
Polymeric Polylactic Acid–Glycolic Acid-Based Nanoparticles Deliver Nintedanib Across the Blood–Brain Barrier to Inhibit Glioblastoma Growth
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
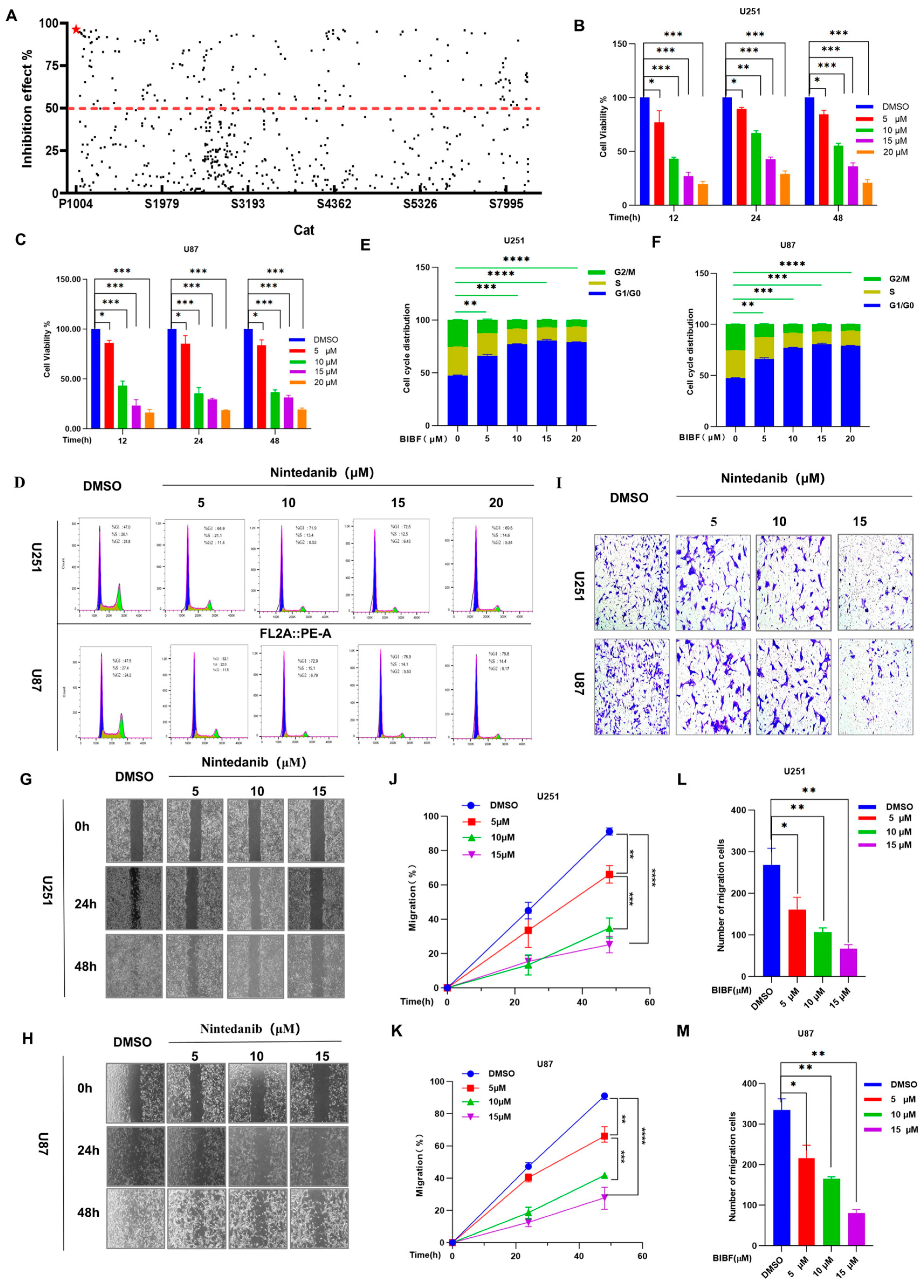
2.1. Effects of Screened Nintedanib on Glioblastoma Cell Biology: Proliferation, Migration, and Invasion
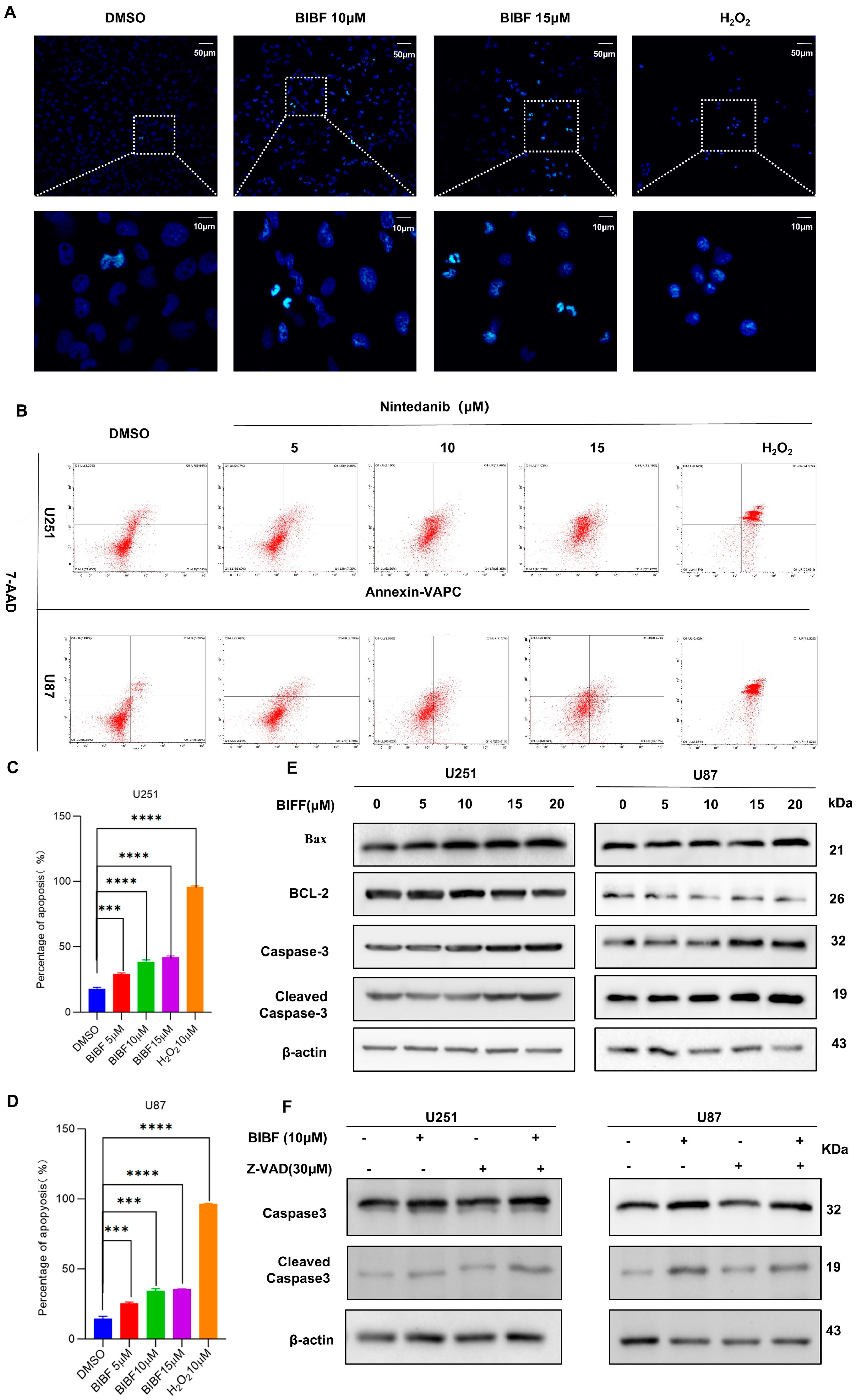
2.2. Effect of Nintedanib on Apoptosis in Glioblastoma
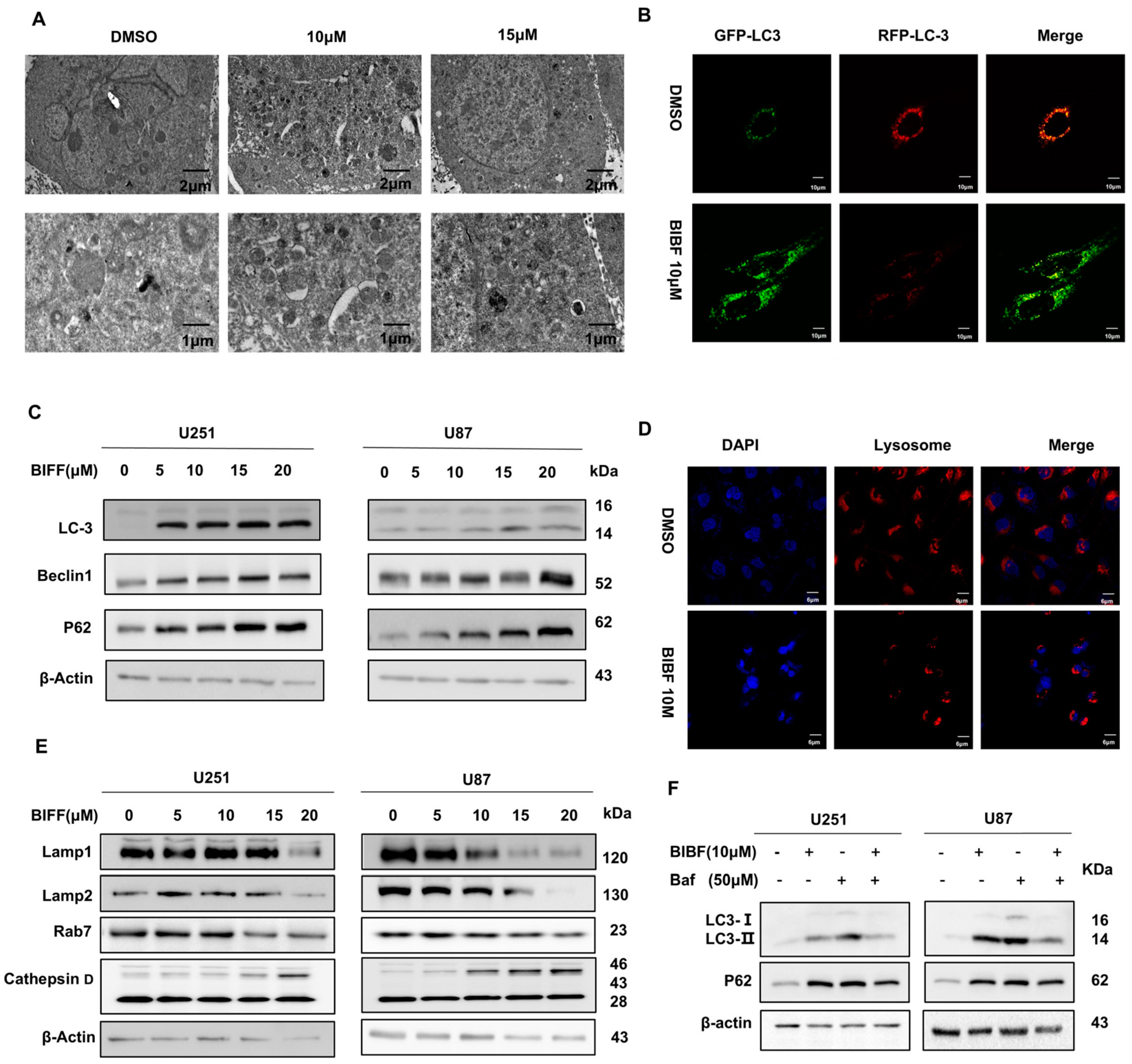
2.3. Inhibition of Glioblastoma Autophagy by Nintedanib
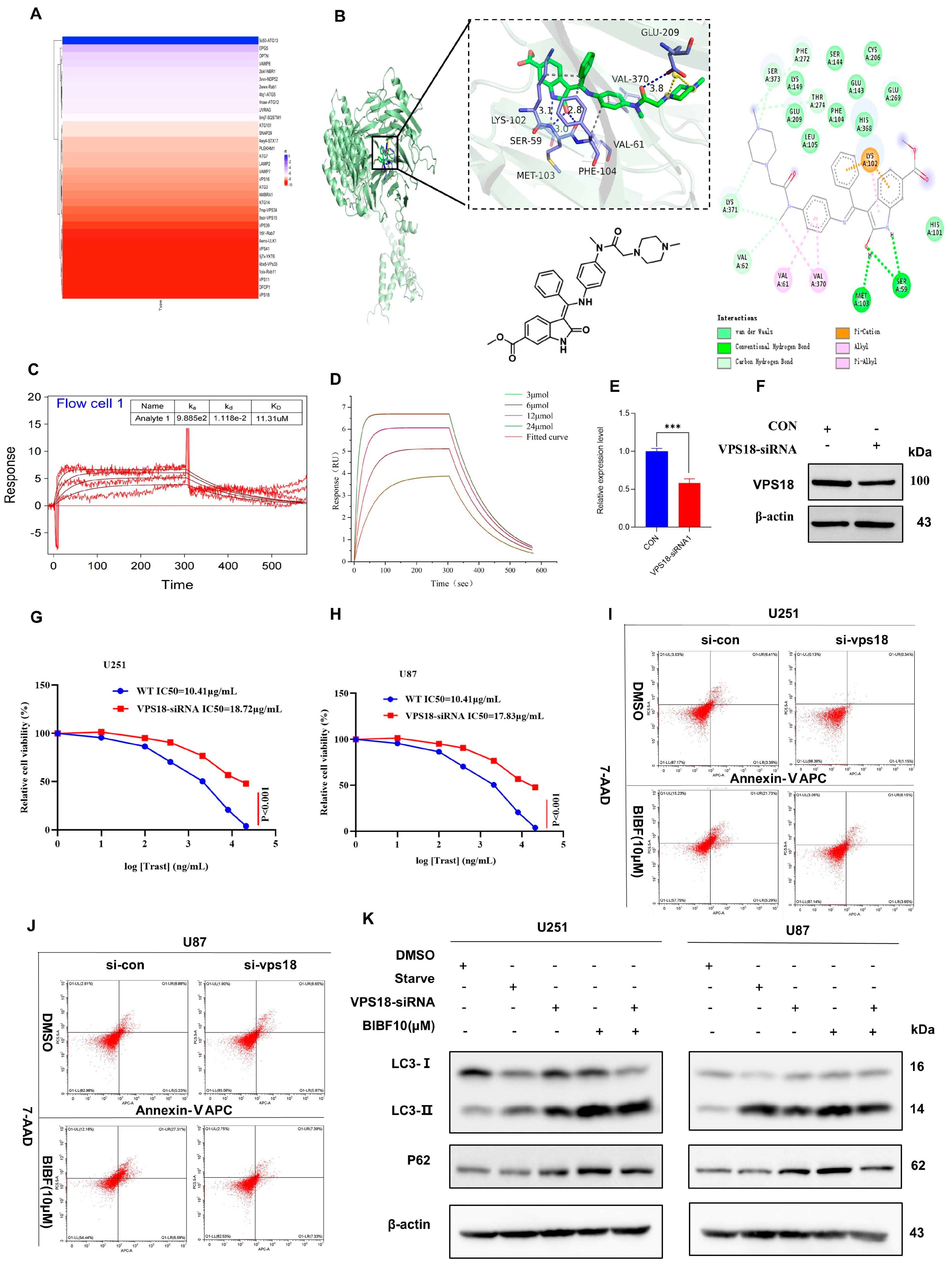
2.4. Nintedanib-Mediated VPS18 Pathway Affects Autophagy
2.5. PLGA-Polymrized BIBF NPs Solve the Problem of BIBF’s Inability to Cross the Blood–Brain Barrier
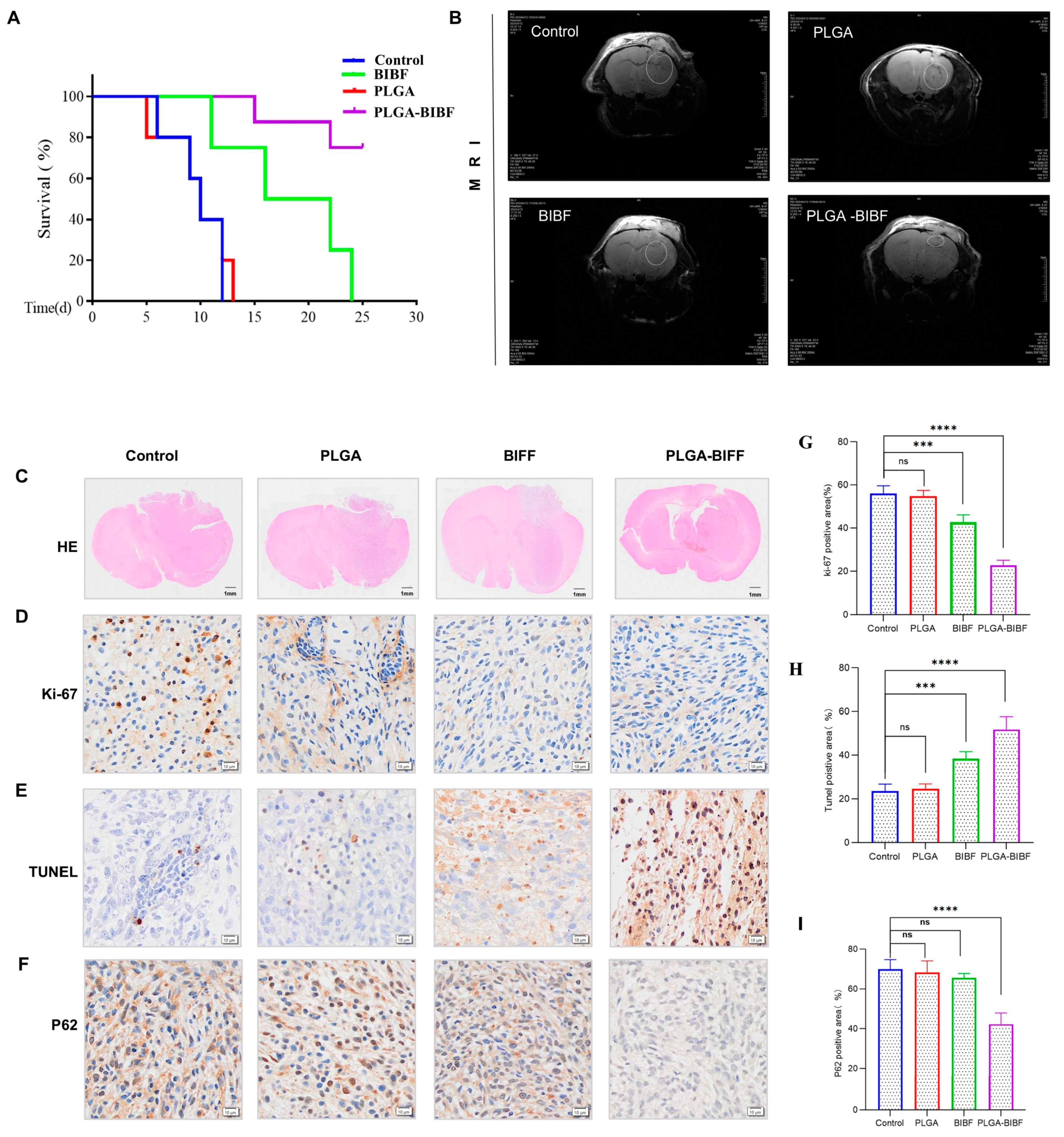
2.6. PLGA-Delivered Nintedanib Inhibits GBM Growth to Improve Survival in BALB/c Nude Mice
3. Discussion
4. Materials and Methods
4.1. Drug Preparation and Reagents
4.2. Cells and Culture Conditions
4.3. Small-Molecule Drug Library Screening
4.4. Cell Viability and Cytotoxicity Assays
4.5. Cell Cycle Assay
4.6. Monolayer Wound-Healing and Transwell Assays
4.7. Apoptosis Assay
4.8. Transmission Electron Microscopy
4.9. Plasmid Infection
4.10. Western Blot Analysis
4.11. Molecular Docking
4.12. Protein and Small Molecule Interaction Analysis
4.13. PLGA for Dintedanib Delivery
4.14. High-Performance Liquid Chromatography Analysis
4.15. Animals Studies
4.16. In Vivo Imaging of Small Animals
4.17. Immunohistochemical and Immunofluorescence Assessment
4.18. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Ballard, C.; Benedetti, J.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S.; Ostrom, Q.T. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2017–2021. Neuro Oncol. 2024, 26, vi1–vi85. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Choudhury, A.; Keough, M.B.; Seo, K.; Ni, L.; Kakaizada, S.; Lee, A.; Aabedi, A.; Popova, G.; Lipkin, B.; et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature 2023, 617, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Thakor, A.S.; Gambhir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA Cancer J. Clin. 2013, 63, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Kashani, G.K.; Naghib, S.M.; Soleymani, S.; Mozafari, M.R. A review of DNA nanoparticles-encapsulated drug/gene/protein for advanced controlled drug release: Current status and future perspective over emerging therapy approaches. Int. J. Biol. Macromol. 2024, 268, 131694. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.W.; Chan, L.W.; Li, W.; Wong, T.W. Critical clinical gaps in cancer precision nanomedicine development. J. Control Release 2022, 345, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Velagacherla, V.; Suresh, A.; Mehta, C.H.; Nayak, U.Y. Advances and challenges in nintedanib drug delivery. Expert. Opin. Drug Deliv. 2021, 18, 1687–1706. [Google Scholar] [CrossRef]
- Baldini, C.; Danlos, F.X.; Varga, A.; Texier, M.; Halse, H.; Mouraud, S.; Cassard, L.; Champiat, S.; Signolle, N.; Vuagnat, P.; et al. Safety, recommended dose, efficacy and immune correlates for nintedanib in combination with pembrolizumab in patients with advanced cancers. J. Exp. Clin. Cancer Res. 2022, 41, 217. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ren, T.; Huang, Y.; Sun, K.; Bao, X.; Wang, S.; Zheng, B.; Guo, W. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 2017, 8, e3015. [Google Scholar] [CrossRef]
- Prashanth Goud, M.; Bale, S.; Pulivendala, G.; Godugu, C. Therapeutic effects of Nimbolide, an autophagy regulator, in ameliorating pulmonary fibrosis through attenuation of TGF-β1 driven epithelial-to-mesenchymal transition. Int. Immunopharmacol. 2019, 75, 105755. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, L.; Szücs, K.D.; Kudla, M.; Heidenreich, J.; Jendrossek, V.; Peña-Llopis, S.; Garay, T.; Czirok, A.; Aigner, C.; Plönes, T.; et al. Nintedanib and Dasatinib Treatments Induce Protective Autophagy as a Potential Resistance Mechanism in MPM Cells. Front. Cell Dev. Biol. 2022, 10, 852812. [Google Scholar] [CrossRef] [PubMed]
- Eilenberger, C.; Rothbauer, M.; Selinger, F.; Gerhartl, A.; Jordan, C.; Harasek, M.; Schädl, B.; Grillari, J.; Weghuber, J.; Neuhaus, W.; et al. A Microfluidic Multisize Spheroid Array for Multiparametric Screening of Anticancer Drugs and Blood-Brain Barrier Transport Properties. Adv. Sci. 2021, 8, e2004856. [Google Scholar] [CrossRef] [PubMed]
- Guntner, A.S.; Peyrl, A.; Mayr, L.; Englinger, B.; Berger, W.; Slavc, I.; Buchberger, W.; Gojo, J. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol. Commun. 2020, 8, 78. [Google Scholar] [CrossRef]
- Becker, M.L.; Burdick, J.A. Introduction: Polymeric Biomaterials. Chem. Rev. 2021, 121, 10789–10791. [Google Scholar] [CrossRef]
- Polley, E.; Kunkel, M.; Evans, D.; Silvers, T.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Selby, M.; Connelly, J.; et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, M.; Zhang, F.; Zhang, J.; Xuanyuan, T.; Liu, W. A heterotypic tumor-on-a-chip platform for user-friendly combinatorial chemotherapeutic testing. Anal. Chim. Acta 2024, 1330, 343278. [Google Scholar] [CrossRef]
- Shan, X.; Cai, Y.; Zhu, B.; Zhou, L.; Sun, X.; Xu, X.; Yin, Q.; Wang, D.; Li, Y. Rational strategies for improving the efficiency of design and discovery of nanomedicines. Nat. Commun. 2024, 15, 9990. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; He, P.Y.; Du, J.; Zhang, J.Z. Quercetin in combating H2O2 induced early cell apoptosis and mitochondrial damage to normal human keratinocytes. Chin. Med. J. 2010, 123, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Gitego, N.; Agianian, B.; Mak, O.W.; Kumar Mv, V.; Cheng, E.H.; Gavathiotis, E. Chemical modulation of cytosolic BAX homodimer potentiates BAX activation and apoptosis. Nat. Commun. 2023, 14, 8381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Zhu, X.D.; Feng, L.H.; Li, X.L.; Liu, X.F.; Sun, H.C.; Tang, Z.Y. PCSK9 promotes tumor growth by inhibiting tumor cell apoptosis in hepatocellular carcinoma. Exp. Hematol. Oncol. 2021, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Hentzen, N.B.; Mogaki, R.; Otake, S.; Okuro, K.; Aida, T. Intracellular Photoactivation of Caspase-3 by Molecular Glues for Spatiotemporal Apoptosis Induction. J. Am. Chem. Soc. 2020, 142, 8080–8084. [Google Scholar] [CrossRef]
- Park, H.K.; Han, B.R.; Park, W.H. Combination of Arsenic Trioxide and Valproic Acid Efficiently Inhibits Growth of Lung Cancer Cells via G2/M-Phase Arrest and Apoptotic Cell Death. Int. J. Mol. Sci. 2020, 21, 2649. [Google Scholar] [CrossRef]
- Shan, R.; Liu, N.; Yan, Y.; Liu, B. Apoptosis, autophagy and atherosclerosis: Relationships and the role of Hsp27. Pharmacol. Res. 2021, 166, 105169. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yang, J.; Peng, A.; Qian, Y.; Liu, Y.; Pan, P.; Liu, Q. Lysosome Targeted Nanoparticle Aggregation Reverses Immunosuppressive Tumor Microenvironment for Cancer Immunotherapy. Adv. Mater. 2024, 36, e2412730. [Google Scholar] [CrossRef] [PubMed]
- Peña-Martinez, C.; Rickman, A.D.; Heckmann, B.L. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci. Adv. 2022, 8, eabn1702. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Wrobel, L.; Ashkenazi, A.; Fernandez-Estevez, M.; Tan, K.; Bürli, R.W.; Rubinsztein, D.C. VCP/p97 regulates Beclin-1-dependent autophagy initiation. Nat. Chem. Biol. 2021, 17, 448–455. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Terasawa, K.; Kato, Y.; Ikami, Y.; Sakamoto, K.; Ohtake, K.; Kusano, S.; Tomabechi, Y.; Kukimoto-Niino, M.; Shirouzu, M.; Guan, J.L.; et al. Direct homophilic interaction of LAMP2A with the two-domain architecture revealed by site-directed photo-crosslinks and steric hindrances in mammalian cells. Autophagy 2021, 17, 4286–4304. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Z.; Yang, C.; Zhuang, X.X.; Yuan, N.N.; Wu, M.Y.; Tan, J.Q.; Song, J.X.; Cheung, K.H.; Su, H.; Wang, Y.T.; et al. NRBF2 is a RAB7 effector required for autophagosome maturation and mediates the association of APP-CTFs with active form of RAB7 for degradation. Autophagy 2021, 17, 1112–1130. [Google Scholar] [CrossRef] [PubMed]
- Drobny, A.; Boros, F.A.; Balta, D.; Prieto Huarcaya, S.; Caylioglu, D.; Qazi, N.; Vandrey, J.; Schneider, Y.; Dobert, J.P.; Pitcairn, C.; et al. Reciprocal effects of alpha-synuclein aggregation and lysosomal homeostasis in synucleinopathy models. Transl. Neurodegener. 2023, 12, 31. [Google Scholar] [CrossRef]
- Liu, S.; Perez, P.; Sun, X.; Chen, K.; Fatirkhorani, R.; Mammadova, J.; Wang, Z. MLKL polymerization-induced lysosomal membrane permeabilization promotes necroptosis. Cell Death Differ. 2024, 31, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Luo, G.; Ye, D.; She, M.; Sun, N.; Lu, Y.J.; Zheng, J. Network pharmacology, molecular docking integrated surface plasmon resonance technology reveals the mechanism of Toujie Quwen Granules against coronavirus disease 2019 pneumonia. Phytomedicine 2021, 85, 153401. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.; Agarwal, M.; Jamal, S.; Gangwar, R.; Sharma, R.; Mubarak, M.M.; Wani, Z.A.; Ahmad, Z.; Khan, A.; Sheikh, J.A.; et al. Revitalizing antimicrobial strategies: Paromomycin and dicoumarol repurposed as potent inhibitors of M.tb’s replication machinery via targeting the vital protein DnaN. Int. J. Biol. Macromol. 2024, 278, 134652. [Google Scholar] [CrossRef] [PubMed]
- Hsin, K.Y.; Ghosh, S.; Kitano, H. Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology. PLoS ONE 2013, 8, e83922. [Google Scholar] [CrossRef]
- Shakeri, S.; Ashrafizadeh, M.; Zarrabi, A.; Roghanian, R.; Afshar, E.G.; Pardakhty, A.; Mohammadinejad, R.; Kumar, A.; Thakur, V.K. Multifunctional Polymeric Nanoplatforms for Brain Diseases Diagnosis, Therapy and Theranostics. Biomedicines 2020, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nintedanib: A Review in Fibrotic Interstitial Lung Diseases. Drugs 2021, 81, 575–586. [Google Scholar] [CrossRef]
- Quintela-Fandino, M.; Bermejo, B.; Zamora, E.; Moreno, F.; García-Saenz, J.; Pernas, S.; Martínez-Jañez, N.; Jiménez, D.; Adrover, E.; de Andrés, R.; et al. High Mechanical Conditioning by Tumor Extracellular Matrix Stiffness Is a Predictive Biomarker for Antifibrotic Therapy in HER2-Negative Breast Cancer. Clin. Cancer Res. 2024, 30, 5094–5104. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Lester, J.F.; Jackson, R.; Gornall, M.; Qureshi, M.; Elliott, A.; Crabb, S.J.; Huddart, R.A.; Vasudev, N.; Birtle, A.J.; et al. Addition of Nintedanib to Neoadjuvant Chemotherapy Is Safe in Bladder Cancer. Cancer Discov. 2022, 12, Of9. [Google Scholar] [CrossRef]
- Iyer, R.V.; Konda, B.; Fountzilas, C.; Mukherjee, S.; Owen, D.; Attwood, K.; Wang, C.; Maguire, O.; Minderman, H.; Suffren, S.A.; et al. Multicenter phase 2 trial of nintedanib in advanced nonpancreatic neuroendocrine tumors. Cancer 2020, 126, 3689–3697. [Google Scholar] [CrossRef]
- Li, Y.; Liang, X.; Shen, C.; Deng, K.; Zeng, Z.; Guo, B.; Xu, X. Bio-Responsive Macromolecular Drug and Small-Molecular Drug Conjugates: Nanoparticulate Prodrugs for Tumor Microenvironment Heterogeneity Management and Therapeutic Response Enhancement. Small 2023, 19, e2301656. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hu, Y.; Zhao, M.; Hao, K.; He, P.; Tian, H.; Chen, X.; Chen, M. Prodrug-Based Versatile Nanomedicine with Simultaneous Physical and Physiological Tumor Penetration for Enhanced Cancer Chemo-Immunotherapy. Nano Lett. 2021, 21, 3721–3730. [Google Scholar] [CrossRef]
- Herhaus, L.; Gestal-Mato, U.; Eapen, V.V.; Mačinković, I.; Bailey, H.J.; Prieto-Garcia, C.; Misra, M.; Jacomin, A.C.; Ammanath, A.V.; Bagarić, I.; et al. IRGQ-mediated autophagy in MHC class I quality control promotes tumor immune evasion. Cell 2024, 187, 7285–7302.e29. [Google Scholar] [CrossRef]
- Chumpen Ramirez, S.; Gómez-Sánchez, R.; Verlhac, P.; Hardenberg, R.; Margheritis, E.; Cosentino, K.; Reggiori, F.; Ungermann, C. Atg9 interactions via its transmembrane domains are required for phagophore expansion during autophagy. Autophagy 2023, 19, 1459–1478. [Google Scholar] [CrossRef] [PubMed]
- Segala, G.; Bennesch, M.A.; Ghahhari, N.M.; Pandey, D.P.; Echeverria, P.C.; Karch, F.; Maeda, R.K.; Picard, D. Vps11 and Vps18 of Vps-C membrane traffic complexes are E3 ubiquitin ligases and fine-tune signalling. Nat. Commun. 2019, 10, 1833. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Shi, J.; Hao, L.; Wang, Z.; Liao, Y.; Gu, H.; Dong, J.; Dresselhaus, T.; Zhong, S.; Qu, L.J. VPS18-regulated vesicle trafficking controls the secretion of pectin and its modifying enzyme during pollen tube growth in Arabidopsis. Plant Cell 2021, 33, 3042–3056. [Google Scholar] [CrossRef]
- Sanderson, L.E.; Lanko, K.; Alsagob, M.; Almass, R.; Al-Ahmadi, N.; Najafi, M.; Al-Muhaizea, M.A.; Alzaidan, H.; AlDhalaan, H.; Perenthaler, E.; et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain 2021, 144, 769–780. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Liu, X.; Zhong, Q. The hookup model of the HOPS complex in autophagosome-lysosome fusion. Autophagy 2024, 20, 714–715. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Niu, H.; Chu, Z.; Zhou, C.; Gao, Y.; Jia, M.; Sun, B.; Zheng, X.; Zhang, W.; Zhang, J.; et al. Targeting VPS18 hampers retromer trafficking of PD-L1 and augments immunotherapy. Sci. Adv. 2024, 10, eadp4917. [Google Scholar] [CrossRef]
- Niu, H.; Qian, L.; Luo, Y.; Wang, F.; Zheng, H.; Gao, Y.; Wang, H.; Hu, X.; Yuan, H.; Lou, H. Targeting of VPS18 by the lysosomotropic agent RDN reverses TFE3-mediated drug resistance. Signal Transduct. Target. Ther. 2021, 6, 224. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, F.; He, L.; Huang, H.; Chao, M.; Cao, H.; Hu, Y.; Fan, Z.; Zhai, Y.; Zhao, W.; et al. Single-cell dissection of the human blood-brain barrier and glioma blood-tumor barrier. Neuron 2024, 112, 3089–3105.e7. [Google Scholar] [CrossRef] [PubMed]
- Profaci, C.P.; Harvey, S.S.; Bajc, K.; Zhang, T.Z.; Jeffrey, D.A.; Zhang, A.Z.; Nemec, K.M.; Davtyan, H.; O’Brien, C.A.; McKinsey, G.L.; et al. Microglia are not necessary for maintenance of blood-brain barrier properties in health, but PLX5622 alters brain endothelial cholesterol metabolism. Neuron 2024, 112, 2910–2921.e7. [Google Scholar] [CrossRef]
- Correia, A.C.; Monteiro, A.R.; Silva, R.; Moreira, J.N.; Sousa Lobo, J.M.; Silva, A.C. Lipid nanoparticles strategies to modify pharmacokinetics of central nervous system targeting drugs: Crossing or circumventing the blood-brain barrier (BBB) to manage neurological disorders. Adv. Drug Deliv. Rev. 2022, 189, 114485. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiao, N.; Lin, D.; Li, N.; Ma, T.; Tung, S.; Cheng, W.; Wu, A.; Liu, L. Dual-Responsive Nanorobot-Based Marsupial Robotic System for Intracranial Cross-Scale Targeting Drug Delivery. Adv. Mater. 2024, 36, e2306876. [Google Scholar] [CrossRef]
- Wang, L.; Xu, X.; Chu, L.; Meng, C.; Xu, L.; Wang, Y.; Jiao, Q.; Huang, T.; Zhao, Y.; Liu, X.; et al. PEG-modified carbon-based nanoparticles as tumor-targeted drug delivery system reducing doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2023, 168, 115836. [Google Scholar] [CrossRef]
- Jain, A.; Jain, A.; Garg, N.K.; Tyagi, R.K.; Singh, B.; Katare, O.P.; Webster, T.J.; Soni, V. Surface engineered polymeric nanocarriers mediate the delivery of transferrin-methotrexate conjugates for an improved understanding of brain cancer. Acta Biomater. 2015, 24, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Dubernet, C.; Chauvierre, C.; Brigger, I.; Couvreur, P. Drug delivery to resistant tumors: The potential of poly(alkyl cyanoacrylate) nanoparticles. J. Control. Release 2003, 93, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J.; Cotter, J.D.; Knight, B.E.; Sevick-Muraca, E.M.; Sandberg, D.I.; Sirianni, R.W. Intrathecal drug delivery in the era of nanomedicine. Adv. Drug Deliv. Rev. 2020, 165–166, 77–95. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, Y.; Zhao, Z.; Wang, B.; Du, A.; Li, S.; Yuan, G.; Pan, Y. Polymeric Polylactic Acid–Glycolic Acid-Based Nanoparticles Deliver Nintedanib Across the Blood–Brain Barrier to Inhibit Glioblastoma Growth. Int. J. Mol. Sci. 2025, 26, 443. https://doi.org/10.3390/ijms26020443
Dang Y, Zhao Z, Wang B, Du A, Li S, Yuan G, Pan Y. Polymeric Polylactic Acid–Glycolic Acid-Based Nanoparticles Deliver Nintedanib Across the Blood–Brain Barrier to Inhibit Glioblastoma Growth. International Journal of Molecular Sciences. 2025; 26(2):443. https://doi.org/10.3390/ijms26020443
Chicago/Turabian StyleDang, Ying, Zhiwen Zhao, Bo Wang, Aichao Du, Shuangyi Li, Guoqiang Yuan, and Yawen Pan. 2025. "Polymeric Polylactic Acid–Glycolic Acid-Based Nanoparticles Deliver Nintedanib Across the Blood–Brain Barrier to Inhibit Glioblastoma Growth" International Journal of Molecular Sciences 26, no. 2: 443. https://doi.org/10.3390/ijms26020443
APA StyleDang, Y., Zhao, Z., Wang, B., Du, A., Li, S., Yuan, G., & Pan, Y. (2025). Polymeric Polylactic Acid–Glycolic Acid-Based Nanoparticles Deliver Nintedanib Across the Blood–Brain Barrier to Inhibit Glioblastoma Growth. International Journal of Molecular Sciences, 26(2), 443. https://doi.org/10.3390/ijms26020443