
Lessons from Deep Learning Structural Prediction of Multistate Multidomain Proteins—The Case Study of Coiled-Coil NOD-like Receptors

 , and
, and
Abstract
1. Introduction
2. Results
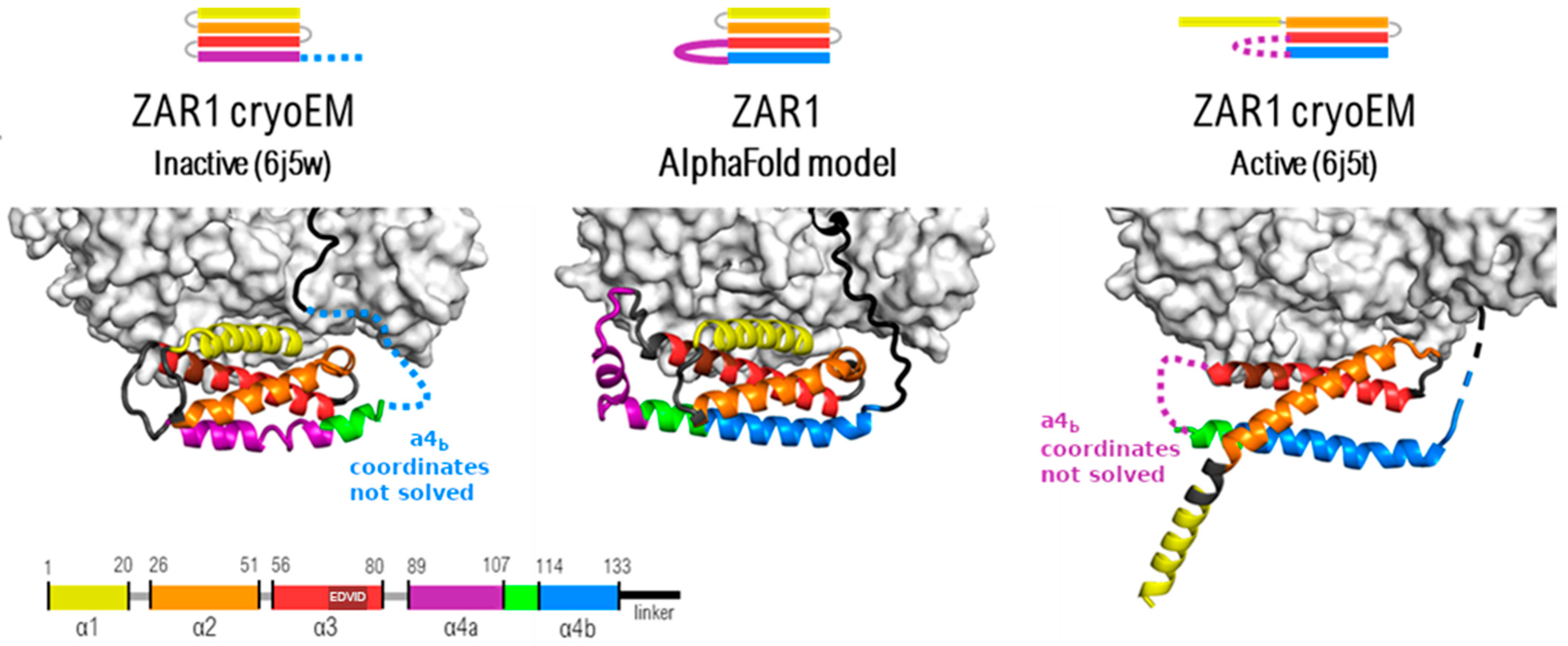
2.1. Gauging the Modeling Performance of AI Platforms by Using ZAR1-Solved Structures
2.1.1. Model Performance at Local Domain Level
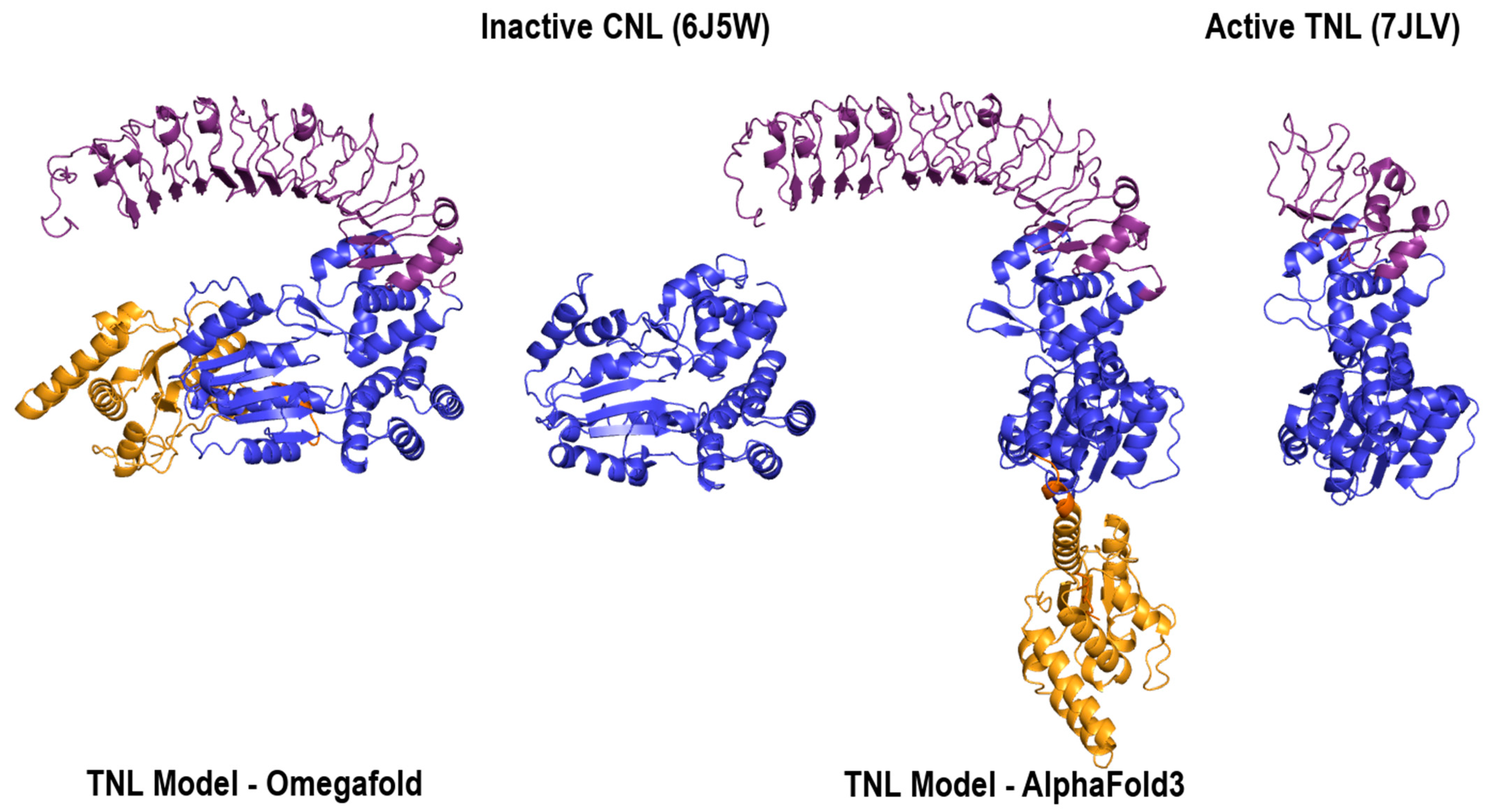
2.1.2. Prediction Performance at the Global Architectural Level
2.2. Modeling the CNL Set
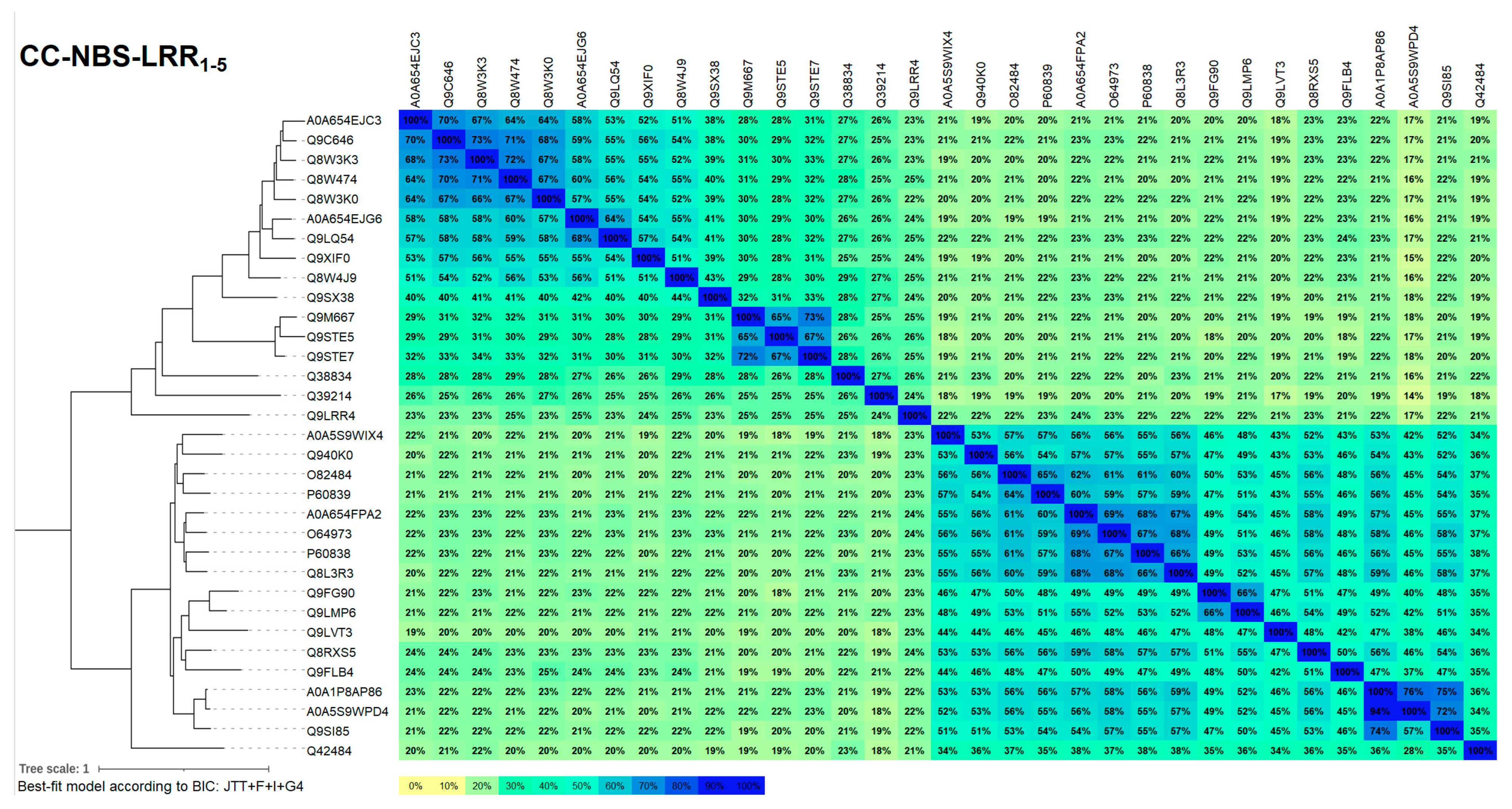
2.2.1. Sequence Selection
2.2.2. Model Generation
2.2.3. Model Refinement and Analysis
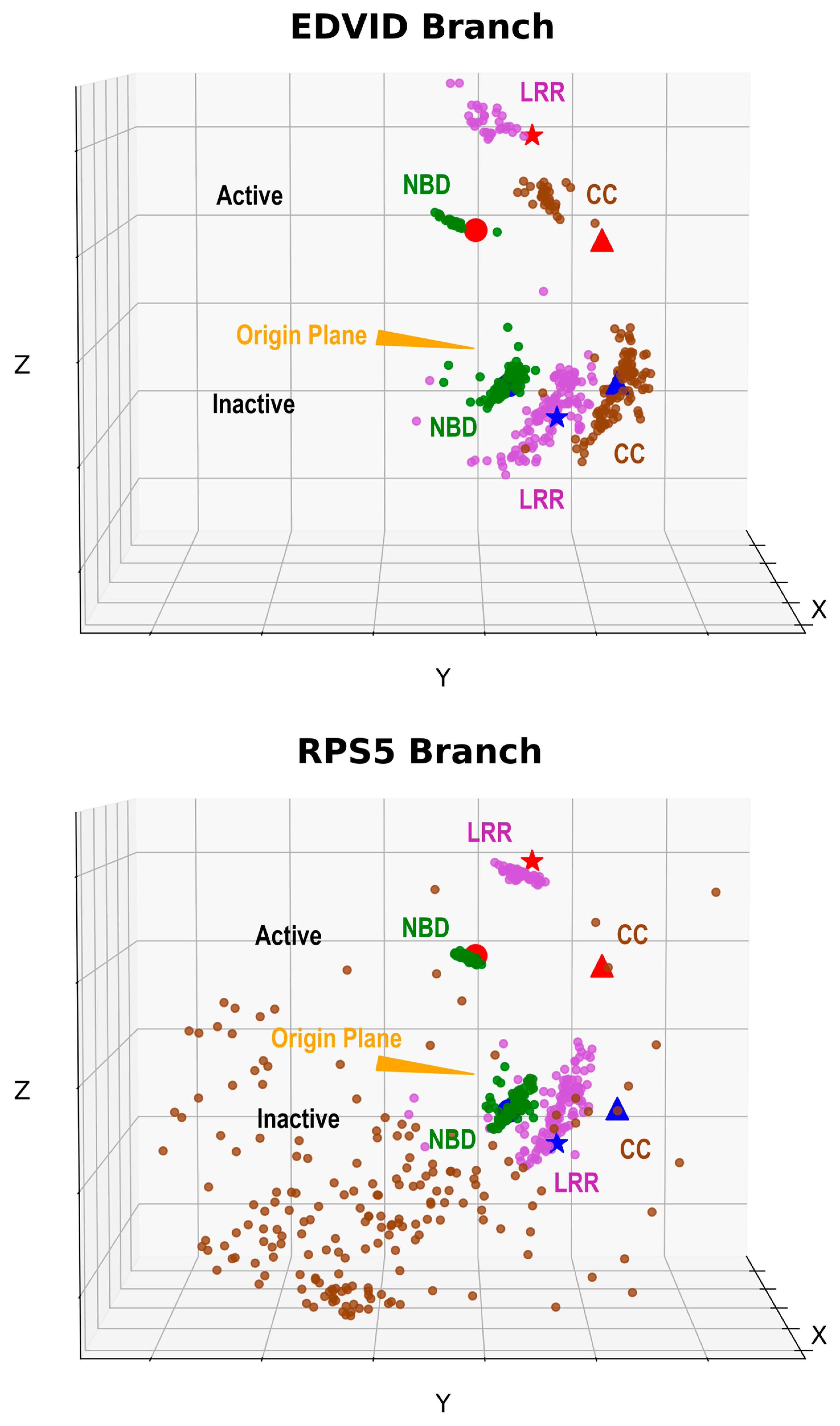
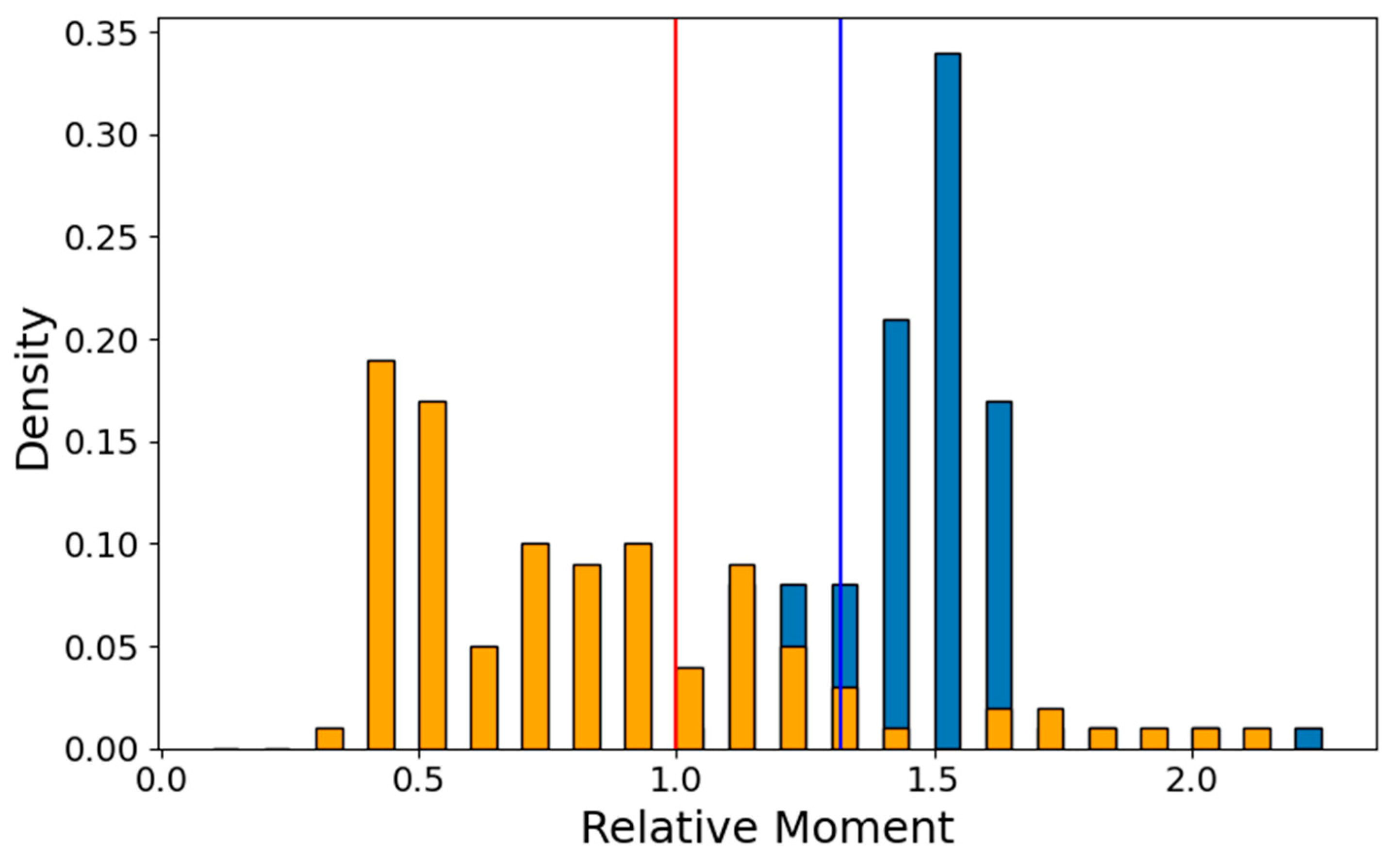
2.2.4. Conformation Preference
3. Discussion
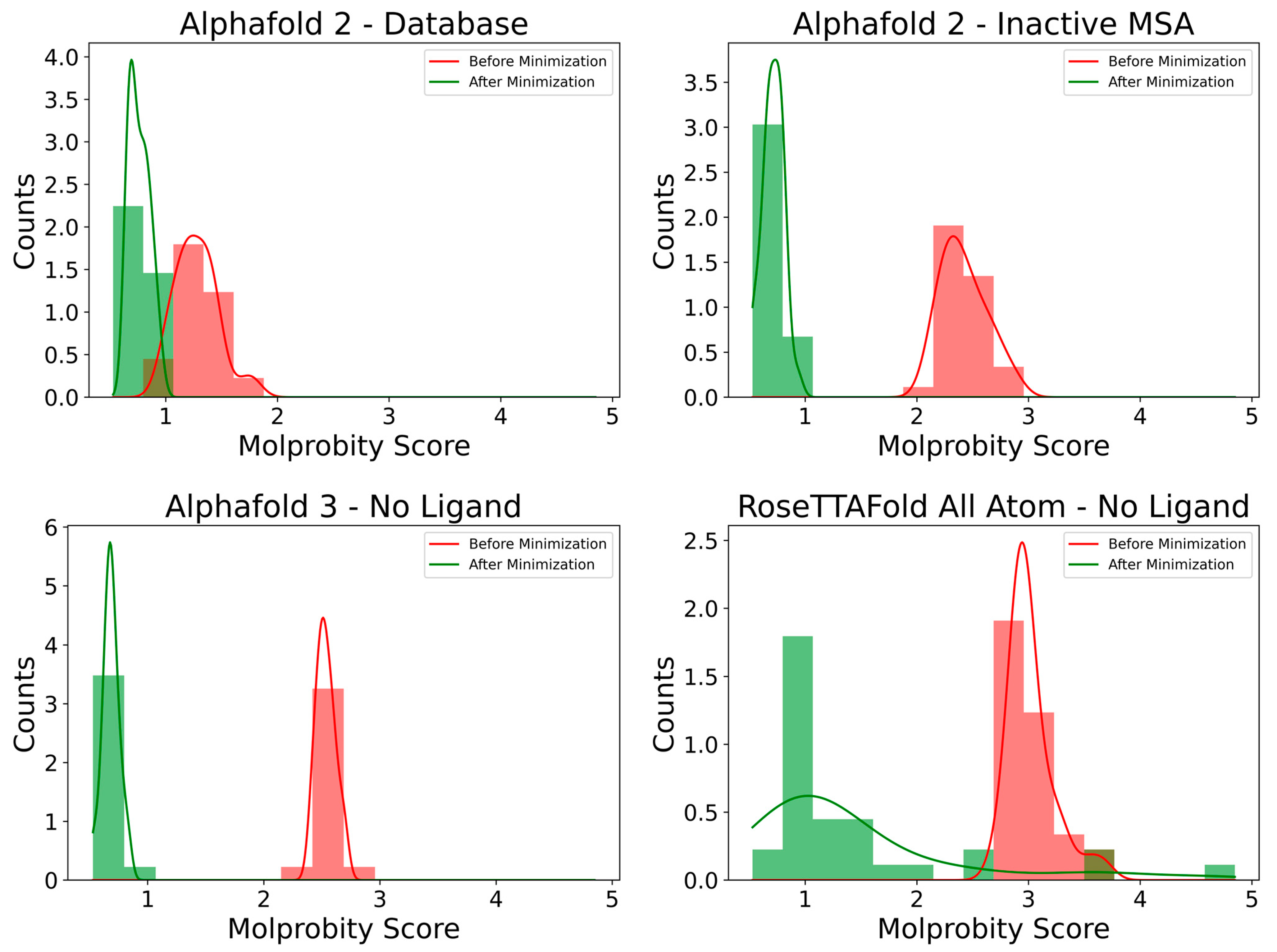
Model Quality
4. Materials and Methods
4.1. Sequence Selection of the A. thaliana Representative CNL Set
4.2. Sequence Analysis of Representative Set Sequences
4.3. Template and MSA Filtering with the Locally Implemented Version of AF2
4.4. Model Generation
- No MSA input, only the “Active” experimental structures from Table 6—“Active Control”.
- No MSA input, only the “Inactive” experimental structures from Table 6—“Inactive Control”.
- MSA consisting of only NLR Proteins retrieved from NLRscape, “Active” experimental structures from Table 6 and structures corresponding to CATH families for CC and LRR architectures—“Active MSA”.
- MSA consisting of only NLR Proteins retrieved from NLRscape, “Inactive” experimental structures from Table 6 and structures corresponding to CATH families for CC and LRR architectures—“Inactive MSA”.
4.5. Model Refinement
4.6. Model Analysis
4.6.1. Model Quality
4.6.2. Assessment of Overall Architecture
4.6.3. Model Stability
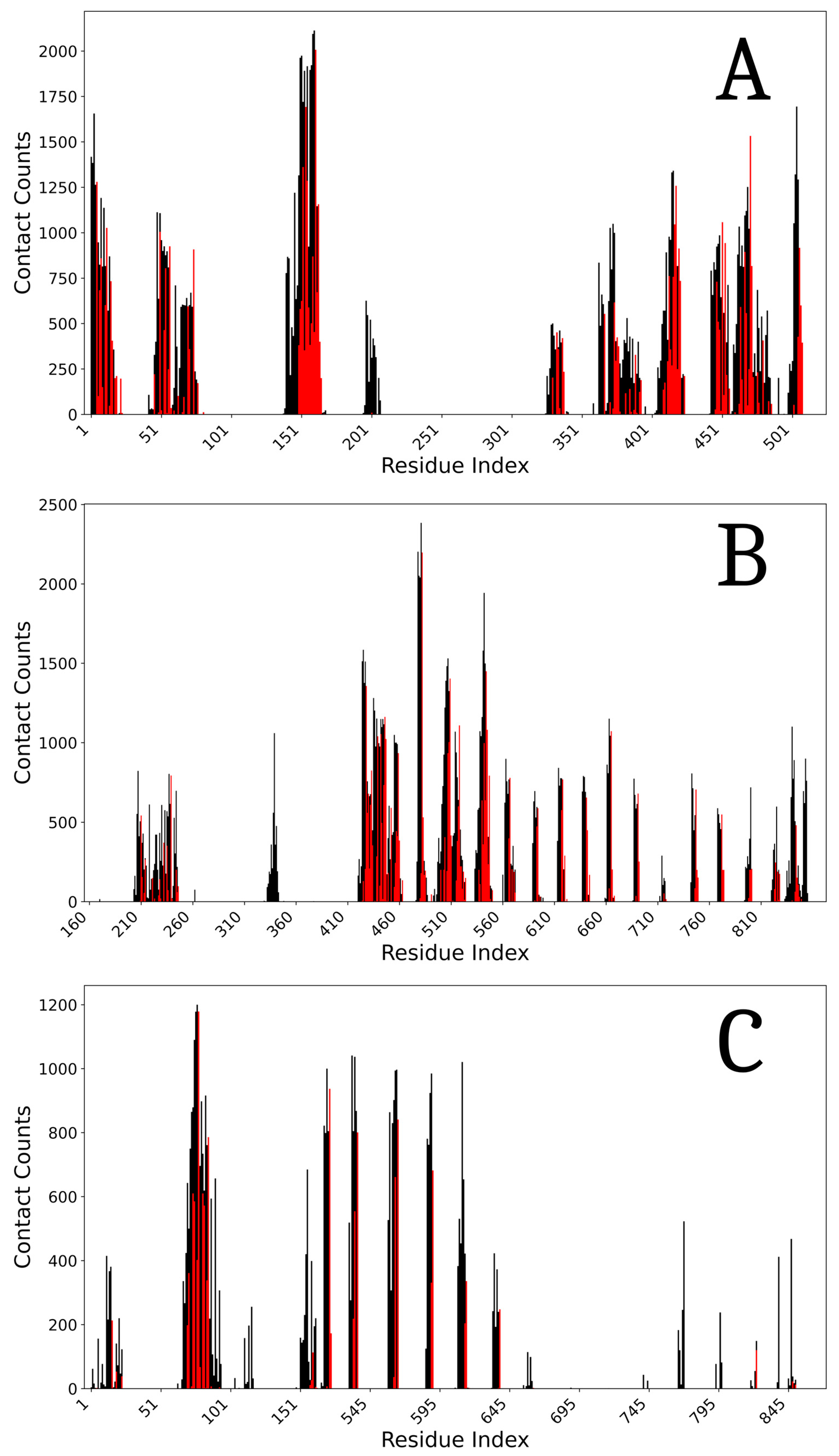
4.6.4. Interface Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perrakis, A.; Sixma, T.K. AI revolutions in biology. Embo Rep. 2021, 22, e54046. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Cambillau, C. Present Impact of AlphaFold2 Revolution on Structural Biology, and an Illustration With the Structure Prediction of the Bacteriophage J-1 Host Adhesion Device. Front. Mol. Biosci. 2022, 9, 907452. [Google Scholar] [CrossRef]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An overview of protein structure prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Wodak, S.J.; Velankar, S. Structural biology: The transformational era. Proteomics 2023, 23, e2200084. [Google Scholar] [CrossRef]
- Read, R.J.; Baker, E.N.; Bond, C.S.; Garman, E.F.; van Raaij, M.J. AlphaFold and the future of structural biology. IUCrJ 2023, 10, 377–379. [Google Scholar] [CrossRef]
- Dahlström, K.M.; Salminen, T.A. Apprehensions and Emerging Solutions in ML-Based Protein Structure Prediction; Elsevier Ltd.: Amsterdam, The Netherlands, 2024. [Google Scholar] [CrossRef]
- Zheng, W.; Schafer, N.P.; Wolynes, P.G. Frustration in the energy landscapes of multidomain protein misfolding. Proc. Natl. Acad. Sci. USA 2013, 110, 1680–1685. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Han, J.-H.; Batey, S.; Nickson, A.A.; Teichmann, S.A.; Clarke, J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 2007, 8, 319–330. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Kragelund, B.B. The molecular basis for cellular function of intrinsically disordered protein regions. Nat. Rev. Mol. Cell Biol. 2024, 25, 187–211. [Google Scholar] [CrossRef]
- Rebeaud, M.E.; Mallik, S.; Goloubinoff, P.; Tawfik, D.S. On the evolution of chaperones and cochaperones and the expansion of proteomes across the Tree of Life. Proc. Natl. Acad. Sci. USA 2021, 118, e2020885118. [Google Scholar] [CrossRef]
- Aziz, M.F.; Caetano-Anollés, G. Evolution of networks of protein domain organization. Sci. Rep. 2021, 11, 12075. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M. Estimation of Effective Concentrations Enforced by Complex Linker Architectures from Conformational Ensembles. Biochemistry 2022, 61, 171–182. [Google Scholar] [CrossRef]
- Harrison, A.; Pearl, F.; Mott, R.; Thornton, J.; Orengo, C. Quantifying the similarities within fold space. J. Mol. Biol. 2002, 323, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Waman, V.P.; Bordin, N.; Alcraft, R.; Vickerstaff, R.; Rauer, C.; Chan, Q.; Sillitoe, I.; Yamamori, H.; Orengo, C. CATH 2024: CATH-AlphaFlow Doubles the Number of Structures in CATH and Reveals Nearly 200 New Folds. J. Mol. Biol. 2024, 436, 168551. [Google Scholar] [CrossRef]
- Andreeva, A.; Kulesha, E.; Gough, J.; Murzin, A.G. The SCOP database in 2020: Expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020, 48, D376–D382. [Google Scholar] [CrossRef]
- Chandonia, J.-M.; Guan, L.; Lin, S.; Yu, C.; Fox, N.K.; Brenner, S.E. SCOPe: Improvements to the structural classification of proteins-extended database to facilitate variant interpretation and machine learning. Nucleic Acids Res. 2022, 50, D553–D559. [Google Scholar] [CrossRef]
- Schaeffer, R.D.; Zhang, J.; Kinch, L.N.; Pei, J.; Cong, Q.; Grishin, N.V. Classification of domains in predicted structures of the human proteome. Proc. Natl. Acad. Sci. USA 2023, 120, e2214069120. [Google Scholar] [CrossRef]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the b 2 adrenergic receptor—Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Slootweg, E.; Roosien, J.; Spiridon, L.N.; Petrescu, A.-J.; Tameling, W.; Joosten, M.; Pomp, R.; van Schaik, C.; Dees, R.; Borst, J.W.; et al. Nucleocytoplasmic distribution is required for activation of resistance by the potato NB-LRR receptor Rx1 and is balanced by its functional domains. Plant Cell 2010, 22, 4195–4215. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Cheng, W.; Spiridon, L.N.; Töller, A.; Lukasik, E.; Saijo, Y.; Liu, P.; Shen, Q.-H.; Micluta, M.A.; Somssich, I.E.; et al. Coiled-coil domain-dependent homodimerization of intracellular barley immune receptors defines a minimal functional module for triggering cell death. Cell Host Microbe 2011, 9, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Sela, H.; Spiridon, L.N.; Petrescu, A.; Akerman, M.; Mandel-Gutfreund, Y.; Nevo, E.; Loutre, C.; Keller, B.; Schulman, A.H.; Fahima, T. Ancient diversity of splicing motifs and protein surfaces in the wild emmer wheat (Triticum dicoccoides) LR10 coiled coil (CC) and leucine-rich repeat (LRR) domains. Mol. Plant Pathol. 2012, 13, 276–287. [Google Scholar] [CrossRef]
- Slootweg, E.J.; Spiridon, L.N.; Roosien, J.; Butterbach, P.; Pomp, R.; Westerhof, L.; Wilbers, R.; Bakker, E.; Bakker, J.; Petrescu, A.-J.; et al. Structural determinants at the interface of the ARC2 and leucine-rich repeat domains control the activation of the plant immune receptors Rx1 and Gpa2. Plant Physiol. 2013, 162, 1510–1528. [Google Scholar] [CrossRef]
- Sela, H.; Spiridon, L.N.; Ashkenazi, H.; Bhullar, N.K.; Brunner, S.; Petrescu, A.-J.; Fahima, T.; Keller, B.; Jordan, T. Three-dimensional modeling and diversity analysis reveals distinct AVR recognition sites and evolutionary pathways in wild and domesticated wheat Pm3 R genes. Mol. Plant-Microbe Interact. 2014, 27, 835–845. [Google Scholar] [CrossRef]
- Sueldo, D.J.; Shimels, M.; Spiridon, L.N.; Caldararu, O.; Petrescu, A.; Joosten, M.H.A.J.; Tameling, W.I.L. Random mutagenesis of the nucleotide-binding domain of NRC1 (NB-LRR Required for Hypersensitive Response-Associated Cell Death-1), a downstream signalling nucleotide-binding, leucine-rich repeat (NB-LRR) protein, identifies gain-of-function mutations in the nucleotide-binding pocket. New Phytol. 2015, 208, 210–223. [Google Scholar] [CrossRef]
- De Oliveira, A.S.; Koolhaas, I.; Boiteux, L.S.; Caldararu, O.F.; Petrescu, A.; Resende, R.d.O.; Kormelink, R. Cell death triggering and effector recognition by Sw-5 SD-CNL proteins from resistant and susceptible tomato isolines to Tomato spotted wilt virus. Mol. Plant Pathol. 2016, 17, 1442–1454. [Google Scholar] [CrossRef]
- Wróblewski, T.; Spiridon, L.; Martin, E.C.; Petrescu, A.-J.; Cavanaugh, K.; Truco, M.J.; Xu, H.; Gozdowski, D.; Pawłowski, K.; Michelmore, R.W.; et al. Genome-wide functional analyses of plant coiled–coil NLR-type pathogen receptors reveal essential roles of their N-terminal domain in oligomerization, networking, and immunity. PLoS Biol. 2018, 16, e2005821. [Google Scholar] [CrossRef]
- Slootweg, E.J.; Spiridon, L.N.; Martin, E.C.; Tameling, W.I.; Townsend, P.D.; Pomp, R.; Roosien, J.; Drawska, O.; Sukarta, O.C.; Schots, A.; et al. Distinct roles of non-overlapping surface regions of the coiled-coil domain in the potato immune receptor Rx1. Plant Physiol. 2018, 178, 1310–1331. [Google Scholar] [CrossRef]
- Baudin, M.; Schreiber, K.J.; Martin, E.C.; Petrescu, A.J.; Lewis, J.D. Structure–function analysis of ZAR1 immune receptor reveals key molecular interactions for activity. Plant J. 2020, 101, 352–370. [Google Scholar] [CrossRef]
- Baudin, M.; Martin, E.C.; Sass, C.; Hassan, J.A.; Bendix, C.; Sauceda, R.; Diplock, N.; Specht, C.D.; Petrescu, A.J.; Lewis, J.D. A natural diversity screen in Arabidopsis thaliana reveals determinants for HopZ1a recognition in the ZAR1-ZED1 immune complex. Plant Cell Environ. 2020, 44, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, Z.; Wu, C.-H.; Ding, P. A Comparative Overview of the Intracellular Guardians of Plants and Animals: NLRs in Innate Immunity and Beyond. Annu. Rev. Plant Biol. 2021, 72, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Hu, M.; Wu, S.; Qi, J.; Wang, G.; Han, Z.; Qi, Y.; Gao, N.; Wang, H.-W.; et al. Ligand-triggered allosteric ADP release primes a plant NLR complex. Science 2019, 364, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, M.; Wang, J.; Qi, J.; Han, Z.; Wang, G.; Qi, Y.; Wang, H.-W.; Zhou, J.-M.; Chai, J. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 2019, 364, 44. [Google Scholar] [CrossRef]
- Cesari, S. Multiple Strategies for Pathogen Perception by Plant Immune Receptors; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Xu, S.; Wei, X.; Yang, Q.; Hu, D.; Zhang, Y.; Yuan, X.; Kang, F.; Wu, Z.; Yan, Z.; Luo, X.; et al. A KNOX Ⅱ transcription factor suppresses the NLR immune receptor BRG8-mediated immunity in rice. Plant Commun. 2024, 5, 101001. [Google Scholar] [CrossRef]
- Contreras, M.P.; Lüdke, D.; Pai, H.; Toghani, A.; Kamoun, S. NLR receptors in plant immunity: Making sense of the alphabet soup. EMBO Rep. 2023, 24, e57495. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Krishna, R.; Wang, J.; Ahern, W.; Sturmfels, P.; Venkatesh, P.; Kalvet, I.; Lee, G.R.; Morey-Burrows, F.S.; Anishchenko, I.; Humphreys, I.R.; et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024, 384, eadl2528. [Google Scholar] [CrossRef]
- Martin, E.C.; Ion, C.F.; Ifrimescu, F.; Spiridon, L.; Bakker, J.; Goverse, A.; Petrescu, A.-J. NLRscape: An atlas of plant NLR proteins. Nucleic Acids Res. 2023, 51, D1470–D1482. [Google Scholar] [CrossRef]
- Martin, E.C.; Spiridon, L.; Goverse, A.; Petrescu, A.-J. NLRexpress—A bundle of machine learning motif predictors—Reveals motif stability underlying plant Nod-like receptors diversity. Front. Plant Sci. 2022, 13, 975888. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Neurosnap Platform. Available online: https://neurosnap.ai/ (accessed on 31 May 2024).
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Spiridon, L.; Şulea, T.A.; Minh, D.D.; Petrescu, A.-J. Robosample: A rigid-body molecular simulation program based on robot mechanics. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2020, 1864, 129616. [Google Scholar] [CrossRef]
- Hao, W.; Collier, S.M.; Moffett, P.; Chai, J. Structural basis for the interaction between the potato virus X resistance protein (Rx) and its cofactor ran GTPase-activating protein 2 (RanGAP2). J. Biol. Chem. 2013, 288, 35868–35876. [Google Scholar] [CrossRef]
- van Grinsven, I.L.; Martin, E.C.; Petrescu, A.-J.; Kormelink, R. Tsw—A case study on structure-function puzzles in plant NLRs with unusually large LRR domains. Front. Plant Sci. 2022, 13, 983693. [Google Scholar] [CrossRef]
- Martin, E.C.; Sukarta, O.C.A.; Spiridon, L.; Grigore, L.G.; Constantinescu, V.; Tacutu, R.; Goverse, A.; Petrescu, A.-J. Lrrpredictor—A new LRR motif detection method for irregular motifs of plant NLR proteins using an ensemble of classifiers. Genes 2020, 11, 286. [Google Scholar] [CrossRef]
- Agarwal, V.; McShan, A.C. The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins. Nat. Chem. Biol. 2024, 20, 950–959. [Google Scholar] [CrossRef]
- Peng, C.-X.; Liang, F.; Xia, Y.-H.; Zhao, K.-L.; Hou, M.-H.; Zhang, G.-J. Recent Advances and Challenges in Protein Structure Prediction. J. Chem. Inf. Model. 2024, 64, 76–95. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Soubrier, J.; Steel, M.; Lee, M.S.; Der Sarkissian, C.; Guindon, S.; Ho, S.Y.; Cooper, A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012, 29, 3345–3358. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessimoz, C.; Gascuel, O. Survey of Branch Support Methods Demonstrates Accuracy, Power, and Robustness of Fast Likelihood-based Approximation Schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.; Toghani, A.; Pai, H.; Sugihara, Y.; Kourelis, J.; Yuen, E.L.H.; Ibrahim, T.; Zhao, H.; Xie, R.; Maqbool, A.; et al. Activation of plant immunity through conversion of a helper NLR homodimer into a resistosome. PLoS Biol. 2024, 22, e3002868. [Google Scholar] [CrossRef]
- Ma, S.; An, C.; Lawson, A.W.; Cao, Y.; Sun, Y.; Tan, E.Y.J.; Pan, J.; Jirschitzka, J.; Kümmel, F.; Mukhi, N.; et al. Oligomerization-mediated autoinhibition and cofactor binding of a plant NLR. Nature 2024, 632, 869–876. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- AlphaFold2 Github Repository. Available online: https://github.com/google-deepmind/alphafold (accessed on 20 May 2024).
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Peng, J. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.D.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. AmberTools. J. Chem. Inf. Model. 2023, 63, 6183–6191. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Meagher, K.L.; Redman, L.T.; Carlson, H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24, 1016–1025. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Spiridon, L.; Minh, D.D.L. Hamiltonian Monte Carlo with Constrained Molecular Dynamics as Gibbs Sampling. J. Chem. Theory Comput. 2017, 13, 4649–4659. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Ficke, A.; Cowger, C.; Bergstrom, G.C.; Brodal, G. Understanding yield loss and pathogen biology to improve disease management: Septoria nodorum blotch—A case study in wheat. Plant Dis. 2018, 102, 696–707. [Google Scholar] [CrossRef]
- Brookes, E.; Rocco, M.; Vachette, P.; Trewhella, J. AlphaFold-predicted protein structures and small-angle X-ray scattering: Insights from an extended examination of selected data in the Small-Angle Scattering Biological Data Bank. J. Appl. Crystallogr. 2023, 56 Pt 4, 910–926. [Google Scholar] [CrossRef]
- Ramelot, T.A.; Tejero, R.; Montelione, G.T. Representing structures of the multiple conformational states of proteins. Curr. Opin. Struct. Biol. 2023, 83, 102703. [Google Scholar] [CrossRef]
- Abdollahi, H.; Prestegard, J.H.; Valafar, H. Computational modeling multiple conformational states of proteins with residual dipolar coupling data. Curr. Opin. Struct. Biol. 2023, 82, 102655. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nédélec, A.; Derenne, A.; Şulea, T.A.; Pecquet, C.; Chachoua, I.; Vertenoeil, G.; Tilmant, T.; Petrescu, A.-J.; Mazzucchelli, G.; et al. Oncogenic CALR mutant C-terminus mediates dual binding to the thrombopoietin receptor triggering complex dimerization and activation. Nat. Commun. 2023, 14, 1881. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Name | CC | NBS-ARC1 | ARC2 | LRR |
|---|---|---|---|---|
| AF2—Database | 14.46 | 1.86 | 1.63 | 1.45 |
| AF2—Active Control | 1.39 | 0.49 | 0.42 | 0.53 |
| AF2—Inactive Control | 18.51 | 1.86 | 1.51 | 0.84 |
| AF2—Active MSA | 1.55 | 0.59 | 0.61 | 0.96 |
| AF2—Inactive MSA | 14.40 | 1.79 | 1.53 | 0.97 |
| AF3—ADP | 14.41 | 1.80 | 1.64 | 1.42 |
| AF3—ATP | 14.39 | 1.89 | 1.58 | 1.33 |
| AF3—No Ligand | 14.45 | 1.92 | 1.59 | 1.51 |
| RFAA—ADP | 14.42 | 2.04 | 1.64 | 1.86 |
| RFAA—ATP | 14.50 | 2.05 | 1.78 | 2.02 |
| RFAA—No Ligand | 14.45 | 1.92 | 1.65 | 1.96 |
| OmegaFold | 14.48 | 1.87 | 1.33 | 3.11 |
| Model Name | CC | NBS-ARC1 | ARC2 | LRR |
|---|---|---|---|---|
| AF2—Database | 12.42 | 1.33 | 0.88 | 1.34 |
| AF2—Active Control | 19.19 | 1.89 | 1.45 | 0.78 |
| AF2—Inactive Control | 0.80 | 0.69 | 0.35 | 0.47 |
| AF2—Active MSA | 18.48 | 1.88 | 1.51 | 0.90 |
| AF2—Inactive MSA | 12.54 | 0.86 | 0.44 | 0.75 |
| AF3—ADP | 11.99 | 1.31 | 0.98 | 1.22 |
| AF3—ATP | 12.11 | 1.35 | 0.87 | 1.16 |
| AF3—No Ligand | 12.08 | 1.26 | 0.93 | 1.37 |
| RFAA—ADP | 12.74 | 1.41 | 1.02 | 1.77 |
| RFAA—ATP | 12.65 | 1.53 | 1.04 | 1.92 |
| RFAA—No Ligand | 12.36 | 1.42 | 1.11 | 1.89 |
| OmegaFold | 12.99 | 1.24 | 0.87 | 3.04 |
| Model Name | RMSD vs. Active | RMSD vs. Inactive |
|---|---|---|
| AF2—Database | 22.158 | 6.004 |
| AF2—Active Control | 0.832 | 23.036 |
| AF2—Inactive Control | 22.612 | 0.675 |
| AF2—Active MSA | 1.271 | 22.873 |
| AF2—Inactive MSA | 21.997 | 5.837 |
| AF3—ADP | 22.119 | 5.785 |
| AF3—ATP | 22.076 | 5.879 |
| AF3—No Ligand | 22.191 | 5.889 |
| RFAA—ADP | 22.406 | 6.756 |
| RFAA—ATP | 22.646 | 6.792 |
| RFAA—No Ligand | 22.424 | 6.027 |
| OmegaFold | 22.374 | 6.442 |
| Model Name | CC/NBD Interface | NBD/LRR Interface | CC/LRR Interface |
|---|---|---|---|
| Inactive Crystal | −8.8 | −14.0 | −7.8 |
| AF2—Database | −10.1 | −15.1 | −8.5 |
| AF2—Inactive Control | −9.7 | −14.7 | −8.3 |
| AF2—Inactive MSA | −10.1 | −14.9 | −8.6 |
| AF3—ADP | −10.9 | −15.6 | −8.9 |
| AF3—ATP | −10.7 | −15.0 | −9.2 |
| AF3—No Ligand | −10.6 | −15.4 | −9.1 |
| RFAA—ADP | −9.7 | −15.0 | −9.6 |
| RFAA—ATP | −9.7 | −16.2 | −9.3 |
| RFAA—No Ligand | −9.7 | −14.4 | −9.2 |
| Protein Name | Length | Cluster | Domains | Publications |
|---|---|---|---|---|
| A0A1P8AP86 | 888 | 1 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q9SI85 | 893 | 2 | CC-NBD-ARC1-ARC2-LRR | 3 |
| Q940K0 | 889 | 3 | CC-NBD-ARC1-ARC2-LRR | 6 |
| Q9M667 | 835 | 4 | CC-NBD-ARC1-ARC2-LRR | 5 |
| Q9C646 | 899 | 5 | CC-NBD-ARC1-ARC2-LRR | 3 |
| Q39214 | 926 | 6 | CC-NBD-ARC1-ARC2-LRR | 10 |
| Q8W474 | 907 | 7 | CC-NBD-ARC1-ARC2-LRR-X | 4 |
| Q8RXS5 | 888 | 8 | CC-NBD-ARC1-ARC2-LRR | 3 |
| Q8W3K3 | 910 | 9 | CC-NBD-ARC1-ARC2-LRR | 3 |
| O64973 | 889 | 10 | CC-NBD-ARC1-ARC2-LRR | 10 |
| A0A654EJG6 | 904 | 11 | CC-NBD-ARC1-ARC2-LRR | 0 |
| Q8L3R3 | 885 | 12 | CC-NBD-ARC1-ARC2-LRR | 4 |
| Q8W4J9 | 908 | 13 | CC-NBD-ARC1-ARC2-LRR | 12 |
| Q9STE5 | 847 | 14 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q9LQ54 | 870 | 15 | CC-NBD-ARC1-ARC2-LRR | 3 |
| A0A7G2ET34 | 1306 | 16 | X-CC-NBD-ARC1-ARC2-LRR | 0 |
| Q9XIF0 | 906 | 17 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q9LVT3 | 948 | 18 | CC-NBD-ARC1-ARC2-LRR-X | 2 |
| Q9STE7 | 847 | 19 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q9FG90 | 862 | 20 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q9LRR5 | 1424 | 21 | CC-NBD-ARC1-ARC2-LRR-X-LRR | 2 |
| Q9FLB4 | 874 | 22 | CC-NBD-ARC1-ARC2-LRR | 2 |
| A0A5S9WIX4 | 875 | 23 | CC-NBD-ARC1-ARC2-LRR | 0 |
| A0A5S9WPD4 | 1025 | 24 | CC-NBD-ARC1-ARC2-LRR-X | 0 |
| A0A654EJV2 | 661 | 25 | CC-NBD-ARC1-ARC2-LRR | 0 |
| A0A654FPA2 | 881 | 26 | CC-NBD-ARC1-ARC2-LRR | 0 |
| O82484 | 892 | 27 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q8W3K0 | 1138 | 28 | CC-NBD-ARC1-ARC2-LRR | 5 |
| Q38834 | 852 | 29 | CC-NBD-ARC1-ARC2-LRR | 10 |
| Q9LRR4 | 1054 | 30 | CC-NBD-ARC1-ARC2-LRR | 2 |
| P60839 | 884 | 31 | CC-NBD-ARC1-ARC2-LRR | 2 |
| P60838 | 894 | 32 | CC-NBD-ARC1-ARC2-LRR | 5 |
| A0A654EJC3 | 921 | 33 | CC-NBD-ARC1-ARC2-LRR | 0 |
| Q9LMP6 | 851 | 34 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q9SX38 | 857 | 35 | CC-NBD-ARC1-ARC2-LRR | 2 |
| Q42484 | 909 | 36 | CC-NBD-ARC1-ARC2-LRR | 17 |
| UniProt Acc. | RCSB Acc | Chain | Domains | Organism | Geometric Classification |
|---|---|---|---|---|---|
| Q38834 | 6J5T | C | CC-NBD-ARC1-ARC2-LRR | A. thaliana | Active |
| Q38834 | 6J6I | C | CC-NBD-ARC1-ARC2-LRR | A. thaliana | Active |
| Q9ZSN5 | 7CRC | A | X-TIR-NBD-ARC1-ARC2-LRR | A. thaliana | Active |
| Q9ZSN5 | 7DFV | A | X-TIR-NBD-ARC1-ARC2-LRR | A. thaliana | Active |
| A0A290U7C4 | 7JLV | A | TIR-NBD-ARC1-ARC2-LRR-X | N. benthamiana | Active |
| S5ABD6 | 7XC2 | A | CC-NBD-ARC1-ARC2-LRR | T. monococcum | Active |
| S5ABD6 | 7XE0 | A | CC-NBD-ARC1-ARC2-LRR | T. monococcum | Active |
| S5ABD6 | 7XX2 | A | CC-NBD-ARC1-ARC2-LRR | T. monococcum | Active |
| Q38834 | 6J5W | A | CC-NBD-ARC1-ARC2-LRR | A. thaliana | Inactive |
| A1X877 | 6S2P | N | CC-NBD-ARC1-ARC2-LRR | S. lycopersicum | Inactive |
| A1X877 | 8BV0 | A | CC-NBD-ARC1-ARC2-LRR | S. lycopersicum | Inactive |
| A0A0S3ANR1 | 8RFH | A | CC-NBD-ARC1-ARC2-LRR | N. benthamiana | Inactive |
| A0A3Q7IF17 | 8XUO | A | CC-NBD-ARC1-ARC2-LRR | S. lycopersicum | Inactive |
| A0A3Q7IF17 | 8XUQ | A | CC-NBD-ARC1-ARC2-LRR | S. lycopersicum | Inactive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Șulea, T.A.; Martin, E.C.; Bugeac, C.A.; Bectaș, F.S.; Iacob, A.-L.; Spiridon, L.; Petrescu, A.-J. Lessons from Deep Learning Structural Prediction of Multistate Multidomain Proteins—The Case Study of Coiled-Coil NOD-like Receptors. Int. J. Mol. Sci. 2025, 26, 500. https://doi.org/10.3390/ijms26020500
Șulea TA, Martin EC, Bugeac CA, Bectaș FS, Iacob A-L, Spiridon L, Petrescu A-J. Lessons from Deep Learning Structural Prediction of Multistate Multidomain Proteins—The Case Study of Coiled-Coil NOD-like Receptors. International Journal of Molecular Sciences. 2025; 26(2):500. https://doi.org/10.3390/ijms26020500
Chicago/Turabian StyleȘulea, Teodor Asvadur, Eliza Cristina Martin, Cosmin Alexandru Bugeac, Floriana Sibel Bectaș, Anca-L Iacob, Laurențiu Spiridon, and Andrei-Jose Petrescu. 2025. "Lessons from Deep Learning Structural Prediction of Multistate Multidomain Proteins—The Case Study of Coiled-Coil NOD-like Receptors" International Journal of Molecular Sciences 26, no. 2: 500. https://doi.org/10.3390/ijms26020500
APA StyleȘulea, T. A., Martin, E. C., Bugeac, C. A., Bectaș, F. S., Iacob, A.-L., Spiridon, L., & Petrescu, A.-J. (2025). Lessons from Deep Learning Structural Prediction of Multistate Multidomain Proteins—The Case Study of Coiled-Coil NOD-like Receptors. International Journal of Molecular Sciences, 26(2), 500. https://doi.org/10.3390/ijms26020500