA glycosylase NEIL2, resulting in increased genome damage and inflammation in gastric epithelial cells. J. Biol. Chem. 2020, 295, 11082–11098. [Google Scholar] [CrossRef]
- Han, T.; Jing, X.; Bao, J.; Zhao, L.; Zhang, A.; Miao, R.; Guo, H.; Zhou, B.; Zhang, S.; Sun, J.; et al. H. pylori infection alters repair of DNA double-strand breaks via SNHG17. J. Clin. Investig. 2020, 130, 3901–3918. [Google Scholar] [CrossRef]
- Beil, W.; Enss, M.L.; Muller, S.; Obst, B.; Sewing, K.-F.; Wagner, S. Role of vacA and cagA in helicobacter pylori inhibition of mucin synthesis in gastric mucous cells. J. Clin. Microbiol. 2000, 38, 2215–2218. [Google Scholar] [CrossRef]
- Butcher, L.D.; den Hartog, G.; Ernst, P.B.; Crowe, S.E. Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 316–322. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Romero-Gallo, J.; Barry, D.P.; Hoge, S.; De Sablet, T.; Delgado, A.G.; Wroblewski, L.E.; Piazuelo, M.B.; Yan, F.; et al. Spermine oxidase mediates the gastric cancer risk associated eith Helicobacter pylori CagA. Gastroenterology 2011, 141, 1696–1708.e2. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates, DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Yao, B.; Dong, T.; Chen, Y.; Yao, J.; Liu, Y.; Li, H.; Bai, H.; Liu, X.; Zhang, Y.; et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 2022, 185, 1356–1372.e26. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Narunsky-Haziza, L.; Sepich-Poore, G.D.; Livyatan, I.; Asraf, O.; Martino, C.; Nejman, D.; Gavert, N.; Stajich, J.E.; Amit, G.; González, A.; et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 2022, 185, 3789–3806.e17. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral microbiota: Roles in cancer initiation, development and therapeutic efficacy. Signal Transduct. Target. Ther. 2023, 8, 35. [Google Scholar] [CrossRef]
- de Miranda, N.F.C.C.; Smit, V.T.H.B.M.; van der Ploeg, M.; Wesseling, J.; Neefjes, J. Absence of lipopolysaccharide (LPS) expression in breast cancer cells. bioRxiv 2023. [Google Scholar] [CrossRef]
- Dohlman, A.B.; Klug, J.; Mesko, M.; Gao, I.H.; Lipkin, S.M.; Shen, X.; Iliev, I.D. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 2022, 185, 3807–3822.e12. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.-A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Danhof, H.A.; Ruan, W.; Engevik, A.C.; Chang-Graham, A.L.; Engevik, K.A.; Shi, Z.; Zhao, Y.; Brand, C.K.; Krystofiak, E.S.; et al. Fusobacterium nucleatum secretes outer membrane vesicles and promotes intestinal inflammation. mBio 2021, 12, e02706-20. [Google Scholar] [CrossRef] [PubMed]
- Melmed, G.; Thomas, L.S.; Lee, N.; Tesfay, S.Y.; Lukasek, K.; Michelsen, K.S.; Zhou, Y.; Hu, B.; Arditi, M.; Abreu, M.T. Human intestinal epithelial cells are broadly unresponsive to toll-like receptor 2-dependent bacterial ligands: Implications for host-microbial interactions in the gut. J. Immunol. 2003, 170, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Abreu, M.T.; Fukata, M. Practical techniques for detection of Toll-like receptor-4 in the human intestine. Methods Mol. Biol. 2009, 517, 345–361. [Google Scholar]
- Liu, L.; Liang, L.; Yang, C.; Zhou, Y.; Chen, Y. Extracellular vesicles of Fusobacterium nucleatum compromise intestinal barrier through targeting RIPK1-mediated cell death pathway. Gut Microbes 2021, 13, 1902718. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Xia, Y.; Sun, J. Bacterial translocation and barrier dysfunction enhance colonic tumorigenesis. Neoplasia 2022, 35, 100847. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.-S.; Derosa, L.; Khan, A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, J.; Yang, Y.; Chen, Y. PD-1/PD-L1 checkpoint inhibitors in advanced hepatocellular carcinoma immunotherapy. Front. Immunol. 2022, 13, 1070961. [Google Scholar] [CrossRef]
- Park, J.S.; Gazzaniga, F.S.; Wu, M.; Luthens, A.K.; Gillis, J.; Zheng, W.; LaFleur, M.W.; Johnson, S.B.; Morad, G.; Park, E.M.; et al. Targeting PD-L2–RGMb overcomes microbiome-related immunotherapy resistance. Nature 2023, 617, 377–385. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2020, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.-M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Erratum: Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2014, 504, 446–450. [Google Scholar] [CrossRef]
- Lee, P.-C.; Wu, C.-J.; Hung, Y.-W.; Lee, C.J.; Chi, C.-T.; Lee, I.-C.; Yu-Lun, K.; Chou, S.-H.; Luo, J.-C.; Hou, M.-C.; et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor–treated unresectable hepatocellular carcinoma. J. Immunother. Cancer 2022, 10, e004779. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K.; Duda, D.G. The current landscape of immune checkpoint blockade in hepatocellular carcinoma: A review. JAMA Oncol. 2021, 7, 113–123. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, X.; Wang, M.; He, Z.; Li, H.; Wang, J.; Li, Q. Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 47. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Long, J.; Yang, X.; Lin, J.; Song, Y.; Xie, F.; Xun, Z.; Wang, Y.; Wang, Y.; et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J. Immunother. Cancer 2021, 9, e003334. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef]
- Jin, Y.; Dong, H.; Xia, L.; Yang, Y.; Zhu, Y.; Shen, Y.; Zheng, H.; Yao, C.; Wang, Y.; Lu, S. The diversity of gut microbiome is associated with favorable responses to anti–programmed death 1 immunotherapy in Chinese patients with NSCLC. J. Thorac. Oncol. 2019, 14, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, D.; Wang, Y.; Liu, L.; Li, J.; Yuan, J.; Jiang, Z.; Jiang, Z.; Hsiao, W.W.; Liu, H.; et al. Ginseng polysaccharides alter the gut microbiota and kynurenine/tryptophan ratio, potentiating the antitumour effect of antiprogrammed cell death 1/programmed cell death ligand 1 (anti-PD-1/PD-L1) immunotherapy. Gut 2022, 71, 734–745. [Google Scholar]
- Zhang, F.; Ferrero, M.; Dong, N.; D’auria, G.; Reyes-Prieto, M.; Herreros-Pomares, A.; Calabuig-Fariñas, S.; Duréndez, E.; Aparisi, F.; Blasco, A.; et al. Analysis of the gut microbiota: An emerging source of biomarkers for immune checkpoint blockade therapy in non-small cell lung cancer. Cancers 2021, 13, 2514. [Google Scholar] [CrossRef] [PubMed]
- Newsome, R.C.; Gharaibeh, R.Z.; Pierce, C.M.; da Silva, W.V.; Paul, S.; Hogue, S.R.; Yu, Q.; Antonia, S.; Conejo-Garcia, J.R.; Robinson, L.A.; et al. Interaction of bacterial genera associated with therapeutic response to immune checkpoint PD-1 blockade in a United States cohort. Genome Med. 2022, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.A.; Wilson, M.; Moran, U.; Pavlick, A.; Izsak, A.; Wechter, T.; Weber, J.S.; Osman, I.; Ahn, J. Relating the gut metagenome and metatranscriptome to immunotherapy responses in melanoma patients. Genome Med. 2019, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Gok Yavuz, B.; Hasanov, E.; Lee, S.S.; Mohamed, Y.I.; Curran, M.A.; Koay, E.J.; Cristini, V.; Kaseb, A.O. Current landscape and future directions of biomarkers for immunotherapy in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2021, 8, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, J.; Li, Q.; Xia, W.; Zhang, C.; Liu, Z.; Xiao, J.; Yi, Z.; Deng, H.; Xiao, Z.; et al. Gut microbiota and tumor immune escape: A new perspective for improving tumor immunotherapy. Cancers 2022, 14, 5317. [Google Scholar] [CrossRef] [PubMed]
- Behary, J.; Amorim, N.; Jiang, X.-T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Mohs, A.; Gui, W.; Galvez, E.J.C.; Candels, L.S.; Hoenicke, L.; Muthukumarasamy, U.; Holland, C.H.; Elfers, C.; Kilic, K.; et al. Imbalanced gut microbiota fuels hepatocellular carcinoma development by shaping the hepatic inflammatory microenvironment. Nat. Commun. 2022, 13, 3964. [Google Scholar] [CrossRef] [PubMed]
- Rossi, T.; Vergara, D.; Fanini, F.; Maffia, M.; Bravaccini, S.; Pirini, F. Microbiota-derived metabolites in tumor progression and metastasis. Int. J. Mol. Sci. 2020, 21, 5786. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef]
- Yamagishi, R.; Kamachi, F.; Nakamura, M.; Yamazaki, S.; Kamiya, T.; Takasugi, M.; Cheng, Y.; Nonaka, Y.; Yukawa-Muto, Y.; Thuy, L.T.T.; et al. Gasdermin D–mediated release of IL-33 from senescent hepatic stellate cells promotes obesity-associated hepatocellular carcinoma. Sci. Immunol. 2022, 7, eabl7209. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, F.; Zhuang, Y.; Xu, J.; Wang, J.; Mao, X.; Zhang, Y.; Liu, X. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kundu, P.; Seow, S.W.; de Matos, C.T.; Aronsson, L.; Chin, K.C.; Kärre, K.; Pettersson, S.; Greicius, G. Gut Microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis 2012, 33, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Huycke, M.M. Microbiome-driven carcinogenesis in colorectal cancer: Models and mechanisms. Free. Radic. Biol. Med. 2017, 105, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, X.; Cao, Y.; Fang, J.; Hong, J.; Chen, H. Fecal Fusobacterium nucleatum for the diagnosis of colorectal tumor: A systematic review and meta-analysis. Cancer Med. 2019, 8, 480–491. [Google Scholar] [CrossRef]
- Nannini, G.; Meoni, G.; Amedei, A.; Tenori, L. Metabolomics profile in gastrointestinal cancers: Update and future perspectives. World J. Gastroenterol. 2020, 26, 2514–2532. [Google Scholar] [CrossRef]
- Lin, Y.; Ma, C.; Bezabeh, T.; Wang, Z.; Liang, J.; Huang, Y.; Zhao, J.; Liu, X.; Ye, W.; Tang, W.; et al. 1H NMR-based metabolomics reveal overlapping discriminatory metabolites and metabolic pathway disturbances between colorectal tumor tissues and fecal samples. Int. J. Cancer 2019, 145, 1679–1689. [Google Scholar] [CrossRef]
- Le Gall, G.; Guttula, K.; Kellingray, L.; Tett, A.J.; Hoopen, R.T.; Kemsley, E.K.; Savva, G.M.; Ibrahim, A.; Narbad, A. Metabolite quantification of faecal extracts from colorectal cancer patients and healthy controls. Oncotarget 2018, 9, 33278–33289. [Google Scholar] [CrossRef]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef]
- Liu, S.; Dai, J.; Lan, X.; Fan, B.; Dong, T.; Zhang, Y.; Han, M. Intestinal bacteria are potential biomarkers and therapeutic targets for gastric cancer. Microb. Pathog. 2021, 151, 104747. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Nagasawa, M.; Ikeda, H.; Yasuo, S.; Koga, Y.; Furuse, M. Gut microbiota of mice putatively modifies amino acid metabolism in the host brain. Br. J. Nutr. 2017, 117, 775–783. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Sardone, R.; Dibello, V.; Di Lena, L.; D’urso, F.; Stallone, R.; Petruzzi, M.; Giannelli, G.; et al. Different cognitive frailty models and health- and cognitive-related outcomes in older age: From epidemiology to prevention. J. Alzheimer’s Dis. 2018, 62, 993–1012. [Google Scholar] [CrossRef]
- Schmidt, T.S.B.; Raes, J.; Bork, P. The human gut microbiome: From association to modulation. Cell 2018, 172, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Lin, C.-H.; Lane, H.-Y. D-glutamate and gut microbiota in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 2676. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, 6349. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sugahara, H.; Shimada, K.; Mitsuyama, E.; Kuhara, T.; Yasuoka, A.; Kondo, T.; Abe, K.; Xiao, J.-Z. Therapeutic potential of Bifidobacterium breve strain A1 for preventing cognitive impairment in Alzheimer’s disease. Sci. Rep. 2017, 7, 13510. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef] [PubMed]
- Nimgampalle, M.; Kuna, Y. Anti-Alzheimer properties of probiotic, Lactobacillus plantarum MTCC 1325 in Alzheimer’s disease induced albino rats. J. Clin. Diagn. Res. 2017, 11, KC01–KC05. [Google Scholar] [CrossRef] [PubMed]
- Azm, S.A.N.; Djazayeri, A.; Safa, M.; Azami, K.; Ahmadvand, B.; Sabbaghziarani, F.; Sharifzadeh, M.; Vafa, M. Lactobacilli and bifidobacteria ameliorate memory and learning deficits and oxidative stress in β-amyloid (1–42) injected rats. Appl. Physiol. Nutr. Metab. 2018, 43, 718–726. [Google Scholar] [CrossRef]
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: A randomized, double-blind and controlled trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Leblhuber, F.; Steiner, K.; Schuetz, B.; Fuchs, D.; Gostner, J.M. Probiotic supplementation in patients with Alzheimer’s dementia—An explorative intervention study. Curr. Alzheimer Res. 2018, 15, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Cushing, K.; Alvarado, D.M.; Ciorba, M.A. Butyrate and mucosal inflammation: New scientific evidence supports clinical observation. Clin. Transl. Gastroenterol. 2015, 6, e108. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.-H.; Cheng, Y.; Park, H.; Davidson, L.A.; Callaway, E.S.; Chapkin, R.S.; Jayaraman, A.; Asante, A.; Allred, C.; Weaver, E.A.; et al. Short chain fatty acids enhance aryl hydrocarbon (Ah) responsiveness in mouse colonocytes and Caco-2 human colon cancer cells. Sci. Rep. 2017, 7, 10163. [Google Scholar] [CrossRef]
- Sun, M.-F.; Shen, Y.-Q. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s disease. Ageing Res. Rev. 2018, 45, 53–61. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Galland, L. The Gut Microbiome and the brain. J. Med. Food 2014, 17, 1261–1272. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Abdel-Haq, R.; Schlachetzki, J.C.M.; Glass, C.K.; Mazmanian, S.K. Microbiome-microglia connections via the gut-brain axis. J. Exp. Med. 2019, 216, 41–59. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Tengeler, A.C.; Dam, S.A.; Wiesmann, M.; Naaijen, J.; van Bodegom, M.; Belzer, C.; Dederen, P.J.; Verweij, V.; Franke, B.; Kozicz, T.; et al. Gut microbiota from persons with attention-deficit/hyperactivity disorder affects the brain in mice. Microbiome 2020, 8, 44. [Google Scholar] [CrossRef]
- Xu, R.; Wu, B.; Liang, J.; He, F.; Gu, W.; Li, K.; Luo, Y.; Chen, J.; Gao, Y.; Wu, Z.; et al. Altered gut microbiota and mucosal immunity in patients with schizophrenia. Brain Behav. Immun. 2020, 85, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; He, S.; Fang, L.; Wang, B.; Bai, S.-J.; Xie, J.; Zhou, C.-J.; Wang, W.; Xie, P. Age-specific differential changes on gut microbiota composition in patients with major depressive disorder. Aging 2020, 12, 2764–2776. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Watling, M.; Imbimbo, B.P. Time to test antibacterial therapy in Alzheimer’s disease. Brain 2019, 142, 2905–2929. [Google Scholar] [CrossRef] [PubMed]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-García, J.; Osca-Verdegal, R.; Pallardó, F.V.; Ferreres, J.; Rodríguez, M.; Mulet, S.; Sanchis-Gomar, F.; Carbonell, N.; García-Giménez, J.L. Oxidative stress and inflammation in COVID-19-associated sepsis: The potential role of anti-oxidant therapy in avoiding disease progression. Antioxidants 2020, 9, 936. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The role of NLRP3 inflammasome in Alzheimer’s disease and potential therapeutic targets. Front. Pharmacol. 2022, 13, 845185. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Kapoor, R.; Felts, P.A. Demyelination: The role of reactive oxygen and nitrogen species. Brain Pathol. 1999, 9, 69–92. [Google Scholar] [CrossRef]
- Duarte-Silva, E.; Meuth, S.G.; Peixoto, C.A. Microbial metabolites in multiple sclerosis: Implications for pathogenesis and treatment. Front. Neurosci. 2022, 16, 885031. [Google Scholar] [CrossRef]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Do, J.-S.; Velankanni, P.; Lee, C.-G.; Kwon, H.-K. Gut microbial metabolites on host immune responses in health and disease. Immune Netw. 2023, 23, e6. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.-H.; May, C.; Wilck, N.; et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Bai, S.; Hao, Y.; Guan, Y. Fatty acids role in multiple sclerosis as “metabokines”. J. Neuroinflamm. 2022, 19, 157. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.D.; Lam, A.D.; Chiu, C.; Tran, G.T.; Hall, B.M.; Hodgkinson, S.J. Multiple sclerosis patients have reduced resting and increased activated CD4+CD25+FOXP3+T regulatory cells. Sci. Rep. 2021, 11, 10476. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Bitzer-Quintero, O.K.; Ramírez-Anguiano, A.C.; Flores-Alvarado, L.J.; Ramírez-Ramírez, V.; Macias-Islas, M.A.; Torres-Sánchez, E.D. Immunology and oxidative stress in multiple sclerosis: Clinical and basic approach. J. Immunol. Res. 2013, 2013, 708659. [Google Scholar] [CrossRef] [PubMed]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. CC chemokine receptor 6–regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Melzer, N.; Meuth, S.G.; Wiendl, H. CD8+T cells and neuronal damage: Direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J. 2009, 23, 3659–3673. [Google Scholar] [CrossRef]
- Maciak, K.; Pietrasik, S.; Dziedzic, A.; Redlicka, J.; Saluk-Bijak, J.; Bijak, M.; Włodarczyk, T.; Miller, E. Th17-related cytokines as potential discriminatory markers between neuromyelitis optica (Devic’s disease) and multiple sclerosis—A review. Int. J. Mol. Sci. 2021, 22, 8946. [Google Scholar] [CrossRef]
- Botía-Sánchez, M.; Alarcón-Riquelme, M.E.; Galicia, G. B cells and microbiota in autoimmunity. Int. J. Mol. Sci. 2021, 22, 4846. [Google Scholar] [CrossRef]
- van Langelaar, J.; Rijvers, L.; Janssen, M.; Wierenga-Wolf, A.F.; Melief, M.J.; Siepman, T.A.; de Vries, H.E.; Unger, P.P.; van Ham, S.M.; Hintzen, R.Q.; et al. Induction of brain-infiltrating T-bet-expressing B cells in multiple sclerosis. Ann. Neurol. 2019, 86, 264–278. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Kinnunen, T.; Chamberlain, N.; Morbach, H.; Cantaert, T.; Lynch, M.; Preston-Hurlburt, P.; Herold, K.C.; Hafler, D.A.; O’connor, K.C.; Meffre, E. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J. Clin. Investig. 2013, 123, 2737–2741. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Zorov, D.B.; Plotnikov, E.Y.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Zorov, S.D.; Babenko, V.A.; Jankauskas, S.S.; Popkov, V.A.; Savina, P.S. Microbiota and mitobiota. Putting an equal sign between mitochondria and bacteria. Biochemistry 2014, 79, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, T.P.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX dependent ROS generation and cell metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef]
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox signaling at the crossroads of human health and disease. MedComm 2022, 3, e127. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Hoedt, E.C.; Khushi, S.; Salim, A.A.; Bergot, A.S.; Schreiber, V.; Thomas, R.; McGuckin, M.A.; Florin, T.H.; Morrison, M.; et al. Secreted NF-κB suppressive microbial metabolites modulate gut inflammation. Cell Rep. 2022, 39, 110646. [Google Scholar] [CrossRef]
- Zhang, S.; Paul, S.; Kundu, P. NF-κB regulation by gut microbiota decides homeostasis or disease outcome during ageing. Front. Cell Dev. Biol. 2022, 10, 874940. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, B.J.; Liu, K.H.; Owens, J.A.; Hunter-Chang, S.; Camacho, M.C.; Eboka, R.U.; Chandrasekharan, B.; Baker, N.F.; Darby, T.M.; Robinson, B.S.; et al. Gut-resident lactobacilli activate hepatic Nrf2 and protect against oxidative liver injury. Cell Metab. 2020, 31, 956–968.e5. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.J.; De Plaen, I.G. Inflammatory signaling in NEC: Role of NF-κB, cytokines and other inflammatory mediators. Pathophysiology 2014, 21, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Conway, S. Bacterial modulation of mucosal innate immunity. Mol. Immunol. 2005, 42, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Dopkins, N.; Becker, W.; Miranda, K.; Walla, M.; Nagarkatti, P.; Nagarkatti, M. Tryptamine attenuates experimental multiple sclerosis through activation of aryl hydrocarbon receptor. Front. Pharmacol. 2020, 11, 619265. [Google Scholar] [CrossRef]
- Melnikov, M.; Kasatkin, D.; Lopatina, A.; Spirin, N.; Boyko, A.; Pashenkov, M. Serotonergic drug repurposing in multiple sclerosis: A new possibility for disease-modifying therapy. Front. Neurol. 2022, 13, 920408. [Google Scholar] [CrossRef]
- Hernandez, A.M.S.; Singh, C.; Valero, D.J.; Nisar, J.; Ramirez, J.I.T.; Kothari, K.K.; Isola, S.; Gordon, D.K. Multiple sclerosis and serotonin: Potential therapeutic applications. Cureus 2020, 12, e11293. [Google Scholar] [CrossRef]
- Muñoz-Jurado, A.; Escribano, B.M.; Caballero-Villarraso, J.; Galván, A.; Agüera, E.; Santamaría, A.; Túnez, I. Melatonin and multiple sclerosis: Antioxidant, anti-inflammatory and immunomodulator mechanism of action. Inflammopharmacology 2022, 30, 1569–1596. [Google Scholar] [CrossRef]
- Neveu, V.; Nicolas, G.; Amara, A.; Salek, R.M.; Scalbert, A. The human microbial exposome: Expanding the exposome-explorer database with gut microbial metabolites. Sci. Rep. 2023, 13, 1946. [Google Scholar] [CrossRef]
- Ng, A.C.T.; Delgado, V.; Borlaug, B.A.; Bax, J.J. Diabesity: The combined burden of obesity and diabetes on heart disease and the role of imaging. Nat. Rev. Cardiol. 2020, 18, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Rathod, P.; Yadav, R.P. Anti-diabesity potential of various multi functional natural molecules. J. Herb. Med. 2021, 27, 100430. [Google Scholar] [CrossRef]
- Andoh, A.; Nishida, A.; Takahashi, K.; Inatomi, O.; Imaeda, H.; Bamba, S.; Kito, K.; Sugimoto, M.; Kobayashi, T. Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J. Clin. Biochem. Nutr. 2016, 59, 65–70. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Ghebretatios, M.; Schaly, S.; Prakash, S. Nanoparticles in the foodindustry and their impact on human gut microbiome and diseases. Int. J. Mol. Sci. 2021, 22, 1942. [Google Scholar] [CrossRef] [PubMed]
- Iatcu, C.O.; Steen, A.; Covasa, M. Gut microbiota and complications of type-2 diabetes. Nutrients 2021, 14, 166. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Hartstra, A.V.; Bouter, K.E.; Bäckhed, F.; Nieuwdorp, M. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care 2015, 38, 159–165. [Google Scholar] [CrossRef]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 2016, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, B.; Wang, W.-Z.; Lee, L.-M.; Zhang, S.-H.; Kong, L.-Z. Stroke in China: Epidemiology, prevention, and management strategies. Lancet Neurol. 2007, 6, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.W.; Wong, C.H.Y. An unexplored brain-gut microbiota axis in stroke. Gut Microbes 2017, 8, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Roth, S.; Llovera, G.; Sadler, R.; Garzetti, D.; Stecher, B.; Dichgans, M.; Liesz, A. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J. Neurosci. 2016, 36, 7428–7440. [Google Scholar] [CrossRef] [PubMed]
- Houlden, A.; Goldrick, M.; Brough, D.; Vizi, E.S.; Lénárt, N.; Martinecz, B.; Roberts, I.S.; Denes, A. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav. Immun. 2016, 57, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Mudd, A.T.; Berding, K.; Wang, M.; Donovan, S.M.; Dilger, R.N. Serum cortisol mediates the relationship between fecal Ruminococcus and brain N-acetyl aspartate in the young pig. Gut Microbes 2017, 8, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Chen, N.; Sun, H.; Li, Z.; Chen, X.; Zhou, J.; Zhang, Y. Roseburia abundance associates with severity, evolution and outcome of acute ischemic stroke. Front. Cell. Infect. Microbiol. 2021, 11, 669322. [Google Scholar] [CrossRef]
- Haak, B.W.; Westendorp, W.F.; van Engelen, T.S.R.; Brands, X.; Brouwer, M.C.; Vermeij, J.-D.; Hugenholtz, F.; Verhoeven, A.; Derks, R.J.; Giera, M.; et al. Disruptions of anaerobic gut bacteria are associated with stroke and post-stroke infection: A prospective case–Control study. Transl. Stroke Res. 2020, 12, 581–592. [Google Scholar] [CrossRef]
- Xia, G.H.; You, C.; Gao, X.X.; Zeng, X.L.; Zhu, J.J.; Xu, K.Y.; Tan, C.H.; Xu, R.T.; Wu, Q.H.; Zhou, H.W.; et al. Stroke dysbiosis index (SDI) in gut microbiome are associated with brain injury and prognosis of stroke. Front. Neurol. 2019, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lawson, M.A.; Dantzer, R.; Kelley, K.W. LPS induced indoleamine 2,3-dioxygenase is regulated in an evidence-based complementary and alternative medicine 11 interferon-c-independent manner by a JNK signaling pathway in primary murine microglia. Brain Behav. Immun. 2010, 24, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Packer, C.S.; Rice, A.E.; Johnson, T.C.; Pelaez, N.J.; Temm, C.J.; Potter, G.V.; White, W.A.; Roth, A.H.; Dominguez, J.H.; Peterson, R.G. Oxidized lowdensity lipoprotein (OX-LDL) induced arterial muscle contraction signaling mechanisms. Open Hypertens. J. 2014, 6, 20–26. [Google Scholar] [CrossRef]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond gut feelings: How the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef]
- Wang, H.-X.; Wang, Y.-P. Gut microbiota-brain axis. Chin. Med. J. 2016, 129, 2373–2380. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dicks, L.M.T. Gut Bacteria Provide Genetic and Molecular Reporter Systems to Identify Specific Diseases. Int. J. Mol. Sci. 2024, 25, 4431. https://doi.org/10.3390/ijms25084431
Dicks LMT. Gut Bacteria Provide Genetic and Molecular Reporter Systems to Identify Specific Diseases. International Journal of Molecular Sciences. 2024; 25(8):4431. https://doi.org/10.3390/ijms25084431
Chicago/Turabian StyleDicks, Leon M. T. 2024. "Gut Bacteria Provide Genetic and Molecular Reporter Systems to Identify Specific Diseases" International Journal of Molecular Sciences 25, no. 8: 4431. https://doi.org/10.3390/ijms25084431