



The CaMK Family Differentially Promotes Necroptosis and Mouse Cardiac Graft Injury and Rejection

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
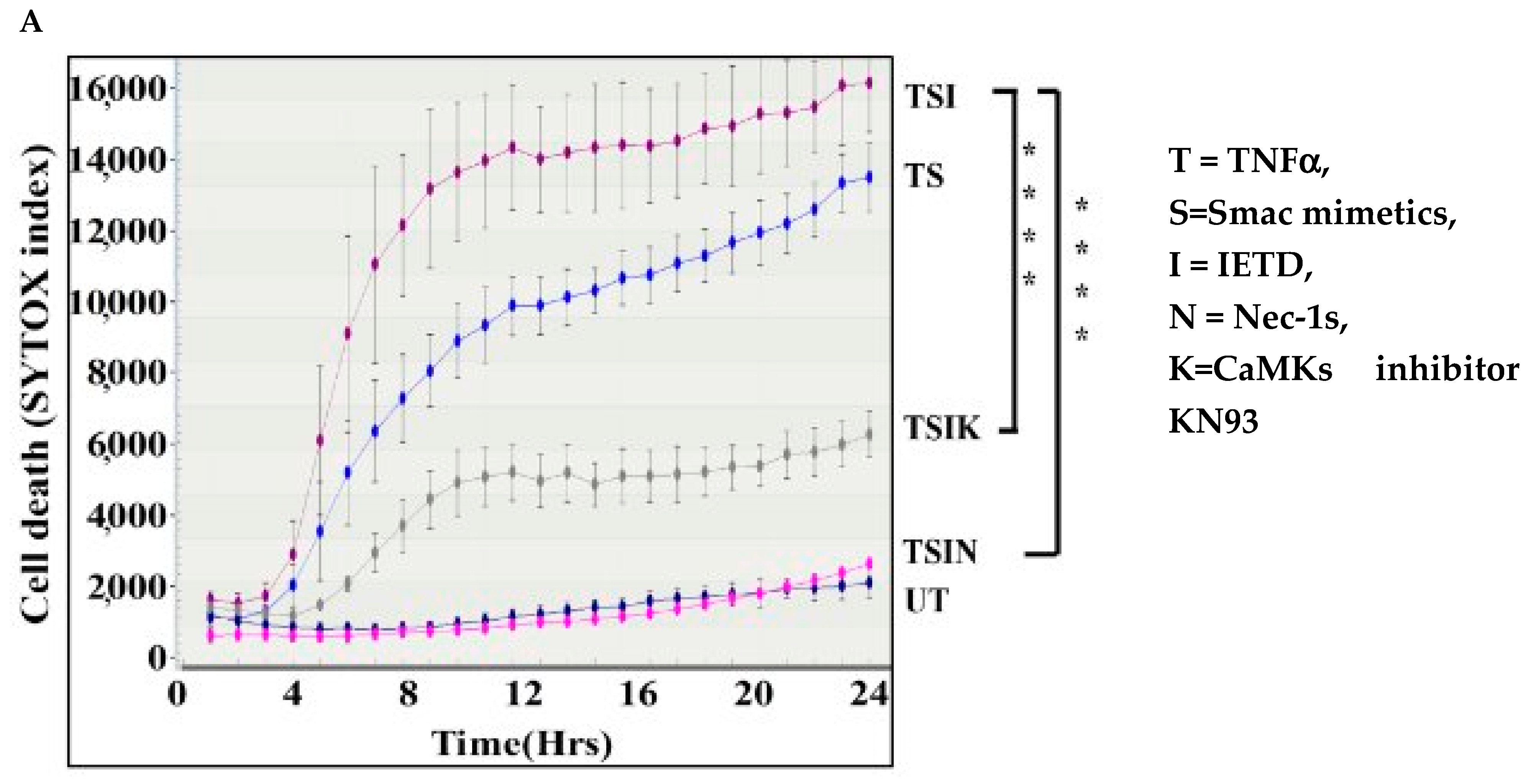
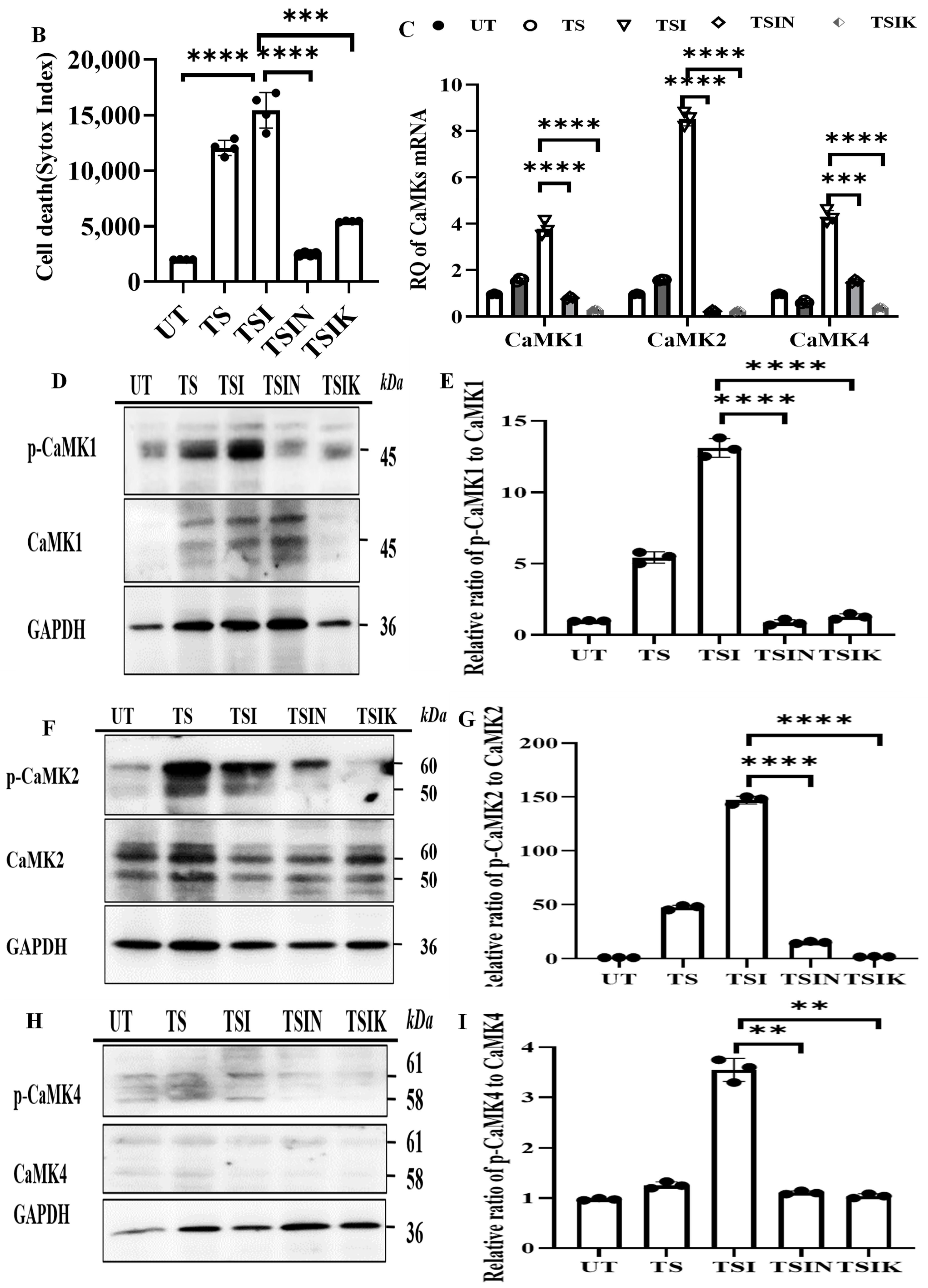
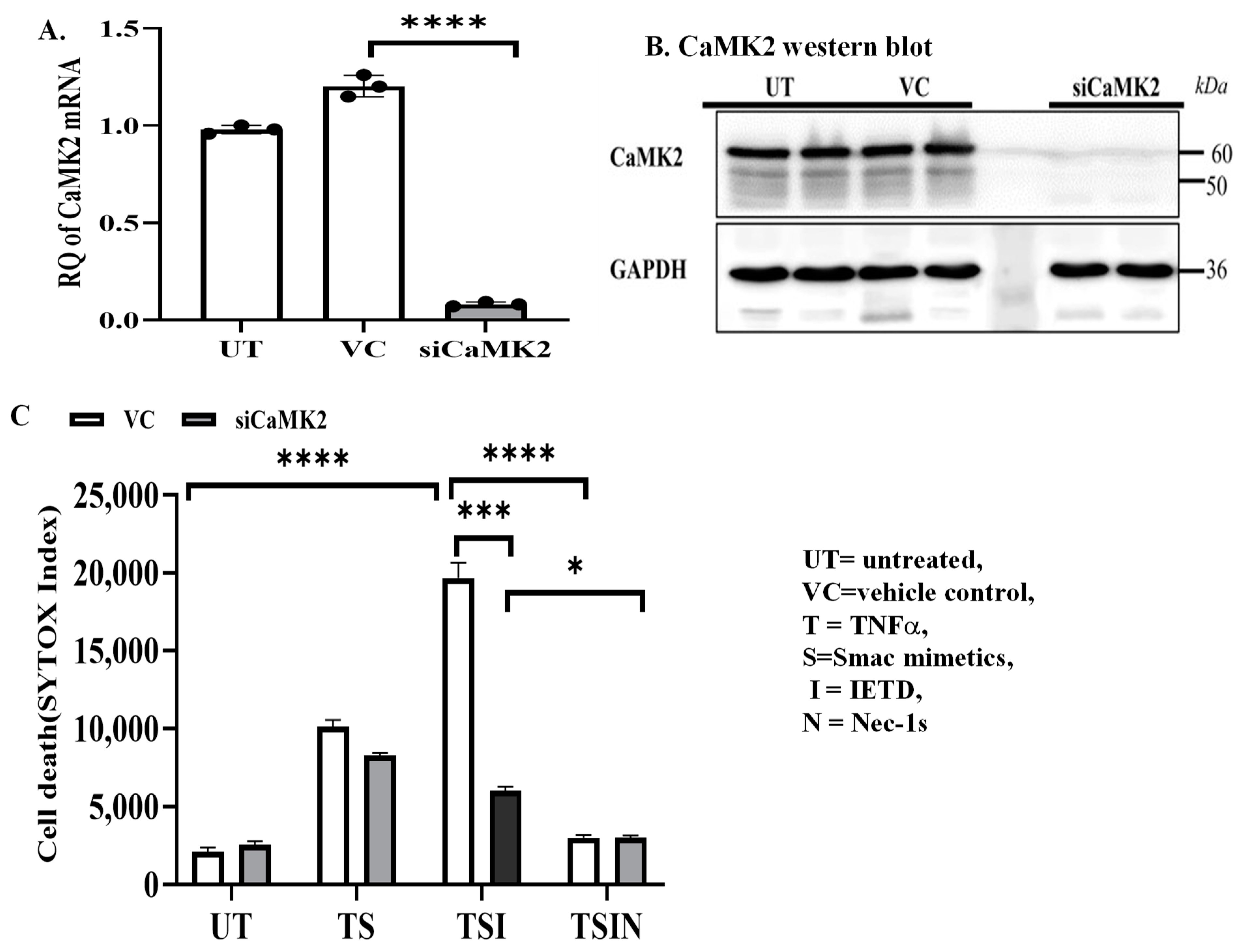
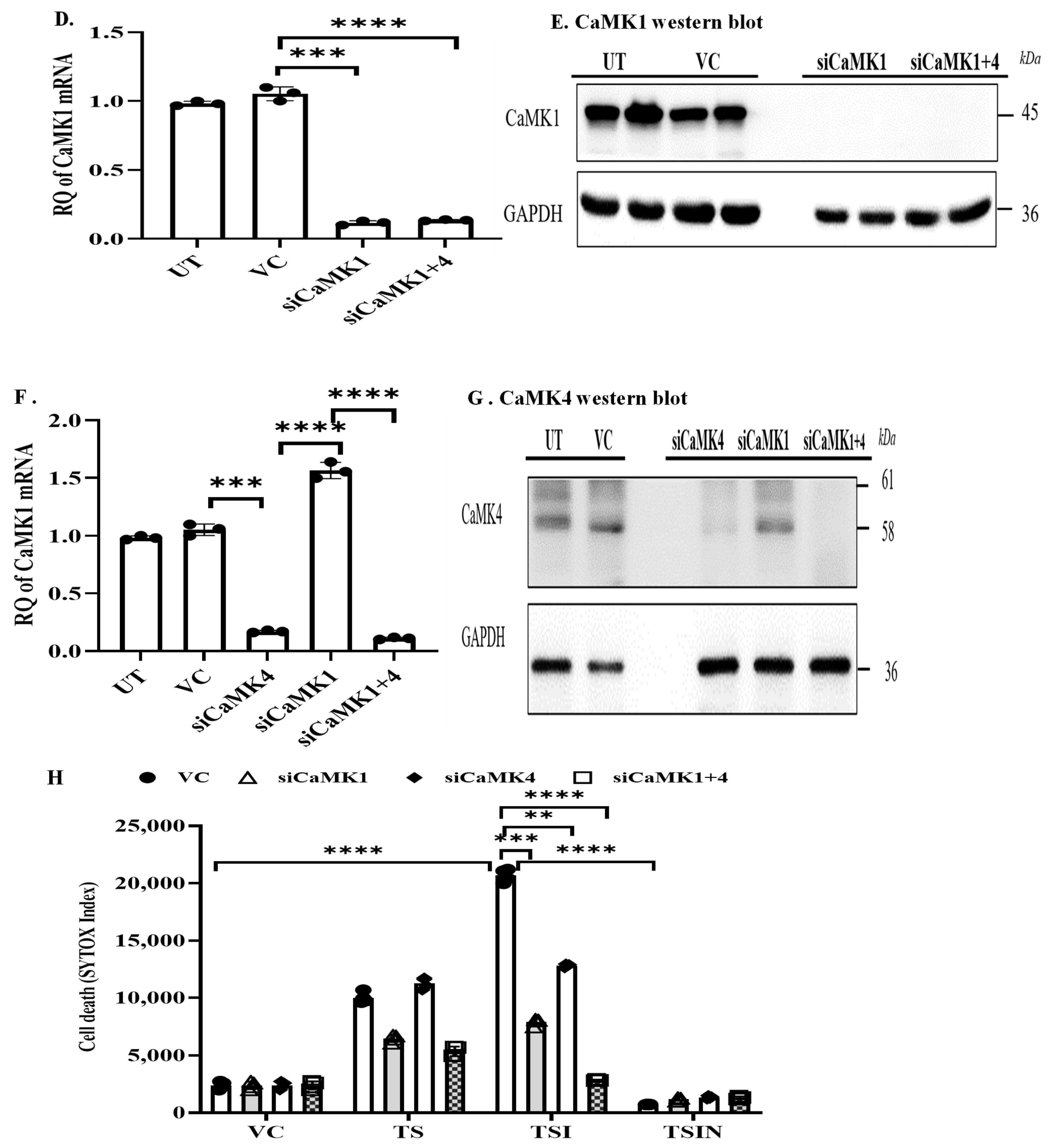
2.1. The CaMK Family Participates in MVEC Necroptosis
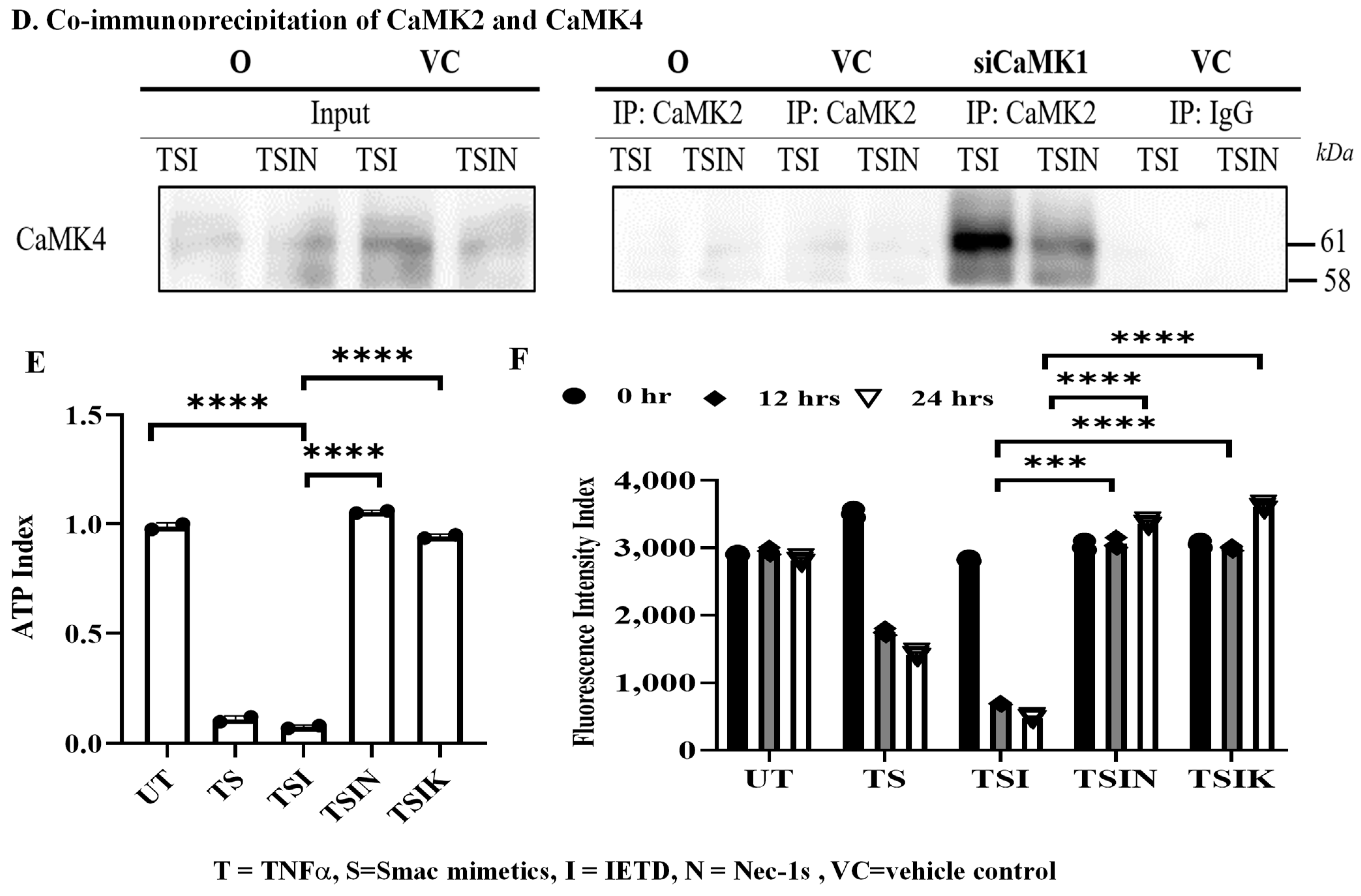
2.2. RIPK3 and the CaMK Family Members Form a Complex in Necroptosis
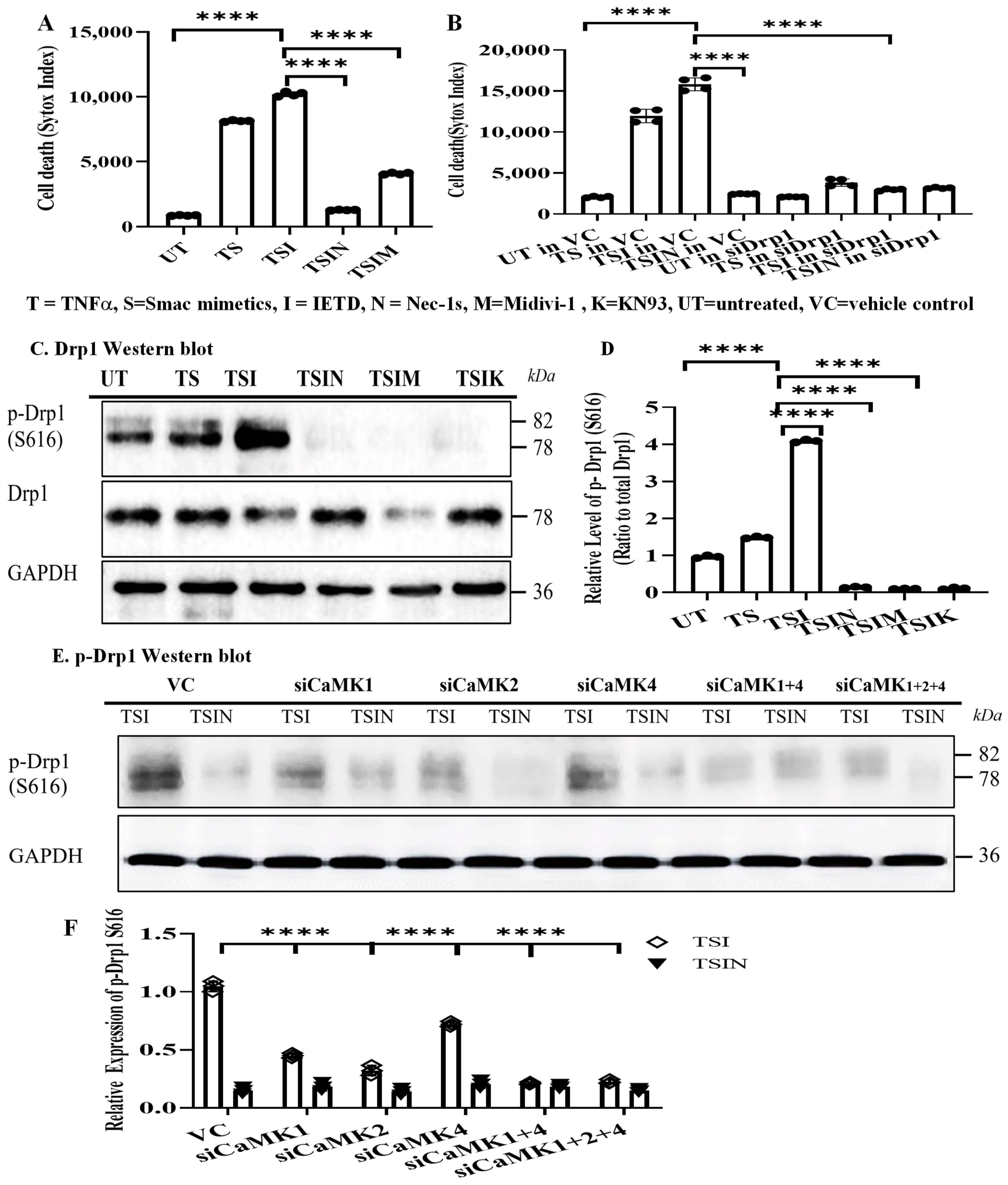
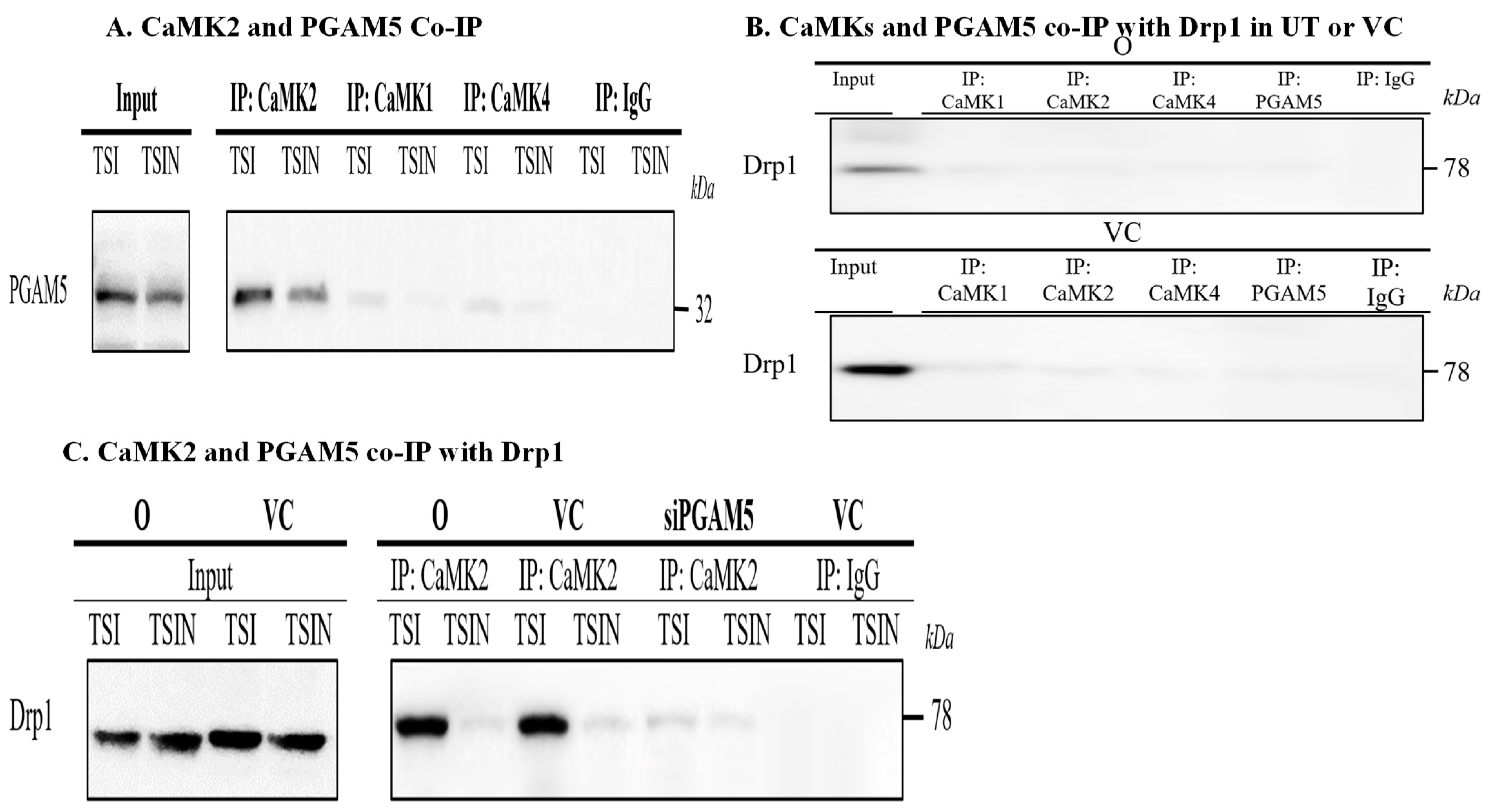
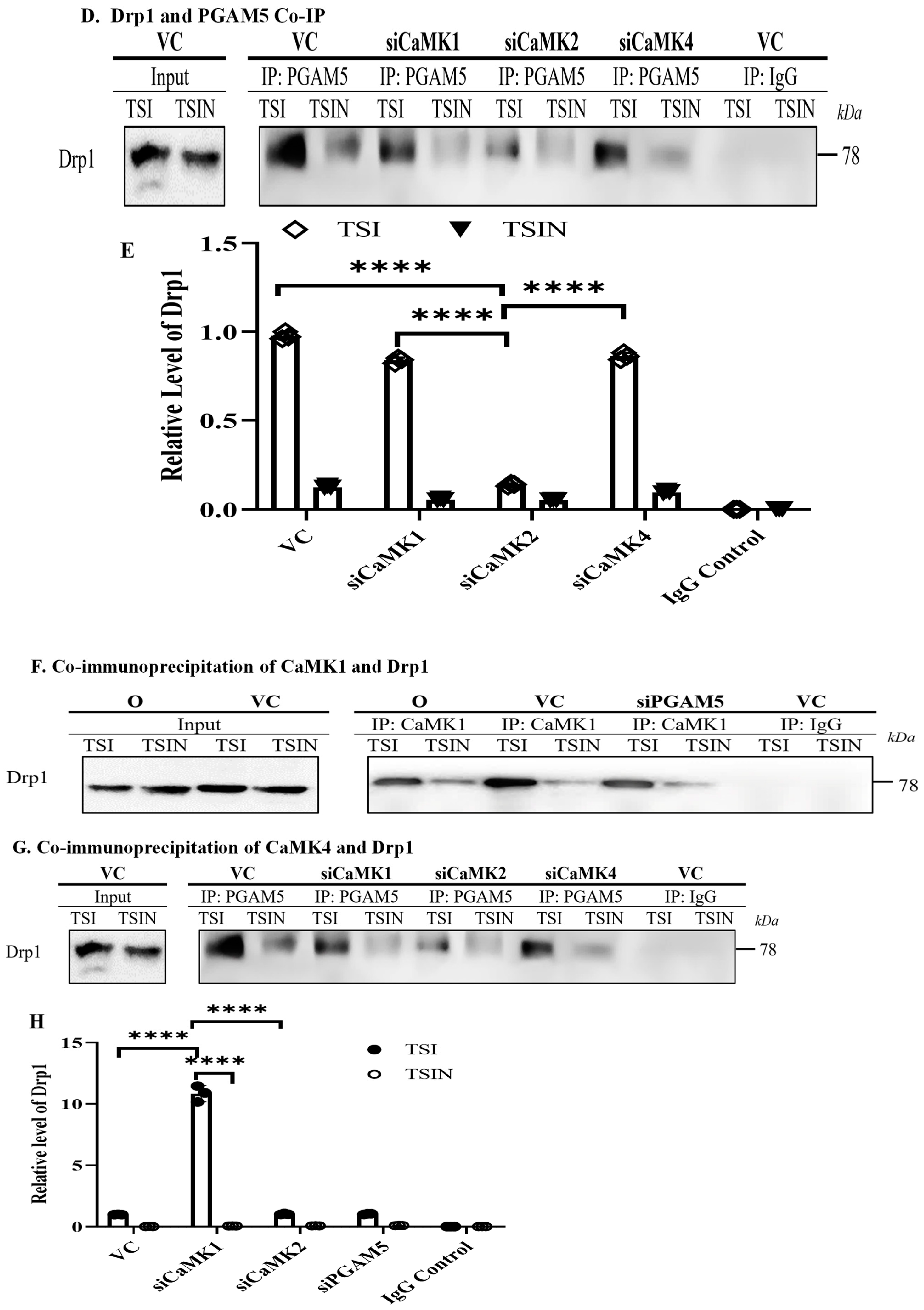
2.3. CaMK1 and CaMK4 Directly Bind to and Phosphorylate Drp1 while CaMK2 Indirectly Regulates Drp1 Phosphorylation via PGAM5
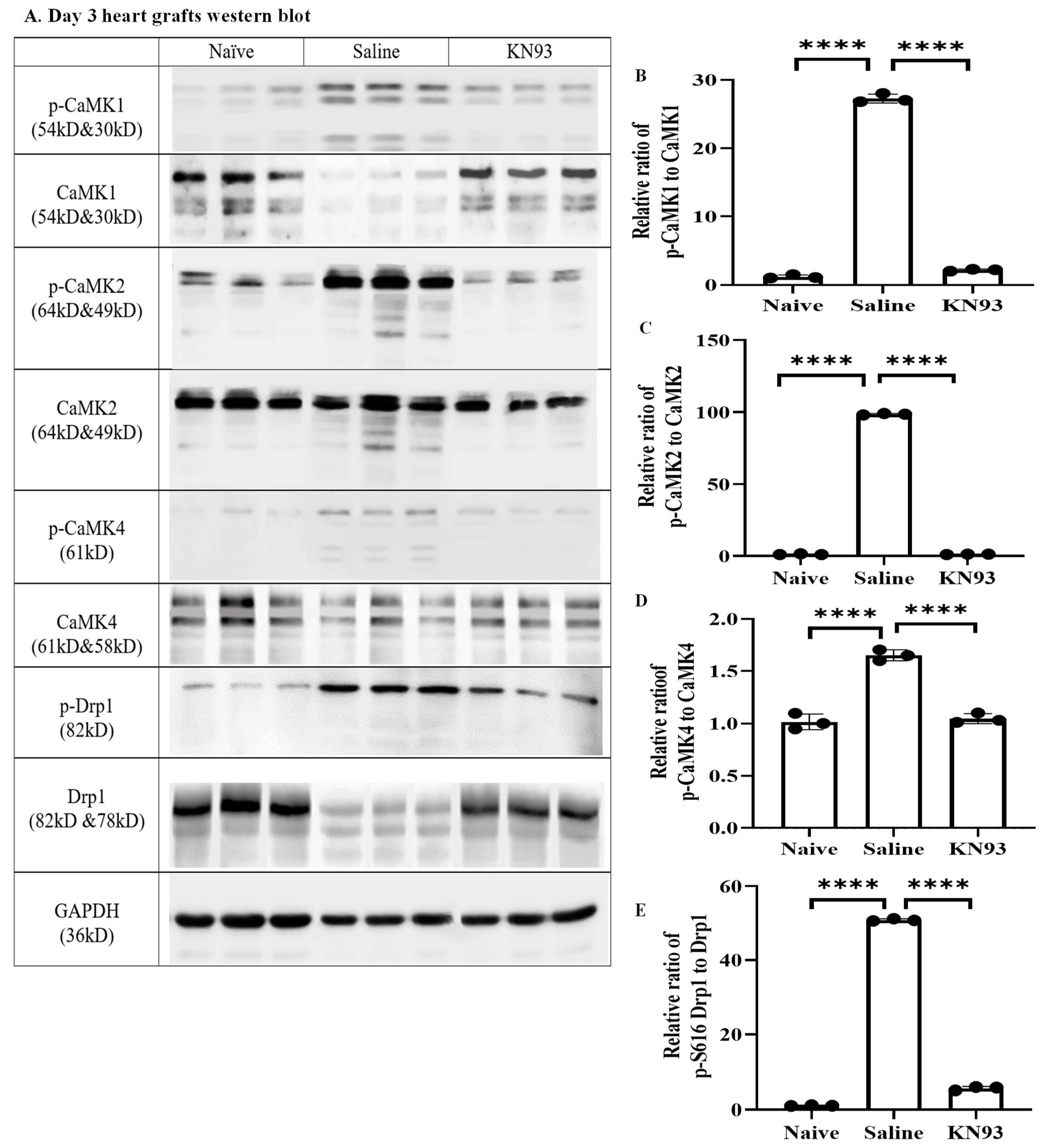
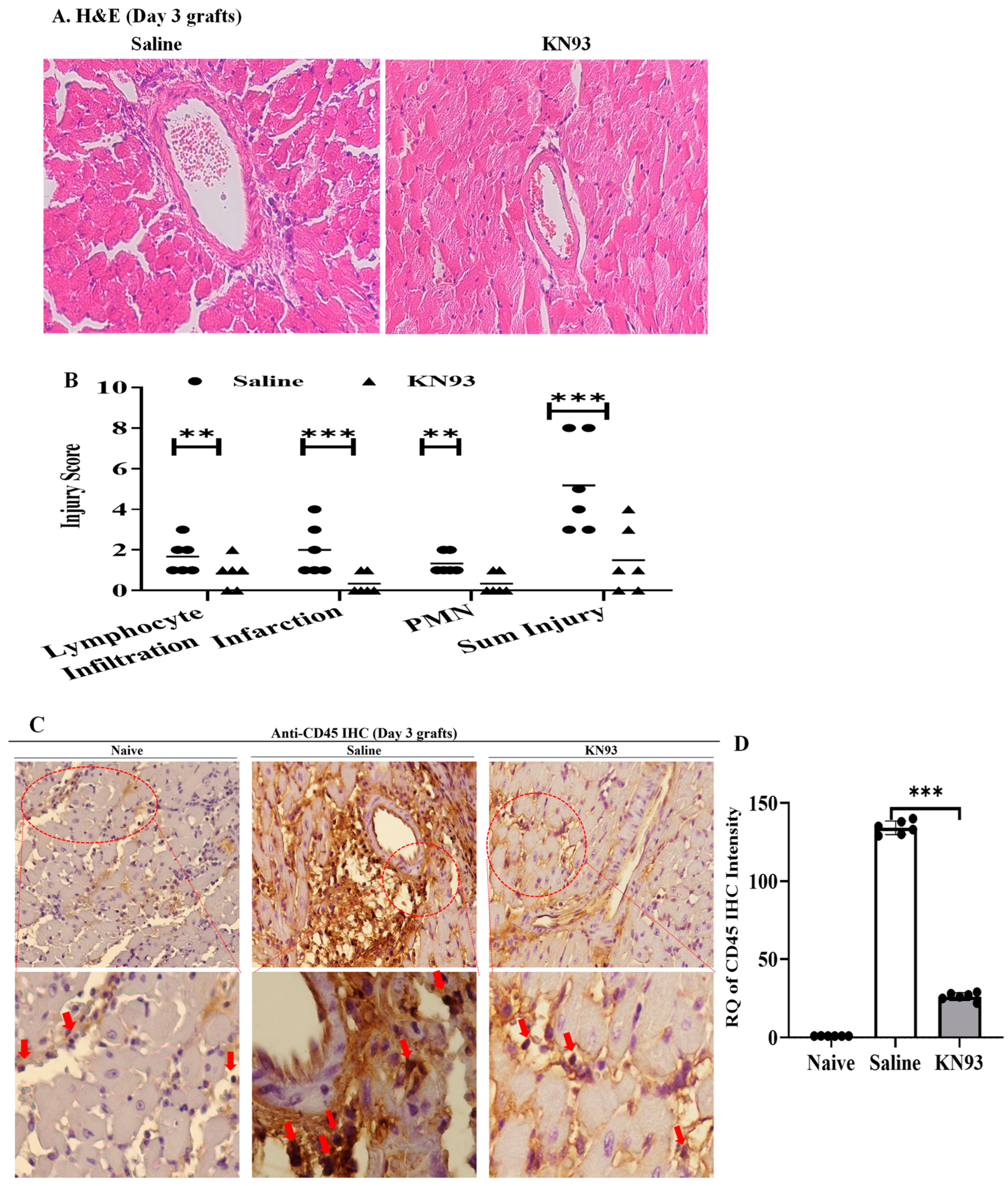
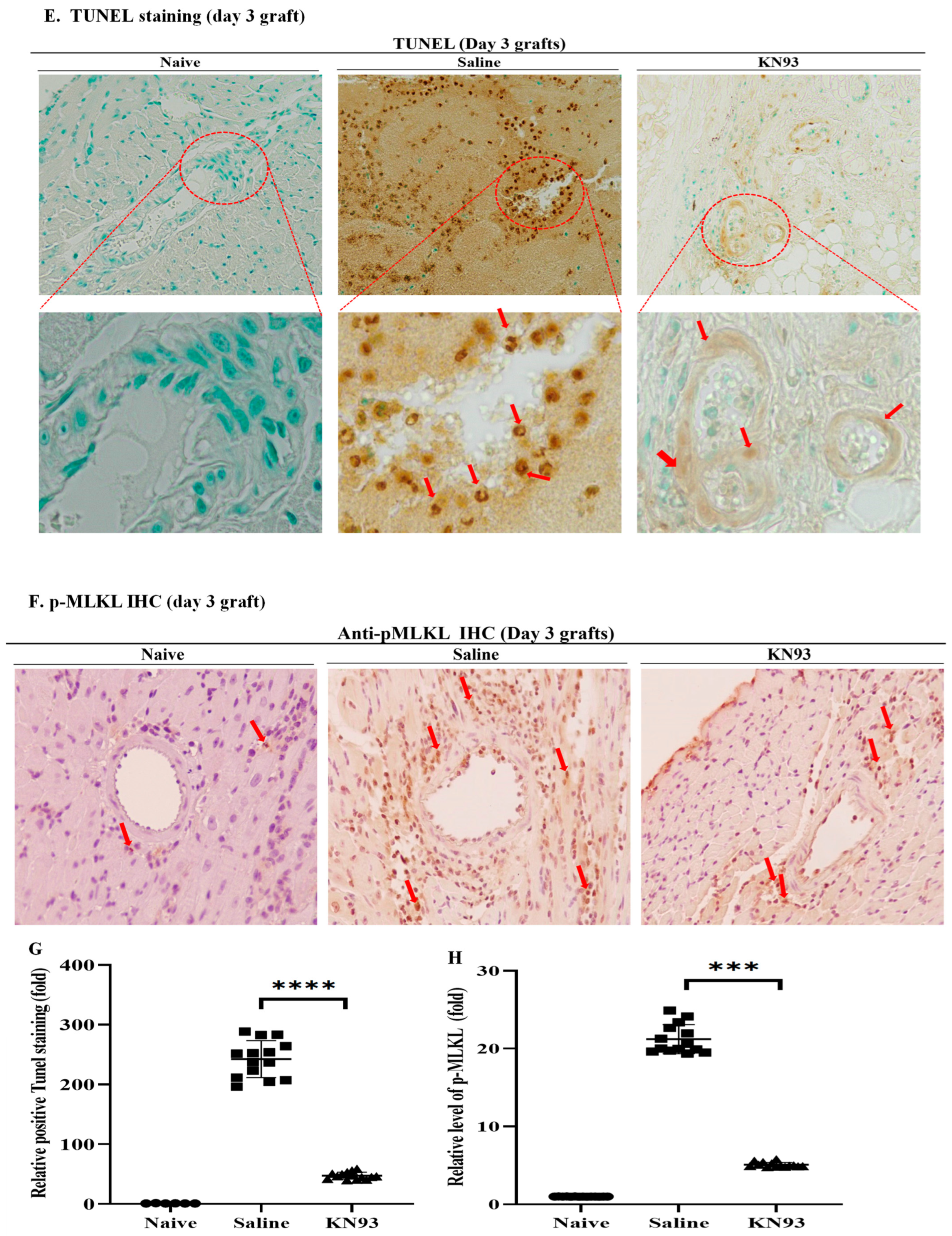
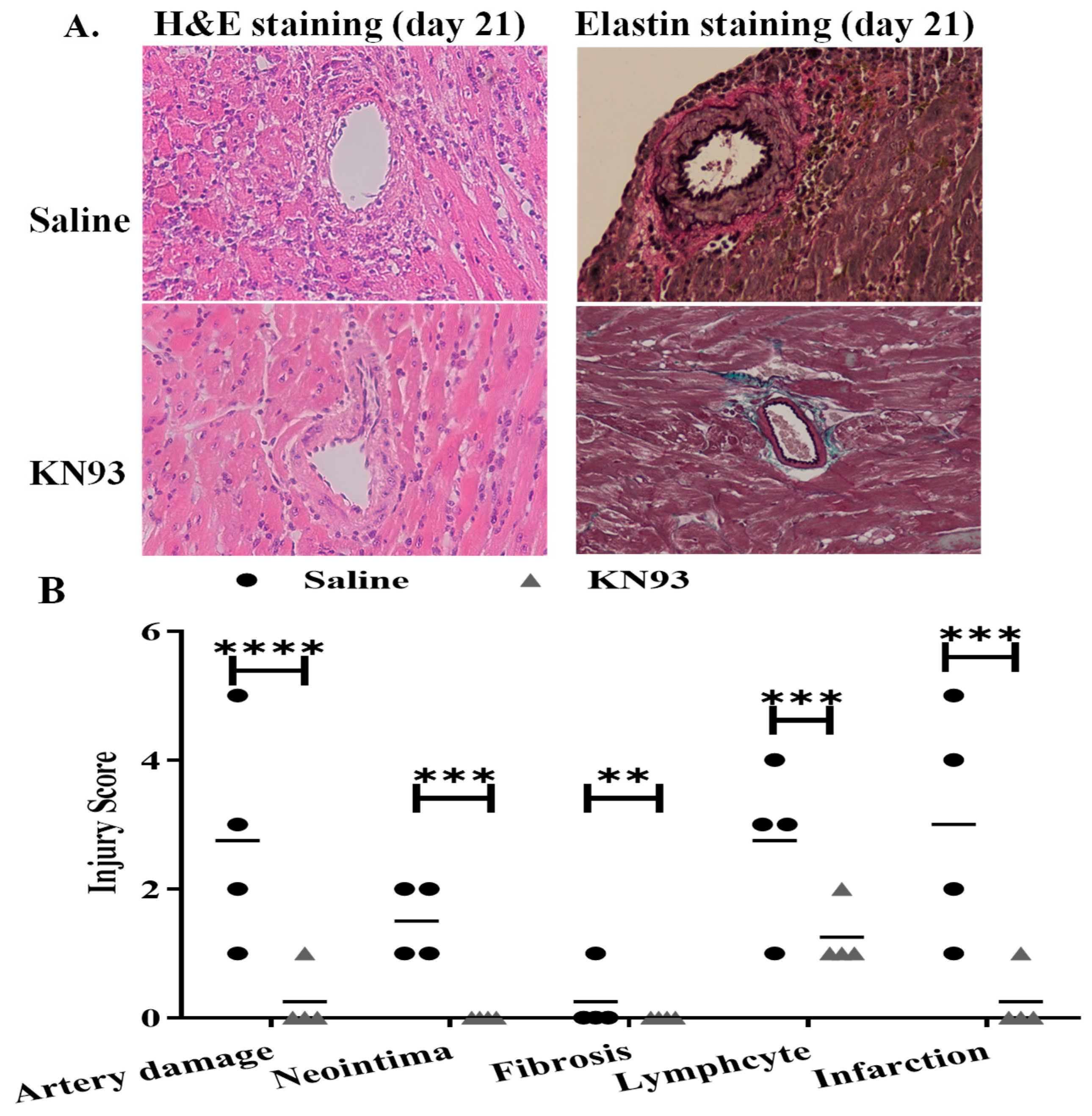
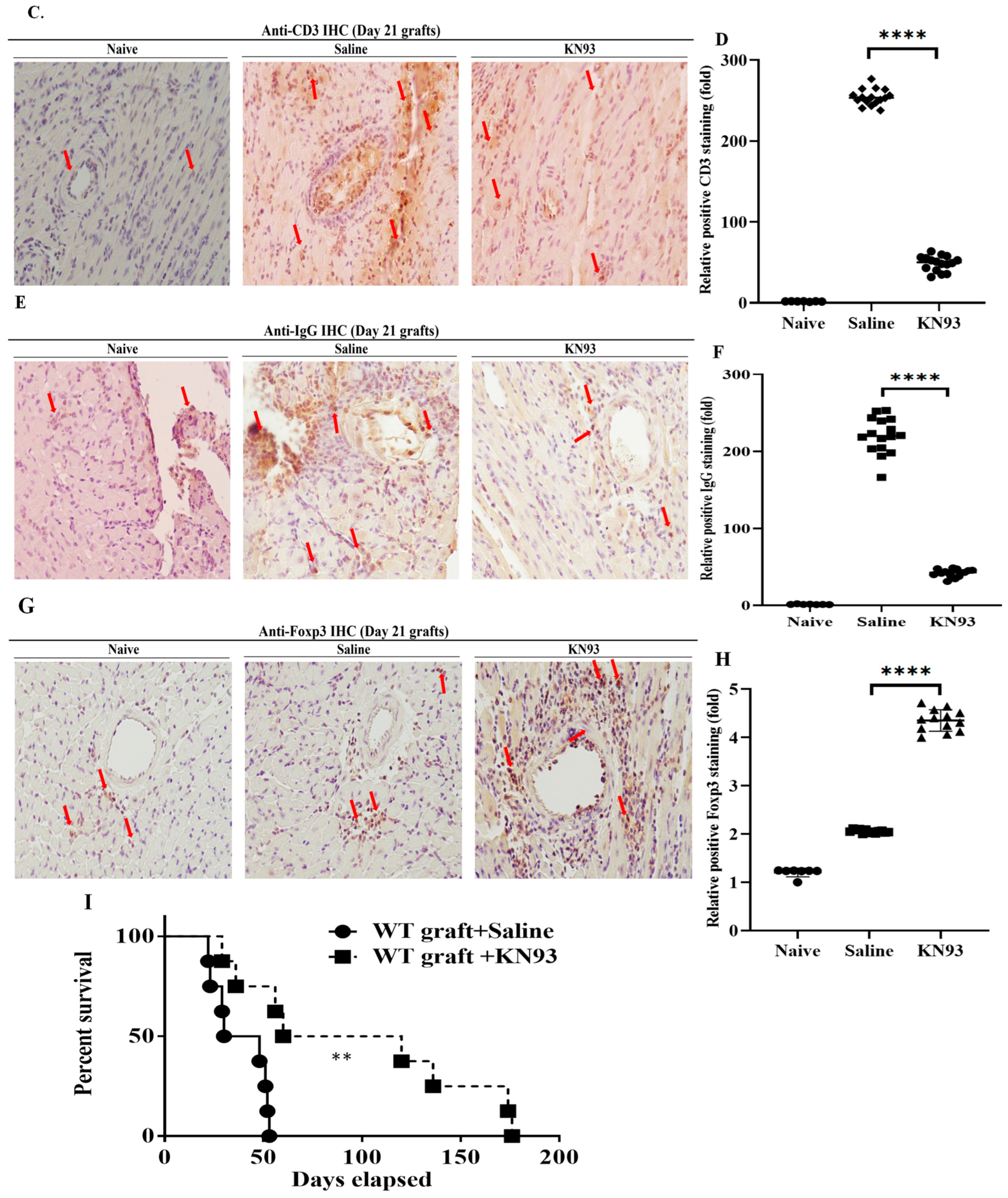
2.4. Inhibition of CaMKs Attenuated Heart Transplant Injury and Rejection
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Death Assay
4.3. Mitochondrial Analysis
4.4. Small Interference RNA (siRNA)
4.5. Quantitative PCR
4.6. Immunoprecipitation
4.7. Western Blot
4.8. Heterotopic Cardiac Transplantation and Post-Operative Monitoring
4.9. Histology
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CaMK | calmodulin-dependent protein kinase |
| Drp1 | dynamin-related protein-1 |
| IRI | Ischemia reperfusion injury |
| MVEC | mouse microvascular endothelial cells |
| Nec-1s | Necrostatin-1 |
| PCR | polymerase chain reaction |
| PGAM5 | phosphoglycerate mutase 5 |
| pMLKL | phosphorylated mixed lineage kinase domain like protein |
| RIPK | receptor interacting protein kinase |
| TUNEL | TdT-mediated dUTP nick-ended labeling |
| Z-IETD | Z-Ile-Glu-Thr-Asp-Fluoromethylketoe |
References
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [PubMed]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Padanilam, B.J. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell. Mol. Life Sci. 2016, 73, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Pavlosky, A.; Lau, A.; Su, Y.; Lian, D.; Huang, X.; Yin, Z.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. RIPK3-Mediated Necroptosis Regulates Cardiac Allograft Rejection. Am. J. Transplant. 2014, 14, 1778–1790. [Google Scholar] [CrossRef]
- Kwok, C.P.A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.; Zhang, Z.X. Necroptosis is Involved in CD4+ T-cell Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037. [Google Scholar] [CrossRef]
- Qamar, A.; Zhao, J.; Xu, L.; McLeod, P.; Huang, X.; Jiang, J.; Liu, W.; Haig, A.; Zhang, Z.X. Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. Int. J. Mol. Sci. 2021, 22, 1038. [Google Scholar] [CrossRef] [PubMed]
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. 2019, 19, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Praefcke, G.J.; McMahon, H.T. The dynamin superfamily: Universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 2004, 5, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Huang, X.; McLeod, P.; Jiang, J.; Liu, W.; Haig, A.; Jevnikar, A.M.; Jiang, Z.; Zhang, Z.X. Toll-like receptor 3 is an endogenous sensor of cell death and a potential target for induction of long-term cardiac transplant survival. Am. J. Transplant. 2021, 21, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Mayford, M.; Bach, M.E.; Huang, Y.Y.; Wang, L.; Hawkins, R.D.; Kandel, E.R. Control of memory formation through regulated expression of a CaMKII transgene. Science 1996, 274, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Takemoto-Kimura, S.; Suzuki, K.; Horigane, S.I.; Kamijo, S.; Inoue, M.; Sakamoto, M.; Fujii, H.; Bito, H. Calmodulin kinases: Essential regulators in health and disease. J. Neurochem. 2017, 141, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Tokumitsu, H.; Davare, M.A.; Soderling, T.R. Analysis of CaM-kinase signaling in cells. Cell Calcium 2011, 50, 1–8. [Google Scholar] [CrossRef]
- Gray, C.B.; Heller Brown, J. CaMKIIdelta subtypes: Localization and function. Front. Pharmacol. 2014, 5, 15. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Chen, S.; Guan, S.; Yan, Z.; Ouyang, F.; Li, S.; Liu, L.; Zhong, J. Role of RIPK3-CaMKII-mPTP signaling pathway-mediated necroptosis in cardiovascular diseases (Review). Int. J. Mol. Med. 2023, 52, 98. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, E.; Zhang, J.; Zhang, Y. CaMKII, ‘jack of all trades’ in inflammation during cardiac ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2023, 184, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef] [PubMed]
- Bo, T.; Yamamori, T.; Suzuki, M.; Sakai, Y.; Yamamoto, K.; Inanami, O. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine 616. Biochem. Biophys. Res. Commun. 2018, 495, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, B.; Hook, S.S.; Selbert, M.A.; Goldstein, E.G.; Tomhave, E.D.; Edelman, A.M.; Snyderman, R.; Means, A.R. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase I kinase. EMBO J. 1995, 14, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Hatano, N.; Inuzuka, H.; Yokokura, S.; Nozaki, N.; Kobayashi, R. Mechanism of the generation of autonomous activity of Ca2+/calmodulin-dependent protein kinase IV. J. Biol. Chem. 2004, 279, 40296–40302. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Soderling, T.R. Requirements for calcium and calmodulin in the calmodulin kinase activation cascade. J. Biol. Chem. 1996, 271, 5617–5622. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.; Anderson, K.A.; Ho, N.; Means, A.R. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J. Biol. Chem. 1996, 271, 21542–21548. [Google Scholar] [CrossRef] [PubMed]
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Griepentrog, J.E.; Zou, B.; Xu, L.; Cyr, A.R.; Chambers, L.M.; Zuckerbraun, B.S.; Shiva, S.; Rosengart, M.R. CaMKIV regulates mitochondrial dynamics during sepsis. Cell Calcium 2020, 92, 102286. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Goossens, V.; Grootjans, S.; Van den Haute, C.; Vanlangenakker, N.; Dondelinger, Y.; Roelandt, R.; Bruggeman, I.; Goncalves, A.; Bertrand, M.J.; et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014, 5, e1004. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014, 5, e1086. [Google Scholar] [CrossRef]
- Moriwaki, K.; Farias Luz, N.; Balaji, S.; De Rosa, M.J.; O’Donnell, C.L.; Gough, P.J.; Bertin, J.; Welsh, R.M.; Chan, F.K. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J. Immunol. 2016, 196, 407–415. [Google Scholar] [CrossRef]
- Tran, D.T.; Tu, Z.; Alawieh, A.; Mulligan, J.; Esckilsen, S.; Quinn, K.; Sundararaj, K.; Wallace, C.; Finnegan, R.; Allen, P.; et al. Modulating donor mitochondrial fusion/fission delivers immunoprotective effects in cardiac transplantation. Am. J. Transplant. 2022, 22, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Vila-Petroff, M.; Salas, M.A.; Said, M.; Valverde, C.A.; Sapia, L.; Portiansky, E.; Hajjar, R.J.; Kranias, E.G.; Mundina-Weilenmann, C.; Mattiazzi, A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc. Res. 2007, 73, 689–698. [Google Scholar] [CrossRef]
- Ling, H.; Gray, C.B.; Zambon, A.C.; Grimm, M.; Gu, Y.; Dalton, N.; Purcell, N.H.; Peterson, K.; Brown, J.H. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ. Res. 2013, 112, 935–944. [Google Scholar] [CrossRef]
- Zhu, W.; Woo, A.Y.; Yang, D.; Cheng, H.; Crow, M.T.; Xiao, R.P. Activation of CaMKIIδC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 10833–10839. [Google Scholar] [CrossRef]
- Joiner, M.L.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Jain, S.; Ashra, S.Y.; Furness, P.N.; Nicholson, M.L. Apoptosis and caspase-3 in long-term renal ischemia/reperfusion injury in rats and divergent effects of immunosuppressants. Transplantation 2006, 81, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, Y.; Chen, S.; Cai, J.; Luo, Z.; Yu, S.; Lu, J. Activation of Ca(2+)/Calmodulin-Dependent Protein Kinase II (CaMKII) with Lidocaine Provokes Pyroptosis of Glioblastoma Cells. Bull. Exp. Biol. Med. 2021, 171, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Li, Y.; Zhang, X.; Wu, B.; Li, M.; Gao, J.; Xia, H.; Xu, L. FGF5 protects heart from sepsis injury by attenuating cardiomyocyte pyroptosis through inhibiting CaMKII/NFkappaB signaling. Biochem. Biophys. Res. Commun. 2022, 636, 104–112. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Jiang, J.; Min, J.; Huang, X.; McLeod, P.; Liu, W.; Haig, A.; Gunaratnam, L.; Jevnikar, A.M.; Zhang, Z.-X. The CaMK Family Differentially Promotes Necroptosis and Mouse Cardiac Graft Injury and Rejection. Int. J. Mol. Sci. 2024, 25, 4428. https://doi.org/10.3390/ijms25084428
Lu H, Jiang J, Min J, Huang X, McLeod P, Liu W, Haig A, Gunaratnam L, Jevnikar AM, Zhang Z-X. The CaMK Family Differentially Promotes Necroptosis and Mouse Cardiac Graft Injury and Rejection. International Journal of Molecular Sciences. 2024; 25(8):4428. https://doi.org/10.3390/ijms25084428
Chicago/Turabian StyleLu, Haitao, Jifu Jiang, Jeffery Min, Xuyan Huang, Patrick McLeod, Weihua Liu, Aaron Haig, Lakshman Gunaratnam, Anthony M. Jevnikar, and Zhu-Xu Zhang. 2024. "The CaMK Family Differentially Promotes Necroptosis and Mouse Cardiac Graft Injury and Rejection" International Journal of Molecular Sciences 25, no. 8: 4428. https://doi.org/10.3390/ijms25084428
APA StyleLu, H., Jiang, J., Min, J., Huang, X., McLeod, P., Liu, W., Haig, A., Gunaratnam, L., Jevnikar, A. M., & Zhang, Z.-X. (2024). The CaMK Family Differentially Promotes Necroptosis and Mouse Cardiac Graft Injury and Rejection. International Journal of Molecular Sciences, 25(8), 4428. https://doi.org/10.3390/ijms25084428