
SLCO1B1 Exome Sequencing and Statin Treatment Response in 64,000 UK Biobank Patients


Abstract
:1. Introduction
2. Results
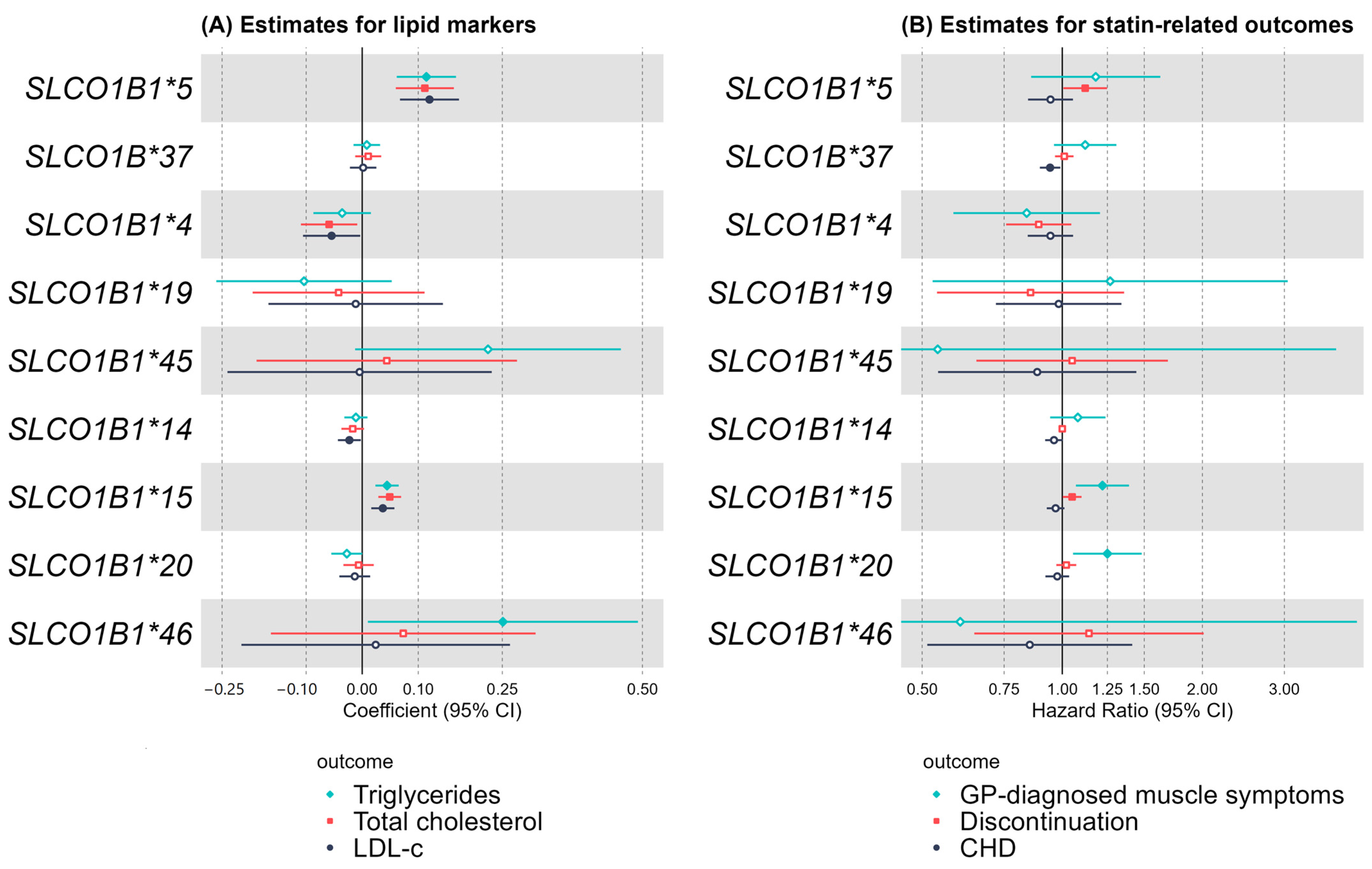
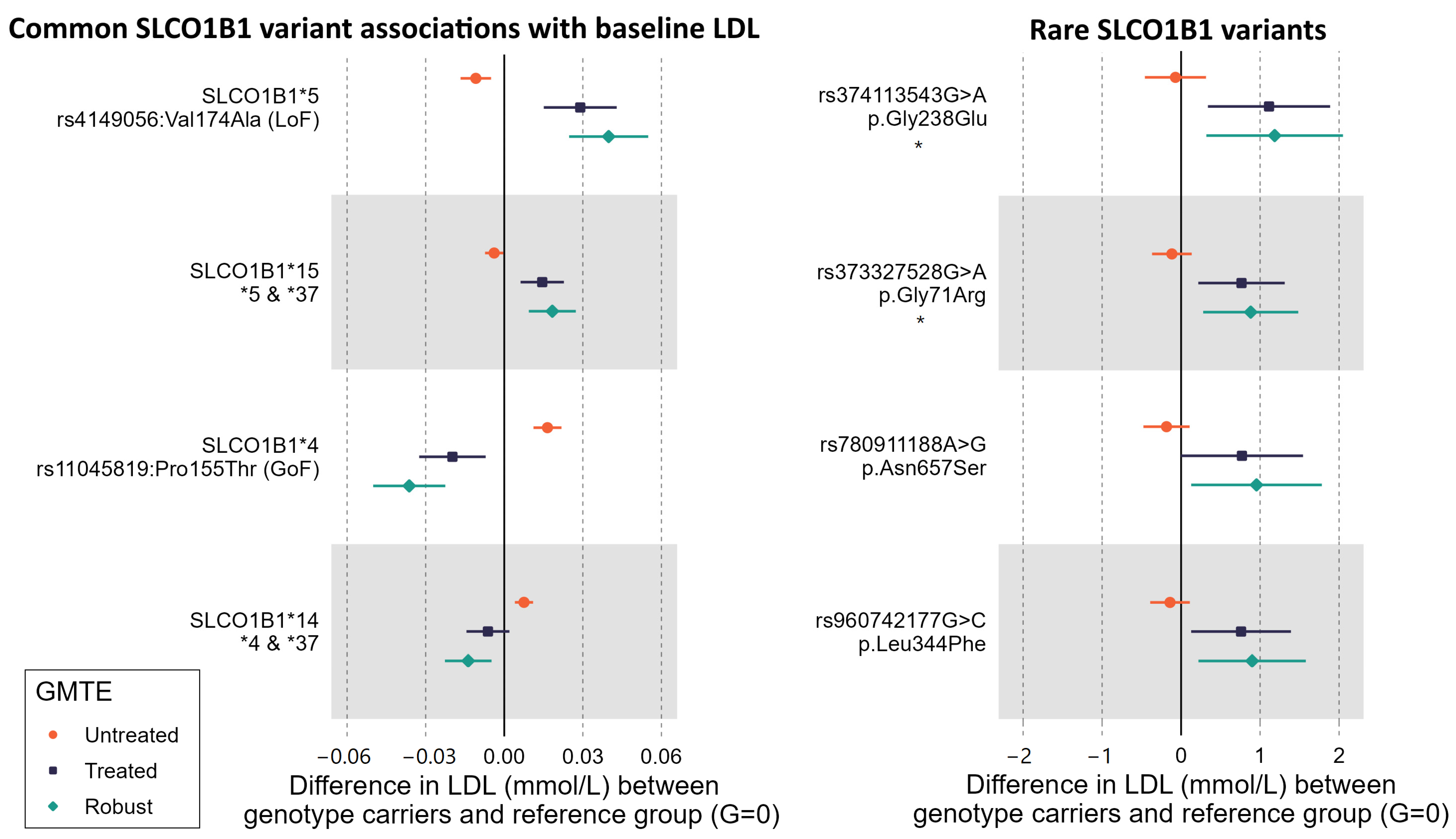
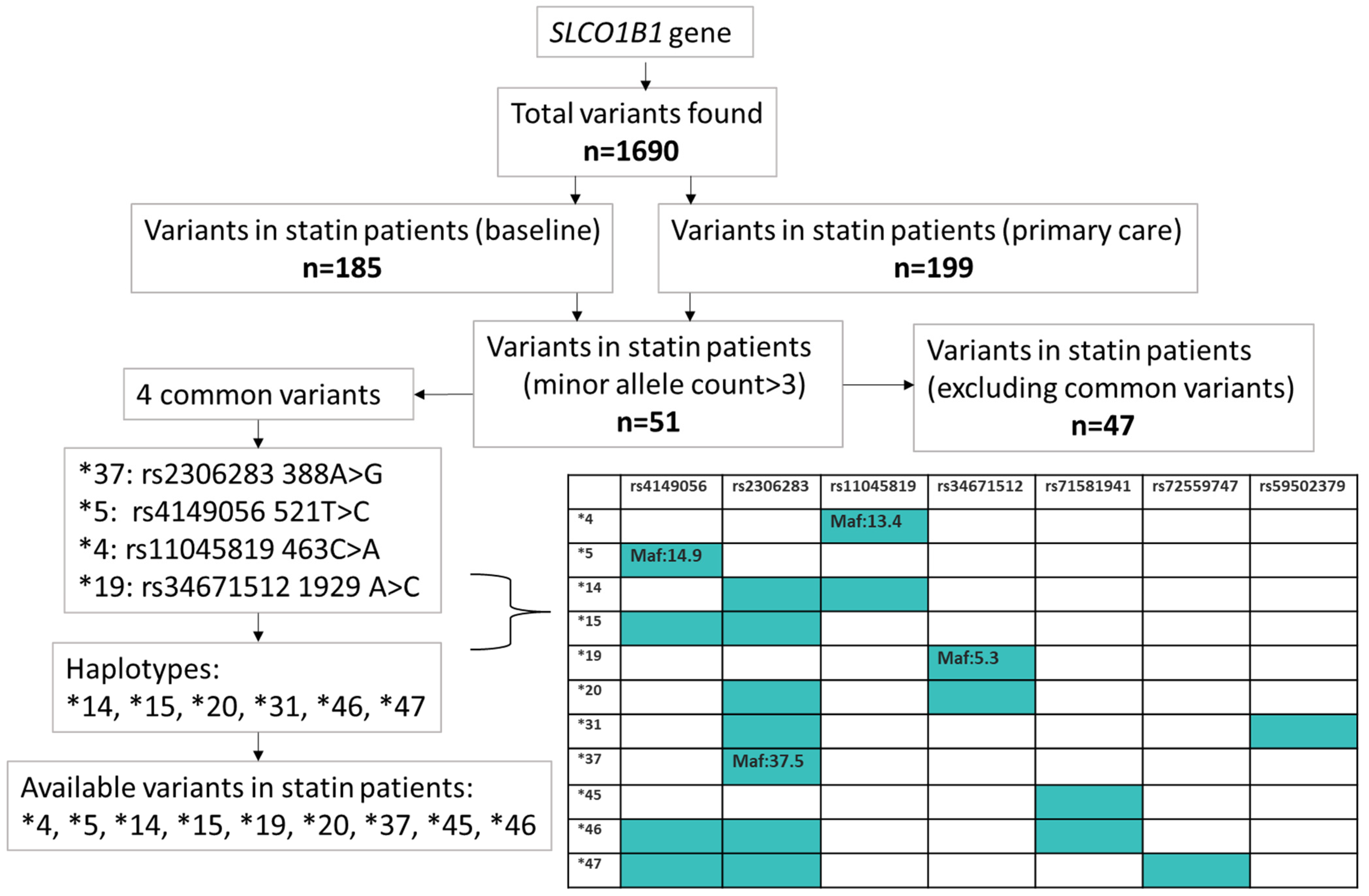
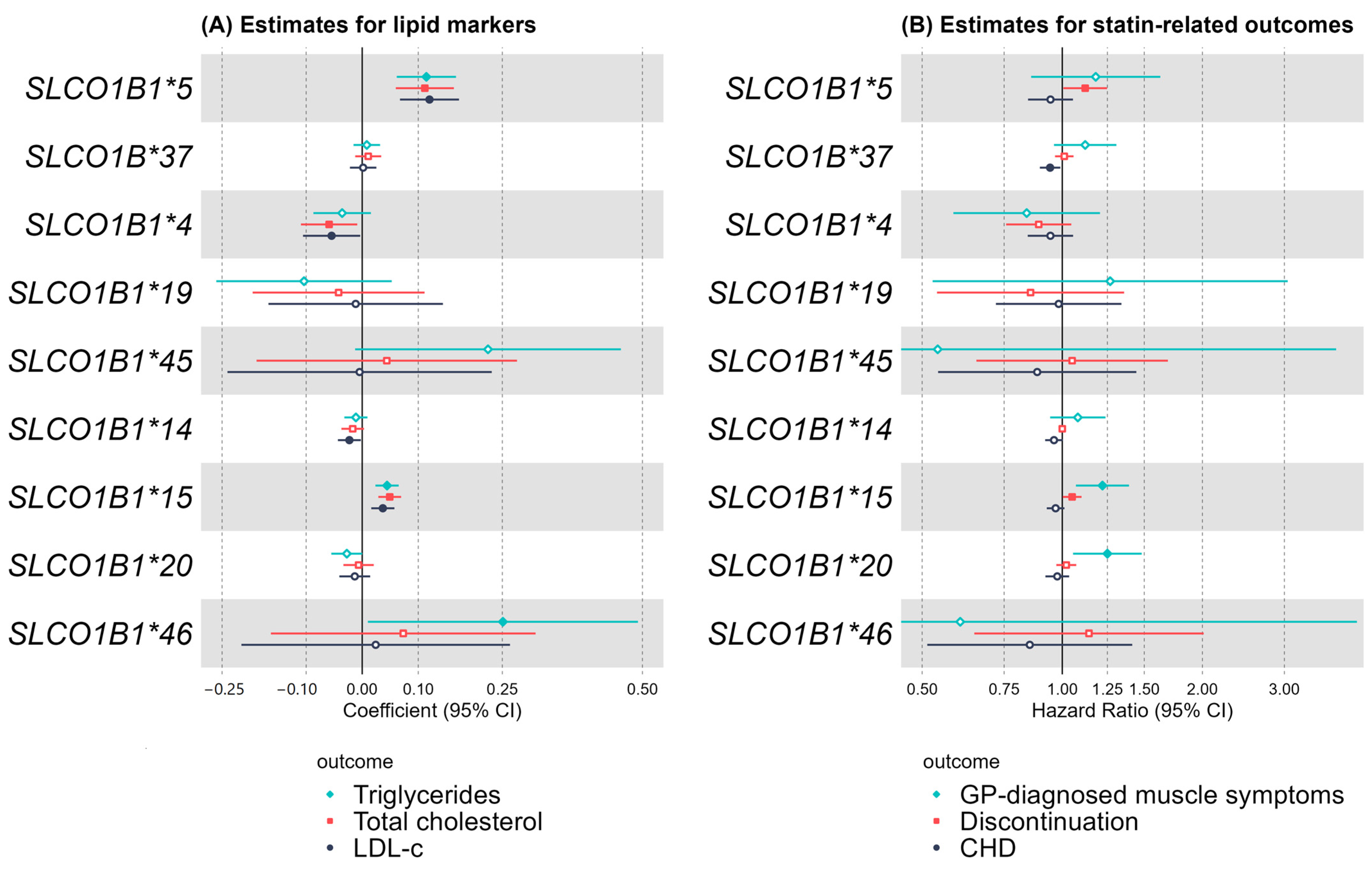
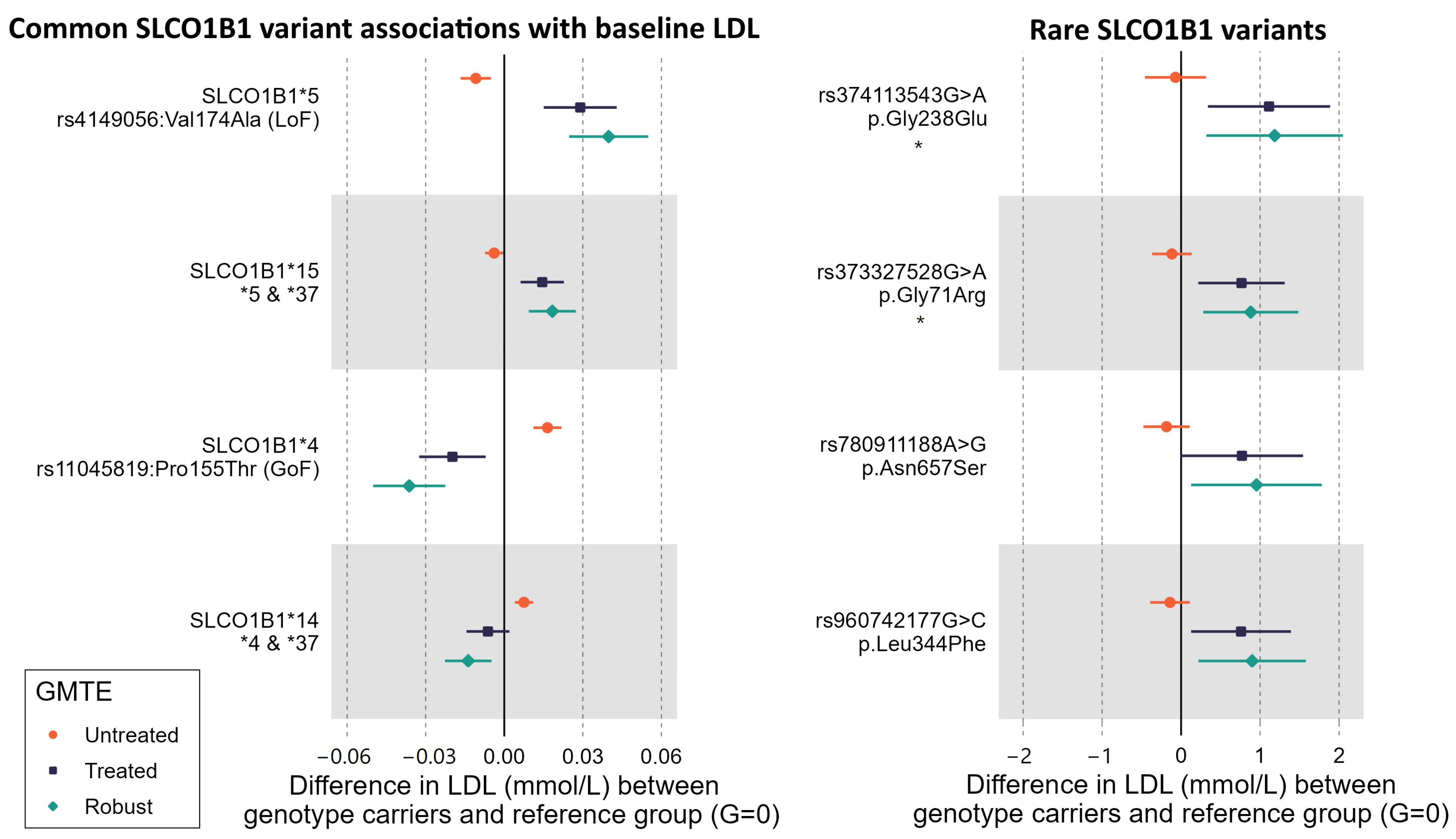
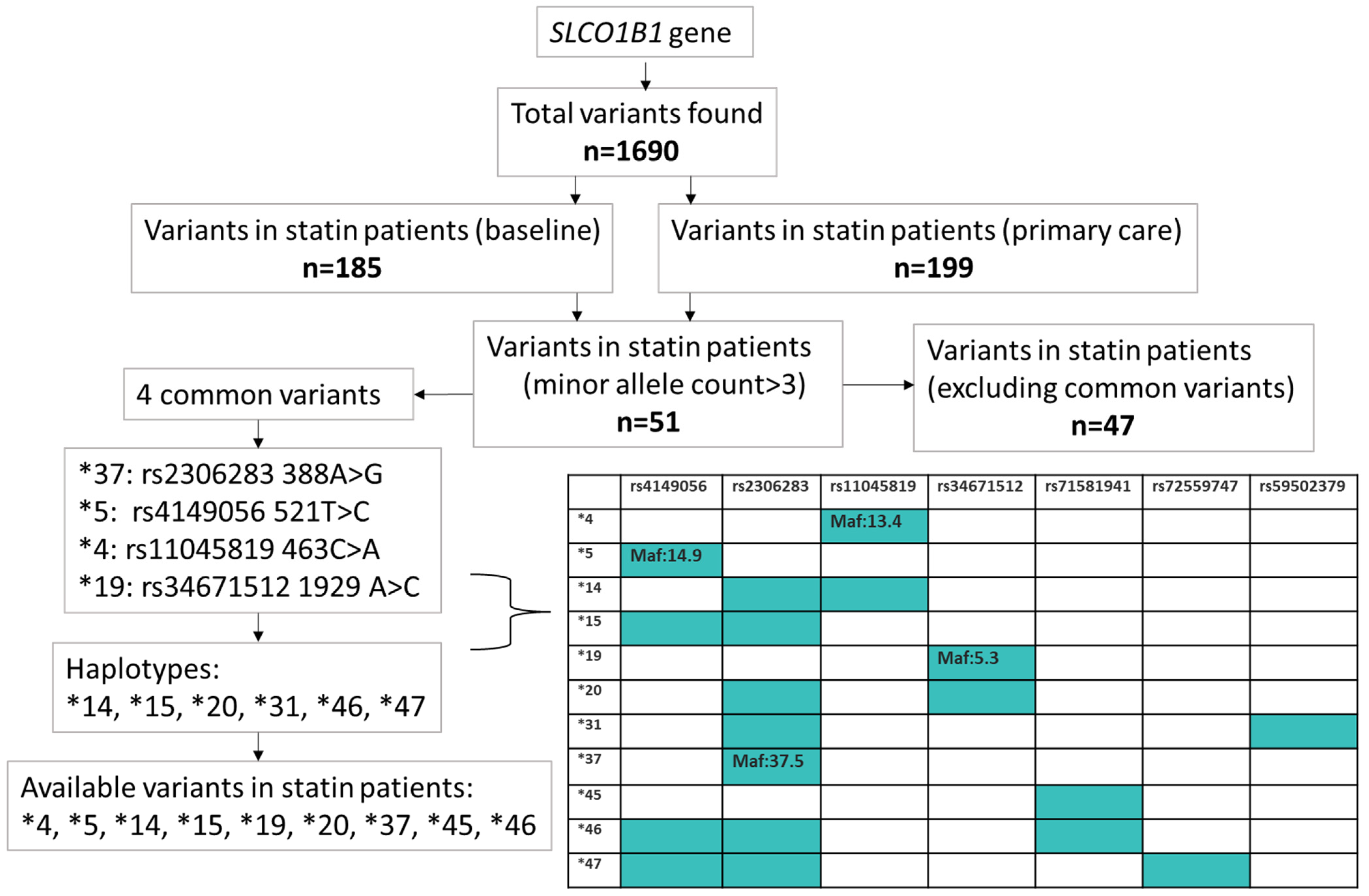
2.1. Common SLCO1B1 Variants
Baseline Self-Reported Statin Use and Biomarkers
2.2. General Practice (GP) Prescribing and Statin-Related Adverse Outcomes
2.3. Statin Gene Risk Score
2.4. Rare Variants in SLCO1B1 Identified by Exome Sequencing
2.5. SLCO1B1 “High Risk” Gene Score plus Rare Variants
3. Discussion
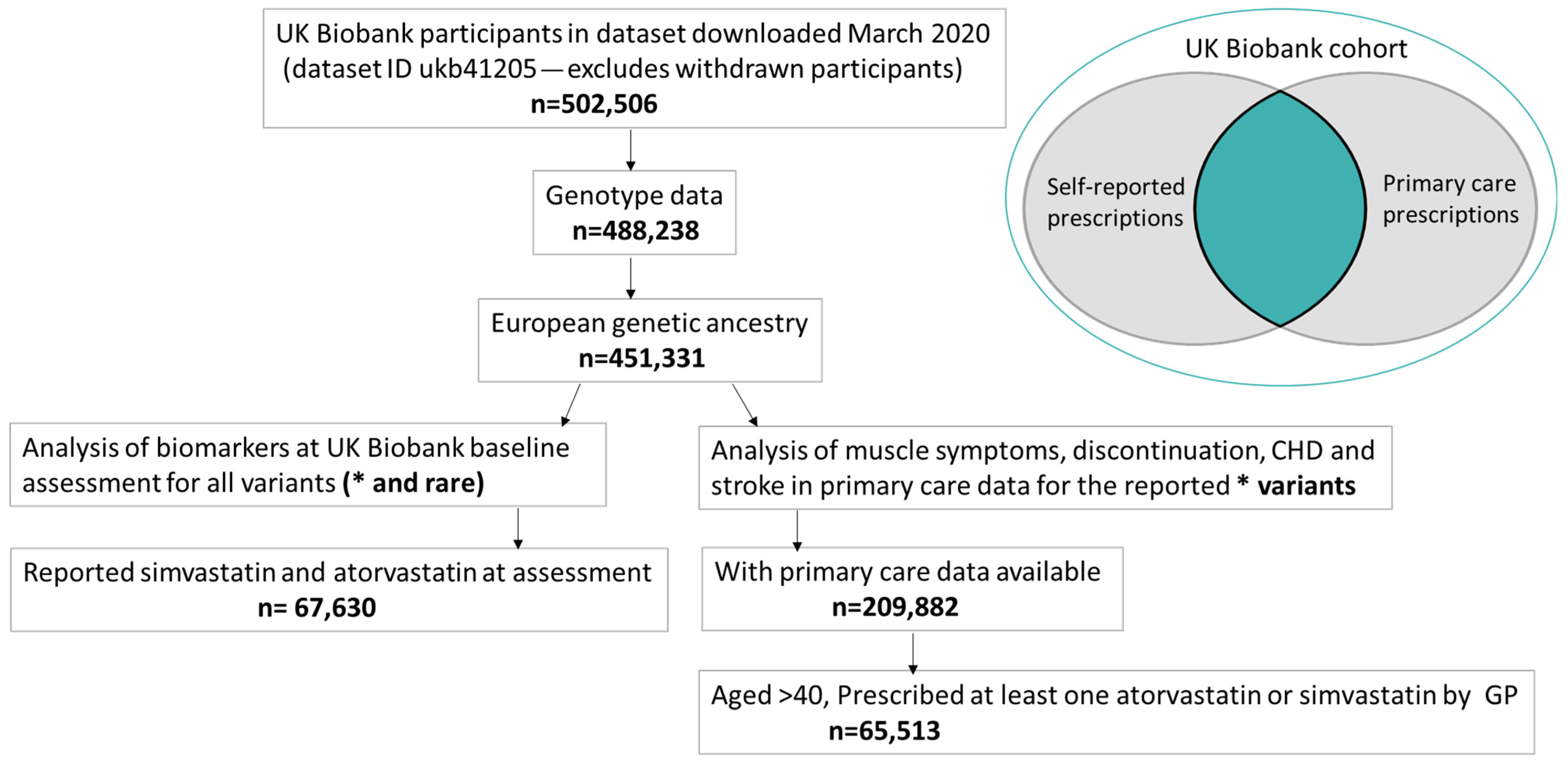
4. Materials and Methods
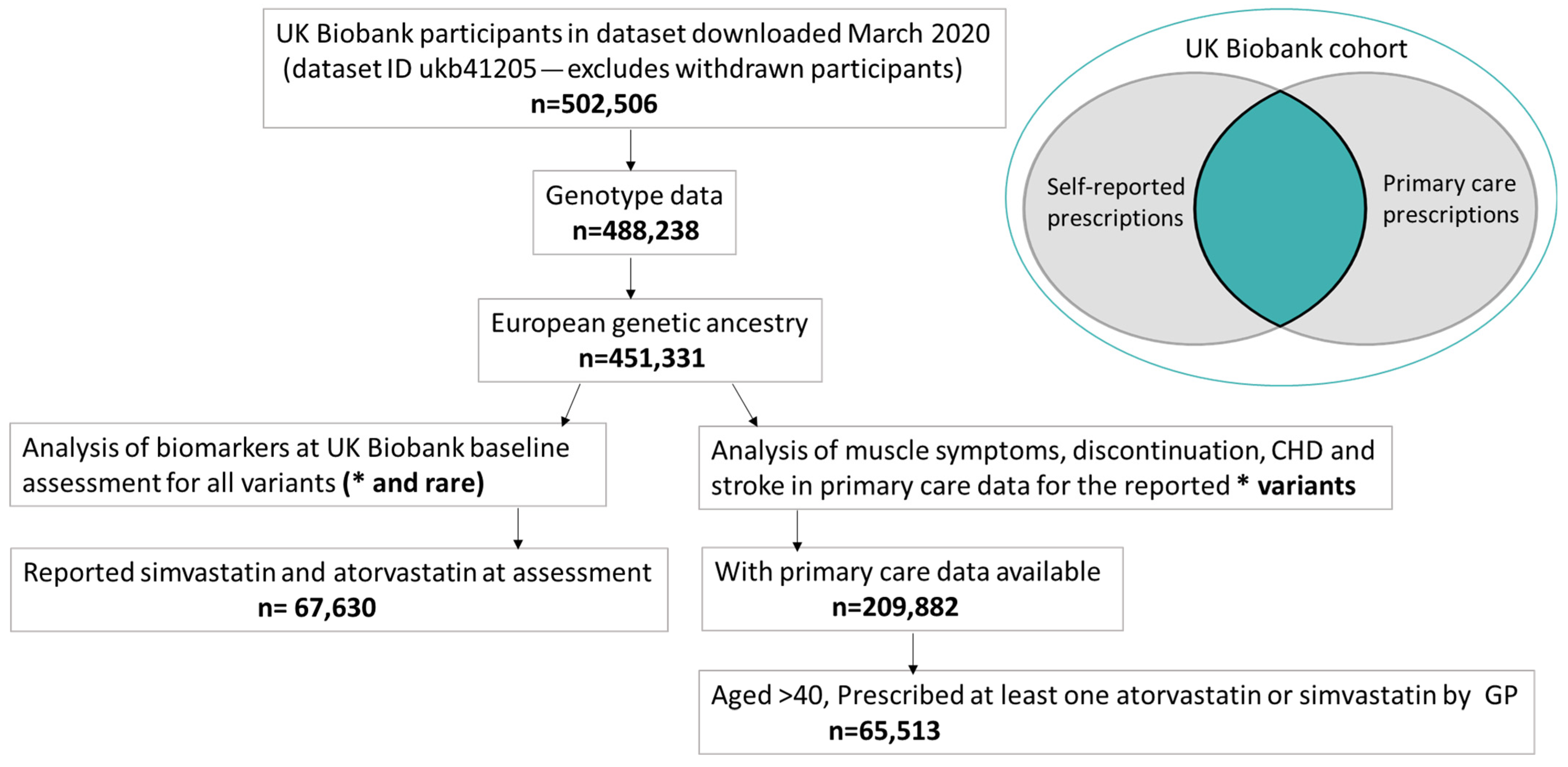
4.1. UK Biobank (UKB)
4.2. Whole-Exome Sequencing (WES)
4.3. SLCO1B1 Variants
4.4. Statin Gene Risk Score (GRS)
4.5. Outcomes
- (1)
- Baseline
- (2)
- General Practice (GP) data
4.6. Statistical Analysis
- (1)
- Baseline assessment data for all variants
- (2)
- GP data for common variants
- (3)
- TWIST
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oshiro, C.; Mangravite, L.; Klein, T.; Altman, R. PharmGKB very important pharmacogene: SLCO1B1. Pharmacogenetics Genom. 2010, 20, 211–216. [Google Scholar] [CrossRef] [PubMed]
- SLCO1B1: A VIP Gene. Available online: https://www.pharmgkb.org/vip/PA166169500 (accessed on 30 March 2023).
- PharmGKB. Gene-Specific Information Tables for SLCO1B1. Available online: https://www.pharmgkb.org/page/slco1b1RefMaterials (accessed on 11 July 2023).
- Cooper-DeHoff, R.M.; Niemi, M.; Ramsey, L.B.; Luzum, J.A.; Tarkiainen, E.K.; Straka, R.J.; Gong, L.; Tuteja, S.; Wilke, R.A.; Wadelius, M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and Statin-Associated Musculoskeletal Symptoms. Clin. Pharmacol. Ther. 2022, 111, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Voora, D.; Shah, S.H.; Spasojevic, I.; Ali, S.; Reed, C.R.; Salisbury, B.A.; Ginsburg, G.S. The SLCO1B1*5Genetic Variant Is Associated With Statin-Induced Side Effects. J. Am. Coll. Cardiol. 2009, 54, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.A.; Doney, A.S.F.; Tavendale, R.; Lang, C.C.; Pearson, E.R.; Colhoun, H.M.; McCarthy, M.I.; Hattersley, A.T.; Morris, A.D.; Palmer, C.N.A. Common Nonsynonymous Substitutions in SLCO1B1 Predispose to Statin Intolerance in Routinely Treated Individuals With Type 2 Diabetes: A Go-DARTS Study. Clin. Pharmacol. Ther. 2010, 89, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Voora, D.; Baye, J.; McDermaid, A.; Gowda, S.N.; Wilke, R.A.; Myrmoe, A.N.; Hajek, C.; Larson, E.A. SLCO1B1*5 Allele Is Associated With Atorvastatin Discontinuation and Adverse Muscle Symptoms in the Context of Routine Care. Clin. Pharmacol. Ther. 2022, 111, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- SEARCH Collaborative Group; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1Variants and Statin-Induced Myopathy—A Genomewide Study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.F.; Francis, B.; Jorgensen, A.L.; Zhang, E.; Chinoy, H.; Heckbert, S.R.; Bis, J.C.; Brody, J.A.; Floyd, J.S.; Psaty, B.M.; et al. Genomewide Association Study of Statin-Induced Myopathy in Patients Recruited Using the UK Clinical Practice Research Datalink. Clin. Pharmacol. Ther. 2019, 106, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Vrablík, M.; Hubáček, J.A.; Dlouhá, D.; Adámková, V.; Zlatohlavek, L.; Viklický, O.; Hrubá, P.; Češka, R. SLCO1B1 Polymorphism is not associated with Risk of Statin-Induced Myalgia/Myopathy in a Czech Population. Med. Sci. Monit. 2015, 21, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Türkmen, D.; Masoli, J.A.H.; Kuo, C.; Bowden, J.; Melzer, D.; Pilling, L.C. Statin treatment effectiveness and the SLCO1B1*5 reduced function genotype: Long-term outcomes in women and men. Br. J. Clin. Pharmacol. 2022, 88, 3230–3240. [Google Scholar] [CrossRef] [PubMed]
- Khine, H.; Yuet, W.C.; Adams-Huet, B.; Ahmad, Z. Statin-associated muscle symptoms and SLCO1B1 rs4149056 genotype in patients with familial hypercholesterolemia. Am. Heart J. 2016, 179, 1–9. [Google Scholar] [CrossRef] [PubMed]
- PharmGKB. Clinical Annotation for rs4149056 (SLCO1B1); Simvastatin; Statin-Related Myopathy (Level 1A Toxicity). Available online: https://www.pharmgkb.org/clinicalAnnotation/655384011 (accessed on 4 June 2023).
- Peyser, B.; Perry, E.P.; Singh, K.; Gill, R.D.; Mehan, M.R.; Haga, S.B.; Musty, M.D.; Milazzo, N.A.; Savard, D.; Li, Y.-J.; et al. Effects of Delivering SLCO1B1 Pharmacogenetic Information in Randomized Trial and Observational Settings. Circ. Genom. Precis. Med. 2018, 11, e002228. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, S.; Rader, D.J. SLCO1B1 and Statin Therapy. Circ. Genom. Precis. Med. 2018, 11, e002320. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, M.K.; Fredrikson, H.; Neuvonen, P.J.; Niemi, M. Different Effects of SLCO1B1 Polymorphism on the Pharmacokinetics of Atorvastatin and Rosuvastatin. Clin. Pharmacol. Ther. 2007, 82, 726–733. [Google Scholar] [CrossRef] [PubMed]
- PharmGKB. Clinical Annotation for SLCO1B1*1, SLCO1B1*5, SLCO1B1*15; Fluvastatin; Statin-Related Myopathy (Level 1A Toxicity). Available online: https://www.pharmgkb.org/clinicalAnnotation/1451678626 (accessed on 27 July 2023).
- Mykkänen, A.J.H.; Taskinen, S.; Neuvonen, M.; Paile-Hyvärinen, M.; Tarkiainen, E.K.; Lilius, T.; Tapaninen, T.; Backman, J.T.; Tornio, A.; Niemi, M. Genomewide Association Study of Simvastatin Pharmacokinetics. Clin. Pharmacol. Ther. 2022, 112, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Bigossi, M.; Maroteau, C.; Dawed, A.Y.; Taylor, A.; Srinivasan, S.; Melhem, A.L.; Pearson, E.R.; Pola, R.; Palmer, C.N.A.; Siddiqui, M.K. A gene risk score using missense variants in SLCO1B1 is associated with earlier onset statin intolerance. Eur. Heart J. Cardiovasc. Pharmacother. 2023, 9, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sarangi, V.; Ho, M.-F.; Moon, I.; Kalari, K.R.; Wang, L.; Weinshilboum, R.M. SLCO1B1: Application and Limitations of Deep Mutational Scanning for Genomic Missense Variant Function. Drug Metab. Dispos. 2021, 49, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lauschke, V.M. Genetic variability and population diversity of the human SLCO (OATP) transporter family. Pharmacol. Res. 2018, 139, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Anesth. Analg. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Michalska-Kasiczak, M.; Sahebkar, A.; Mikhailidis, D.P.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Hovingh, G.K.; Farnier, M.; et al. Analysis of vitamin D levels in patients with and without statin-associated myalgia—A systematic review and meta-analysis of 7 studies with 2420 patients. Int. J. Cardiol. 2014, 178, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Asl, E.S.; Taheraghdam, A.; Rahmani, F.; Javadrashid, R.; Golzari, S.E.J.; Ghaemian, N.; Sadeghpour, Y.; Esfanjani, R.M.; Soleimanpour, H. Determination of the Predictive Value of Serum Bilirubin in Patients with Ischemic Stroke: A Prospective Descriptive Analytical Study. Adv. Pharm. Bull. 2018, 8, 715–719. [Google Scholar] [CrossRef]
- Mirjanic-Azaric, B.; Rizzo, M.; Jürgens, G.; Hallstroem, S.; Srdic, S.; Marc, J.; Cerne, D. Atorvastatin treatment increases plasma bilirubin but not HMOX1 expression in stable angina patients. Scand. J. Clin. Lab. Investig. 2015, 75, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Pilling, L.C.; Türkmen, D.; Kuo, C.-L.; Melzer, D. The Triangulation WIthin a STudy (TWIST) framework for causal inference within pharmacogenetic research. PLoS Genet. 2021, 17, e1009783. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Treviño, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2011, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- PharmGKB. Clinical Annotation for SLCO1B1*1, SLCO1B1*5, SLCO1B1*15, SLCO1B1*37; Atorvastatin (Level 1A Metabolism/PK). Available online: https://www.pharmgkb.org/clinicalAnnotation/1043880630 (accessed on 5 July 2023).
- PharmGKB. Clinical Annotation for SLCO1B1*1, SLCO1B1*5, SLCO1B1*15; Simvastatin or Simvastatin Acid (Level 1A Metabolism/PK). Available online: https://www.pharmgkb.org/clinicalAnnotation/1451681540 (accessed on 5 July 2023).
- Oxford Academic. Effects of Statins on Skeletal Muscle: A Perspective for Physical Therapists. Physical Therapy. Available online: https://academic.oup.com/ptj/article/90/10/1530/2737797 (accessed on 8 March 2024).
- Fry, A.; Littlejohns, T.J.; Sudlow, C.; Doherty, N.; Adamska, L.; Sprosen, T.; Collins, R.; Allen, N.E. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants With Those of the General Population. Am. J. Epidemiol. 2017, 186, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Bytyçi, I.; Penson, P.E.; Mikhailidis, D.P.; Wong, N.D.; Hernandez, A.V.; Sahebkar, A.; Thompson, P.D.; Mazidi, M.; Rysz, J.; Pella, D.; et al. Prevalence of statin intolerance: A meta-analysis. Eur. Heart J. 2022, 43, 3213–3223. [Google Scholar] [CrossRef]
- Our Future Health. Available online: https://ourfuturehealth.org.uk/ (accessed on 3 November 2023).
- Primary Care Linked Data. Available online: https://biobank.ndph.ox.ac.uk/ukb/ukb/docs/primary_care_data.pdf (accessed on 4 June 2023).
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.D.; Liu, D.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Ye, B.; Banerjee, N.; et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020, 586, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Backman, J.D.; Li, A.H.; Marcketta, A.; Sun, D.; Mbatchou, J.; Kessler, M.D.; Benner, C.; Liu, D.; Locke, A.E.; Balasubramanian, S.; et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 2021, 599, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Pilling, L.C.; Tamosauskaite, J.; Jones, G.; Wood, A.R.; Jones, L.; Kuo, C.-L.; Kuchel, G.A.; Ferrucci, L.; Melzer, D. Common conditions associated with hereditary haemochromatosis genetic variants: Cohort study in UK Biobank. BMJ 2019, 364, k5222. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Gong, L.; Lee, S.; Wagner, J.B.; Zhou, X.; Sangkuhl, K.; Adams, S.M.; Straka, R.J.; Empey, P.E.; Boone, E.C.; et al. PharmVar GeneFocus: SLCO1B1. Clin. Pharmacol. Ther. 2022, 113, 782–793. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- GWAS Catalog. Available online: https://www.ebi.ac.uk/gwas/genes/SLCO1B1 (accessed on 3 November 2023).
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A. SLCO1B1 Polymorphisms and Statin-Induced Myopathy. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Data | ||
|---|---|---|
| Sex | N Female | 26,185 (38.7) |
| Age | Mean (SD) | 61.5 (6) |
| min-max | 40–70 | |
| Median | 63 | |
| Simvastatin | N (%) | 51,147 (75.5) |
| Weight | Mean (SD) | 84.3 (16.5) |
| BMI | Mean (SD) | 29.4 (5) |
| LDL | Mean (SD) | 2.75 (0.67) |
| LDL, n > 2.86 | N (%) | 28,450 (42) |
| N female (%) | 12,622(44.3) | |
| Total cholesterol | Mean (SD) | 4.6 (0.9) |
| Triglycerides | Mean (SD) | 1.9 (1.1) |
| Direct Bilirubin | Mean (SD) | 9.5 (4.5) |
| ALT | Mean (SD) | 27.1 (14.5) |
| CRP | Mean (SD) | 2.8 (4.7) |
| Cystatin c | Mean (SD) | 0.9 (0.2) |
| Vitamin D | Mean (SD) | 49.7 (21.6) |
| HbA1c | Mean (SD) | 40.5 (10.2) |
| Primary care data | ||
| Sex | N Female | 27,299 (42) |
| Duration on statins^ (year) | Mean (SD) | 6.7 (4.7) |
| min-max | 1 to 25 | |
| Number of prescriptions per year | Mean (SD) | 9 (5) |
| Muscle diagnoses 1 prior to statin ^ | N (%) | 1308 (2) |
| MI/angina diagnoses 2 prior to statin ^ | N (%) | 6718 (10.4) |
| Stroke diagnoses 2 prior to statin ^ | N (%) | 855 (1.3) |
| Muscle diagnoses 1 after first statin ^ | N (%) | 2242 (3.5) |
| MI/angina diagnoses 2 after first statin ^ | N (%) | 14,751 (22.8) |
| Stroke diagnoses 2 after first statin ^ | N (%) | 1629 (2.5) |
| Discontinuation ever | N (%) | 13,889 (22.4) |
| Allele | ||||||
|---|---|---|---|---|---|---|
| Common variants | N | % | Coef | 95% | CI | p |
| *4-CC | 45,798 | 75.58 | ref | |||
| *4-CA | 13,324 | 21.99 | −0.02 | −0.03 | −0.009 | 8 × 10−4 |
| *4-AA | 1475 | 2.43 | −0.03 | −0.067 | −0.0008 | 0.045 |
| *5-TT | 44,821 | 72.47 | ref | |||
| *5-TC | 15,640 | 25.29 | 0.02 | 0.01 | 0.03 | 4 × 10−4 |
| *5-CC | 1383 | 2.24 | 0.08 | 0.05 | 0.12 | 6 × 10−6 |
| *37-AA | 24,413 | 40.03 | ref | |||
| *37-AG | 27,340 | 44.83 | 0.01 | −0.01 | 0.02 | 0.32 |
| *37-GG | 9235 | 15.14 | 2 × 10−3 | −0.01 | 0.02 | 0.77 |
| *19-AA | 55,469 | 89.69 | ref | |||
| *19-AC | 6223 | 10.06 | −0.01 | −0.03 | 0.01 | 0.18 |
| *19-CC | 154 | 0.25 | −0.01 | −0.12 | 0.09 | 0.83 |
| Haplotypes ^ | N | % | Coef | 95% | CI | p |
| No *14 | 23,870 | 39.91 | ref | |||
| *14 | 14,181 | 23.71 | −0.01 | −0.03 | 0.00 | 0.05 |
| No *15 | 22,015 | 36.13 | ref | |||
| *15 | 14,454 | 23.72 | 0.03 | 0.01 | 0.04 | 3 × 10-4 |
| No *20 | 24,254 | 39.80 | ref | |||
| *20 | 6173 | 10.13 | −0.01 | −0.03 | 0.01 | 0.42 |
| No *45 | 60,852 | 99.89 | ref | |||
| *45 ° | 67 | 0.11 | −0.01 | −0.17 | 0.15 | 0.90 |
| No *46 | 21,707 | 36.2 | ref | |||
| *46 | 65 | 0.1 | 0.002 | −0.01 | 0.01 | 0.70 |
| *4 | *5 | *14 | *15 | *20 | *37 | *45 | *19 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | n | % | n | % | n | % | n | % | n | % | n | % | n | % | n | ||
| SAMS | wild | 72.36 | 1542 | 67.81 | 1445 | 35.48 | 756 | 33.36 | 711 | 36.23 | 772 | 36.60 | 780 | 94.79 | 2020 | 85.83 | 1829 |
| heterozygotes | 20.13 | 429 | 26.09 | 556 | 36.65 | 781 | 36.60 | 780 | 48.71 | 1038 | 43.55 | 928 | 0.05 | 1 | 9.95 | 212 | |
| homozygotes | 1.88 | 40 | 2.39 | 51 | 21.16 | 451 | 25.06 | 534 | 9.90 | 211 | 14.88 | 317 | 0.00 | 0.33 | 7 | ||
| No SAMS | wild | 71.98 | 44,126 | 69.70 | 42,730 | 37.84 | 23,194 | 34.78 | 21,319 | 38.43 | 23,559 | 38.67 | 23,707 | 94.82 | 58,129 | 86.57 | 53,070 |
| heterozygotes | 20.17 | 12,364 | 24.34 | 14,920 | 33.81 | 20,728 | 37.63 | 23,069 | 46.99 | 28,808 | 42.02 | 25,762 | 0.11 | 70 | 9.48 | 5810 | |
| homozygotes | 2.24 | 1373 | 2.26 | 1388 | 21.48 | 13,169 | 22.45 | 13,763 | 9.45 | 5792 | 14.26 | 8741 | 94.82 | 0.28 | 169 | ||
| *5 | *37 | rs11045819 | rs34671512 | ||
|---|---|---|---|---|---|
| High risk | CC | ||||
| GRS1 | High risk | TC | AA | CC | AA |
| Low risk | all remaining | ||||
| High risk | CC | ||||
| GRS2 | High risk | TC | GG | CC | AA |
| Low risk | all remaining | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Türkmen, D.; Bowden, J.; Masoli, J.A.H.; Melzer, D.; Pilling, L.C. SLCO1B1 Exome Sequencing and Statin Treatment Response in 64,000 UK Biobank Patients. Int. J. Mol. Sci. 2024, 25, 4426. https://doi.org/10.3390/ijms25084426
Türkmen D, Bowden J, Masoli JAH, Melzer D, Pilling LC. SLCO1B1 Exome Sequencing and Statin Treatment Response in 64,000 UK Biobank Patients. International Journal of Molecular Sciences. 2024; 25(8):4426. https://doi.org/10.3390/ijms25084426
Chicago/Turabian StyleTürkmen, Deniz, Jack Bowden, Jane A. H. Masoli, David Melzer, and Luke C. Pilling. 2024. "SLCO1B1 Exome Sequencing and Statin Treatment Response in 64,000 UK Biobank Patients" International Journal of Molecular Sciences 25, no. 8: 4426. https://doi.org/10.3390/ijms25084426
APA StyleTürkmen, D., Bowden, J., Masoli, J. A. H., Melzer, D., & Pilling, L. C. (2024). SLCO1B1 Exome Sequencing and Statin Treatment Response in 64,000 UK Biobank Patients. International Journal of Molecular Sciences, 25(8), 4426. https://doi.org/10.3390/ijms25084426