
Distinct Driver Pathway Enrichments and a High Prevalence of TSC2 Mutations in Right Colon Cancer in Chile: A Preliminary Comparative Analysis
 , , , , ,
, , , , ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Clinical and General Characteristics of the Patients
2.2. General Landscape
2.3. Mutational Status of Actionable Genes in CRC
2.3.1. KRAS, NRAS, and BRAF
2.3.2. MLH1, PMS2, MSH2, and MSH6
2.4. Mutational Status of Emerging Predictive and Actionable Genes
2.4.1. PIK3CA
2.4.2. TP53
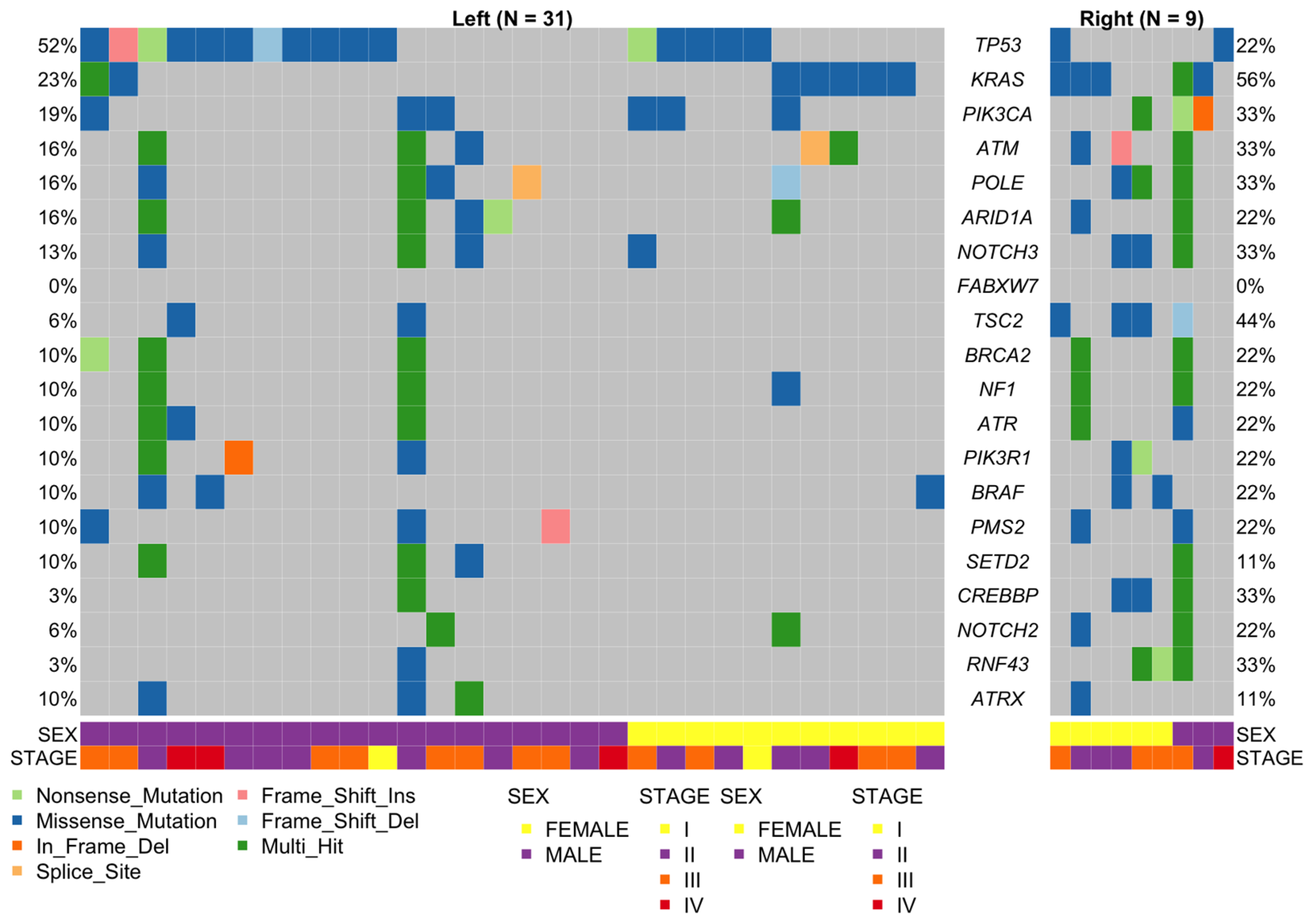
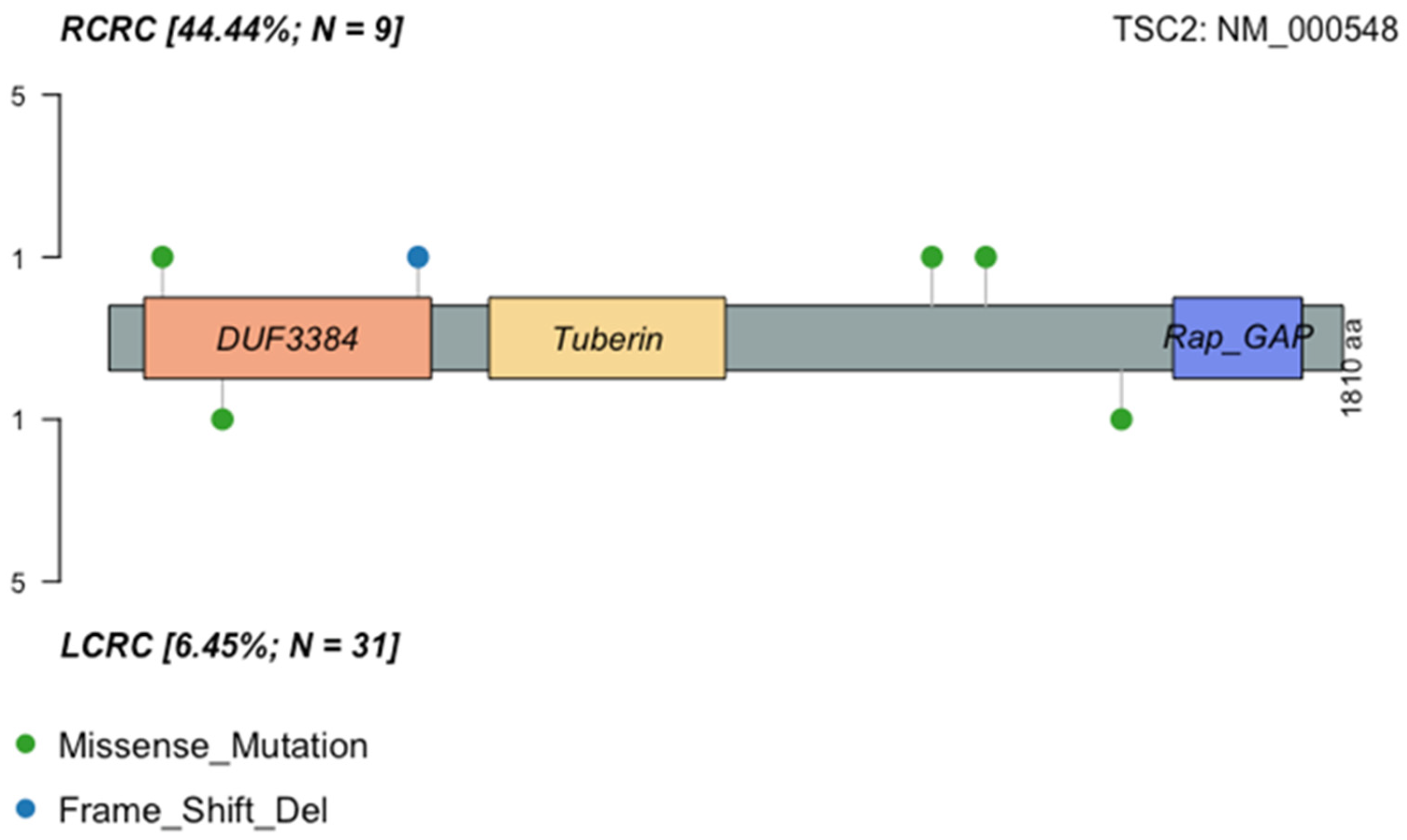
2.5. Left Versus Right-Sided CRC
2.6. Comparison of Mutation Frequencies in Independent Cohorts
2.7. Left Versus Right Sided CRC in MSK-IMPACT and TCGA Cohorts
2.8. Pathway Analyses
2.9. General Characteristics of the PI3K Pathway Altered Samples
3. Discussion
3.1. Frequency of CRC Mutations in LATAM Cohorts
3.2. Comparison of CRC Mutations with TCGA and MSK-IMPACT Cohorts
3.3. Left vs. Right CRC
3.4. Comparison between Oncogenic Pathways
4. Materials and Methods
4.1. Samples and Sequencing
4.2. Bioinformatic Analysis, Variant Calling and Classification
4.3. Public Databases
4.4. Gene Selection
4.5. Pathways Analyses
4.6. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedźwiedzka, E.; Arłukowicz, T.; Przybyłowicz, K.E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers 2021, 13, 2021. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Weinberg, B.A.; Xiu, J.; El-Deiry, W.S.; Hwang, J.J.; Gatalica, Z.; Philip, P.A.; Shields, A.F.; Lenz, H.J.; Marshall, J.L. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget 2017, 8, 86356–86368. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef]
- Baran, B.; Mert Ozupek, N.; Yerli Tetik, N.; Acar, E.; Bekcioglu, O.; Baskin, Y. Difference Between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterol. Res. 2018, 11, 264–273. [Google Scholar] [CrossRef]
- Wielandt, A.M.; Hurtado, C.; Moreno, C.M.; Villarroel, C.; Castro, M.; Estay, M.; Simian, D.; Martinez, M.; Vial, M.T.; Kronberg, U.; et al. Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability. Tumor Biol. 2020, 42, 7–21. [Google Scholar] [CrossRef]
- Heath, E.I.; Lynce, F.; Xiu, J.; Ellerbrock, A.; Reddy, S.K.; Obeid, E.; Liu, S.V.; Bolling-Fischer, A.; Separovic, D.; Vanderwalde, A. Racial disparities in the molecular landscape of cancer. AntiCancer Res. 2018, 38, 2235–2240. [Google Scholar] [CrossRef]
- Hanna, M.C.; Go, C.; Roden, C.; Jones, R.T.; Pochanard, P.; Javed, A.Y.; Javed, A.; Mondal, C.; Palescandolo, E.; Van Hummelen, P.; et al. Colorectal Cancers from Distinct Ancestral Populations Show Variations in BRAF Mutation Frequency. PLoS ONE 2013, 8, e74950. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, J.; Yang, Y.; Lu, J.; Gao, J.; Lu, T.; Sun, J.; Jiang, H.; Zhu, Y.; Zheng, Y.; et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: Analysis of 1110 cases. Sci. Rep. 2015, 5, 18678–18686. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.C.; Thiam, T.K.; Lu, Y.J.; Yeh, C.Y.; Tsai, W.S.; You, J.F.; Hung, H.Y.; Tsai, C.N.; Hsu, A.; Chen, H.C.; et al. Mutations of KRAS/NRAS/BRAF predict cetuximab resistance in metastatic colorectal cancer patients. Oncotarget 2016, 7, 22257–22270. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Moiseyenko, V.M.; Moiseyenko, F.V.; Yanus, G.A.; Kuligina, E.S.; Sokolenko, A.P.; Bizin, I.V.; Kudriavtsev, A.A.; Aleksakhina, S.N.; Volkov, N.M.; Chubenko, V.A.; et al. First-Line Cetuximab Monotherapy in KRAS/NRAS/BRAF Mutation-Negative Colorectal Cancer Patients. Clin. Drug Investig. 2018, 38, 553–562. [Google Scholar] [CrossRef]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Wang, Q.; Shi, Y.L.; Zhou, K.; Wang, L.L.; Yan, Z.X.; Liu, Y.L.; Xu, L.L.; Zhao, S.W.; Chu, H.L.; Shi, T.T.; et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 9, 739–750. [Google Scholar] [CrossRef]
- Mao, C.; Yang, Z.Y.; Hu, X.F.; Chen, Q.; Tang, J.L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2012, 23, 1518–1525. [Google Scholar] [CrossRef]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American society for clinical pathology, college of American pathologists, association for molecular pathology, and American society of clinical oncology. Arch. Pathol. Lab. Med. 2017, 141, 625–657. [Google Scholar] [CrossRef]
- Grady, W.M.; Pritchard, C.C. Molecular alterations and biomarkers in colorectal cancer. Toxicol. Pathol. 2014, 42, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Bartley, A.N.; Mills, A.M.; Konnick, E.; Overman, M.; Ventura, C.B.; Souter, L.; Colasacco, C.; Stadler, Z.K.; Kerr, S.; Howitt, B.E.; et al. Mismatch Repair and Microsatellite Instability Testing for Immune Checkpoint Inhibitor Therapy: Guideline From the College of American Pathologists in Collaboration With the Association for Molecular Pathology and Fight Colorectal Cancer. Arch. Pathol. Lab. Med. 2022, 146, 1194–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ji, Q.; Li, Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: Underlying mechanisms and reversal strategies. J. Exp. Clin. Cancer Res. 2021, 40, 328–345. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425–442. [Google Scholar] [CrossRef]
- Huemer, F.; Thaler, J.; Piringer, G.; Hackl, H.; Pleyer, L.; Hufnagl, C.; Weiss, L.; Greil, R. Sidedness and TP53 mutations impact OS in anti-EGFR but not anti-VEGF treated mCRC—An analysis of the KRAS registry of the AGMT (Arbeitsgemeinschaft Medikamentöse Tumortherapie). BMC Cancer 2018, 18, 11. [Google Scholar] [CrossRef]
- Myer, P.A.; Lee, J.K.; Madison, R.W.; Pradhan, K.; Newberg, J.Y.; Isasi, C.R.; Klempner, S.J.; Frampton, G.M.; Ross, J.S.; Venstrom, J.M.; et al. The Genomics of Colorectal Cancer in Populations with African and European Ancestry. Cancer Discov. 2022, 12, 1282–1293. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Hou, Y.; Zhou, X.; Liang, L.; Zhang, Z.; Shi, H.; Xu, S.; Hu, P.; Zheng, Z.; et al. Performance validation of an amplicon-based targeted next-generation sequencing assay and mutation profiling of 648 Chinese colorectal cancer patients. Virchows Arch. 2018, 472, 959–968. [Google Scholar] [CrossRef]
- Dos Santos, W.; Sobanski, T.; de Carvalho, A.C.; Evangelista, A.F.; Matsushita, M.; Berardinelli, G.N.; de Oliveira, M.A.; Reis, R.M.; Guimarães, D.P. Mutation profiling of cancer drivers in Brazilian colorectal cancer. Sci. Rep. 2019, 9, 13687–13700. [Google Scholar] [CrossRef]
- Torres, Á.; Oliver, J.; Frecha, C.; Montealegre, A.L.; Quezada-Urbán, R.; Díaz-Velásquez, C.E.; Vaca-Paniagua, F.; Perdomo, S. Cancer Genomic Resources and Present Needs in the Latin American Region. Public. Health Genom. 2017, 20, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Rúa, K.A.; Isaza-Jiménez, L.F.; Ahumada-Rodríguez, E.; Ceballos-García, H.; Muñetón-Peña, C.M. Genetic analysis in APC, KRAS, and TP53 in patients with stomach and colon cancer. Rev. Gastroenterol. Mex. 2014, 79, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, I.K.T.M.; Bartelli, T.F.; Defelicibus, A.; Sendoya, J.M.; Golubicki, M.; Robbio, J.; Serpa, M.S.; Branco, G.P.; Santos, L.B.C.; Claro, L.C.L.; et al. Exome and Tissue-Associated Microbiota as Predictive Markers of Response to Neoadjuvant Treatment in Locally Advanced Rectal Cancer. Front. Oncol. 2022, 12, 809441–809457. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo Bermejo, J.; Boekstegers, F.; González Silos, R.; Marcelain, K.; Baez Benavides, P.; Barahona Ponce, C.; Müller, B.; Ferreccio, C.; Koshiol, J.; Fischer, C.; et al. Subtypes of Native American ancestry and leading causes of death: Mapuche ancestry-specific associations with gallbladder cancer risk in Chile. PLoS Genet. 2017, 13, e1006756. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhan, Y.; Lu, J.; Hu, J.; Kong, D. A prognostic predictive model constituted with gene mutations of APC, BRCA2, CDH1, SMO, and TSC2 in colorectal cancer. Ann. Transl. Med. 2021, 9, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Salvo, M.; González-Feliú, E.; Toro, J.; Gallegos, I.; Maureira, I.; Miranda-González, N.; Barajas, O.; Bustamante, E.; Ahumada, M.; Colombo, A.; et al. Validation of an NGS Panel Designed for Detection of Actionable Mutations in Tumors Common in Latin America. J. Pers. Med. 2021, 11, 899. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.Z.; Li, J.L.; Cai, Z.M.; Li, K.Z.; Hu, B.L. Impact of primary colorectal Cancer location on the KRAS status and its prognostic value. BMC Gastroenterol. 2019, 19, 46–55. [Google Scholar] [CrossRef]
- Nawa, T.; Kato, J.; Kawamoto, H.; Okada, H.; Yamamoto, H.; Kohno, H.; Endo, H.; Shiratori, Y. Differences between right- and left-sided colon cancer in patient characteristics, cancer morphology and histology. J. Gastroenterol. Hepatol. 2008, 23, 418–423. [Google Scholar] [CrossRef]
- Scott, N.; Sagar, P.; Stewart, J.; Blair, G.E.; Dixon, M.F.; Quirke, P. p53 in colorectal cancer: Clinicopathological correlation and prognostlc significance. Br. J. Cancer 1991, 63, 317–319. [Google Scholar] [CrossRef]
- Rinne, N.; Christie, E.L.; Ardasheva, A.; Kwok, C.H.; Demchenko, N.; Low, C.; Tralau-Stewart, C.; Fotopoulou, C.; Cunnea, P. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist. 2021, 4, 573–595. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; de Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N.; et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Zureick, A.H.; McFadden, K.A.; Mody, R.; Koschmann, C. Successful treatment of a TSC2-mutant glioblastoma with everolimus. BMJ Case Rep. 2019, 12, 227734–227738. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Lalani, A.K.A.; Jacobus, S.; Wankowicz, S.A.; Polacek, L.; Takeda, D.Y.; Harshman, L.C.; Wagle, N.; Moreno, I.; Lundgren, K.; et al. Everolimus and pazopanib (E/P) benefit genomically selected patients with metastatic urothelial carcinoma. Br. J. Cancer 2018, 119, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Adib, E.; Klonowska, K.; Giannikou, K.; Do, K.T.; Pruitt-Thompson, S.; Bhushan, K.; Milstein, M.I.; Hedglin, J.; Kargus, K.E.; Sholl, L.M.; et al. Phase II clinical trial of everolimus in a pan-cancer cohort of patients with mTOR pathway alterations. Clin. Cancer Res. 2021, 27, 3845–3853. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: The phase 2 CHRONOS trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.H.; Byun, D.S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef] [PubMed]
- Eyheramendy, S.; Martinez, F.I.; Manevy, F.; Vial, C.; Repetto, G.M. Genetic structure characterization of Chileans reflects historical immigration patterns. Nat. Commun. 2015, 6, 6472. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Rimm, E.B.; Curhan, G.C.; Kraft, P.; Hunter, D.J.; Hu, F.B.; van Dam, R.M. Obesity susceptibility loci and uncontrolled eating, emotional eating and cognitive restraint behaviors in men and women. Obesity 2014, 22, E135–E141. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, K.S.; Gjesing, A.P.; Grarup, N.; Justesen, J.M.; Sandholt, C.H.; Witte, D.R.; Jørgensen, T.; Madsbad, S.; Hansen, T.; Pedersen, O. Association studies of novel obesity-related gene variants with quantitative metabolic phenotypes in a population-based sample of 6039 Danish individuals. Diabetologia 2012, 55, 105–113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prazeres, H.; Torres, J.; Rodrigues, F.; Pinto, M.; Pastoriza, M.C.; Gomes, D.; Cameselle-Teijeiro, J.; Vidal, A.; Martins, T.C.; Sobrinho-Simões, M.; et al. Chromosomal, epigenetic and microRNA-mediated inactivation of LRP1B, a modulator of the extracellular environment of thyroid cancer cells. Oncogene 2011, 30, 1302–1317. [Google Scholar] [CrossRef]
- Zhai, X.; Xia, Z.; Du, G.; Zhang, X.; Xia, T.; Ma, D.; Li, X.; Jin, B.; Zhang, H. LRP1B suppresses HCC progression through the NCSTN/PI3K/AKT signaling axis and affects doxorubicin resistance. Genes Dis. 2022, 10, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Zollner, L.; Boekstegers, F.; Barahona Ponce, C.; Scherer, D.; Marcelain, K.; Gárate-Calderón, V.; Waldenberger, M.; Morales, E.; Rojas, A.; Munoz, C.; et al. Gallbladder Cancer Risk and Indigenous South American Mapuche Ancestry: Instrumental Variable Analysis Using Ancestry-Informative Markers. Cancers 2023, 15, 4033. [Google Scholar] [CrossRef]
- Barahona Ponce, C.; Scherer, D.; Brinster, R.; Boekstegers, F.; Marcelain, K.; Gárate-Calderón, V.; Müller, B.; De Toro, G.; Retamales, J.; Barajas, O.; et al. Gallstones, Body Mass Index, C-Reactive Protein, and Gallbladder Cancer: Mendelian Randomization Analysis of Chilean and European Genotype Data. Hepatology 2021, 73, 1783–1796. [Google Scholar] [CrossRef]
- Poli, M.C.; Rebolledo-Jaramillo, B.; Lagos, C.; Orellana, J.; Moreno, G.; Martín, L.M.; Encina, G.; Böhme, D.; Faundes, V.; Zavala, M.J.; et al. Decoding complex inherited phenotypes in rare disorders: The DECIPHERD initiative for rare undiagnosed diseases in Chile. Eur. J. Hum. Genet. 2024. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | RCRC | LCRC | TOTAL | p-Value (LCRC vs. RCRC) * | |
|---|---|---|---|---|---|
| Cases | 9 | 31 | 40 | 0.000001508 | |
| Gender | Males | 3 | 20 | 23 | 0.1338 |
| Females | 6 | 11 | 17 | 0.1338 | |
| Mean age at diagnosis | 71.22 (range 51–86) years | 61.35 (range 25–82) years | 63.58 (range 25–86) years | 0.06236 | |
| Stage | I | 0 | 2 | 2 | 1 |
| II | 4 | 12 | 16 | 1 | |
| III | 4 | 13 | 17 | 1 | |
| IV | 1 | 4 | 5 | 1 | |
| Histological diagnosis | Tubular adenocarcinoma | 3 | 15 | 18 | 0.4761 |
| Adenocarcinoma, NOS | 3 | 10 | 13 | N/A | |
| Papillary-tubular adenocarcinoma | 1 | 1 | 2 | 0.4038 | |
| Poorly differentiated with signet-cells carcinoma | 0 | 1 | 1 | 1 | |
| Mucinous adenocarcinoma | 1 | 0 | 1 | 0.225 | |
| Adenocarcinoma mixed tubular and mucinous | 0 | 1 | 1 | 1 | |
| Not specified | 1 | 3 | 4 | N/A | |
| Ethnicity | Chilean | 2 | 9 | 11 | 1 |
| Mapuche | 2 | 3 | 5 | 0.2677 | |
| European | 0 | 1 | 1 | 1 | |
| Not specified | 6 | 17 | 23 | N/A |
| MSK-IMPACT | TCGA | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene | LCRC (n = 311) | RCRC (n = 199) | p-Value | adj p-Value | LCRC (n = 142) | RCRC (n = 78) | p-Value | adj p-Value |
| ARID1A | 11.58% (36) | 24.12% (48) | 3.140 × 10−4 | 1.13 × 10−3 | 6.34% (9) | 16.67% (13) | 2.725 × 10−2 | 1.18 × 10−1 |
| BRAF | 8.36% (26) | 23.62% (47) | 3.015 × 10−6 | 3.92 × 10−5 | 2.82% (4) | 23.08% (18) | 5.191 × 10−6 | 1.35 × 10−4 |
| BRCA1 | 4.5% (14) | 6.03% (12) | 5.76 × 10−1 | 5.99 × 10−1 | 2.11% (3) | 385% (3) | 7.471 × 10−1 | 9.42 × 10−1 |
| BRCA2 | 6.75% (21) | 22.11% (44) | 7.924 × 10−7 | 2.06 × 10−5 | 8.45% (12) | 12.82% (10) | 4.245 × 10−1 | 8.49 × 10−1 |
| CDK12 | 2.89% (9) | 7.04% (14) | 4.774 × 10−2 | 6.53 × 10−2 | 2.82% (4) | 6.41% (5) | 3.516 × 10−1 | 7.62 × 10−1 |
| CDKN2A | 1.93% (6) | 4.02% (8) | 2.577 × 10−1 | 2.91 × 10−1 | 0% (0) | 1.28% (1) | 7.606 × 10−1 | 9.42 × 10−1 |
| FGFR1 | 1.93% (6) | 4.52% (9) | 1.550 × 10−1 | 1.83 × 10−1 | 2.11% (3) | 1.28% (1) | 1 × 100 | 1 × 10−0 |
| FGFR2 | 2.25% (7) | 5.03% (10) | 1.471 × 10−1 | 1.82 × 10−1 | 3.52% (5) | 1.28% (1) | 5.873 × 10−1 | 9.42 × 10−1 |
| FGFR3 | 1.29% (4) | 6.53% (13) | 3.009 × 10−3 | 6.52 × 10−3 | 0% (0) | 2.56% (2) | 2.402 × 10−1 | 5.68 × 10−1 |
| KRAS | 41.16% (128) | 57.79% (115) | 3.470 × 10−4 | 1.13 × 10−3 | 41.55% (59) | 46.15% (36) | 6.049 × 10−1 | 9.42 × 10−1 |
| MLH1 | 1.93% (6) | 5.53% (11) | 5.053 × 10−2 | 6.57 × 10−2 | 2.82% (4) | 5.13% (4) | 6.173 × 10−1 | 9.42 × 10−1 |
| MSH2 | 2.57% (8) | 8.04% (16) | 8.538 × 10−3 | 1.59 × 10−2 | 3.52% (5) | 2.56% (2) | 1 × 100 | 1 × 10−0 |
| MSH6 | 3.54% (11) | 11.06% (22) | 1.462 × 10−3 | 3.8 × 10−3 | 4.23% (6) | 11.54% (9) | 7.523 × 10−2 | 2.79 × 10−1 |
| MTOR | 3.54% (11) | 14.07% (28) | 2.722 × 10−5 | 1.42 × 10−4 | 5.63% (8) | 11.54% (9) | 1.919 × 10−1 | 5.68 × 10−1 |
| NF1 | 4.18% (13) | 13.07% (26) | 4.442 × 10−4 | 1.28 × 10−3 | 4.23% (6) | 6.41% (5) | 6.98 × 10−1 | 9.42 × 10−1 |
| NRAS | 4.18% (13) | 5.03% (10) | 8.182 × 10−1 | 8.18 × 10−1 | 9.15% (13) | 8.97% (7) | 1 × 100 | 1 × 10−0 |
| NTRK1 | 2.57% (8) | 8.54% (17) | 4.569 × 10−3 | 9.14 × 10−3 | 2.11% (3) | 3.85% (3) | 7.471 × 10−1 | 9.42 × 10−1 |
| NTRK3 | 2.25% (7) | 6.53% (13) | 2.808 × 10−2 | 4.06 × 10−2 | 2.11% (3) | 11.54% (9) | 8.419 × 10−3 | 5.47 × 10−2 |
| PIK3CA | 19.61% (61) | 38.19% (76) | 6.347 × 10−6 | 5.16 × 10−5 | 11.27% (16) | 30.77% (24) | 6.618 × 10−4 | 5.74 × 10−3 |
| PMS2 | 0.96% (3) | 2.51% (5) | 3.139 × 10−1 | 3.4 × 10−1 | 1.41% (2) | 5.13% (4) | 2.349 × 10−1 | 5.68 × 10−1 |
| POLE | 6.75% (21) | 14.07% (28) | 9.839 × 10−3 | 1.71 × 10−2 | 5.63% (8) | 16.67% (13) | 1.534 × 10−2 | 7.98 × 10−2 |
| PTEN | 7.07% (22) | 16.08% (32) | 2.091 × 10−3 | 4.94 × 10−3 | 4.93% (7) | 5.13% (4) | 1 × 100 | 1 × 10−0 |
| RB1 | 3.54% (11) | 8.54% (17) | 2.631 × 10−2 | 4.96 × 10−2 | 2.11% (3) | 6.41% (5) | 2.104 × 10−1 | 5.68 × 10−1 |
| TP53 | 81.03% (252) | 62.81% (125) | 7.944 × 10−6 | 5.16 × 10−5 | 65.49% (93) | 35.9% (28) | 4.515 × 10−5 | 5.87 × 10−4 |
| TSC1 | 1.93% (6) | 6.03% (12) | 2.765 × 10−2 | 4.06 × 10−2 | 1.41% (2) | 2.56% (2) | 9.312 × 10−1 | 1 × 10−0 |
| TSC2 | 1.93% (6) | 10.55% (21) | 5.35 × 10−5 | 2.32 × 10−4 | 1.41% (2) | 0% (0) | 7.562 × 10−1 | 9.42 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapia-Valladares, C.; Valenzuela, G.; González, E.; Maureira, I.; Toro, J.; Freire, M.; Sepúlveda-Hermosilla, G.; Ampuero, D.; Blanco, A.; Gallegos, I.; et al. Distinct Driver Pathway Enrichments and a High Prevalence of TSC2 Mutations in Right Colon Cancer in Chile: A Preliminary Comparative Analysis. Int. J. Mol. Sci. 2024, 25, 4695. https://doi.org/10.3390/ijms25094695
Tapia-Valladares C, Valenzuela G, González E, Maureira I, Toro J, Freire M, Sepúlveda-Hermosilla G, Ampuero D, Blanco A, Gallegos I, et al. Distinct Driver Pathway Enrichments and a High Prevalence of TSC2 Mutations in Right Colon Cancer in Chile: A Preliminary Comparative Analysis. International Journal of Molecular Sciences. 2024; 25(9):4695. https://doi.org/10.3390/ijms25094695
Chicago/Turabian StyleTapia-Valladares, Camilo, Guillermo Valenzuela, Evelin González, Ignacio Maureira, Jessica Toro, Matías Freire, Gonzalo Sepúlveda-Hermosilla, Diego Ampuero, Alejandro Blanco, Iván Gallegos, and et al. 2024. "Distinct Driver Pathway Enrichments and a High Prevalence of TSC2 Mutations in Right Colon Cancer in Chile: A Preliminary Comparative Analysis" International Journal of Molecular Sciences 25, no. 9: 4695. https://doi.org/10.3390/ijms25094695
APA StyleTapia-Valladares, C., Valenzuela, G., González, E., Maureira, I., Toro, J., Freire, M., Sepúlveda-Hermosilla, G., Ampuero, D., Blanco, A., Gallegos, I., Morales, F., Erices, J. I., Barajas, O., Ahumada, M., Contreras, H. R., González, J., Armisén, R., & Marcelain, K. (2024). Distinct Driver Pathway Enrichments and a High Prevalence of TSC2 Mutations in Right Colon Cancer in Chile: A Preliminary Comparative Analysis. International Journal of Molecular Sciences, 25(9), 4695. https://doi.org/10.3390/ijms25094695