
Mitochondrial Genome Insights into Evolution and Gene Regulation in Phragmites australis


Abstract
:1. Introduction
2. Results
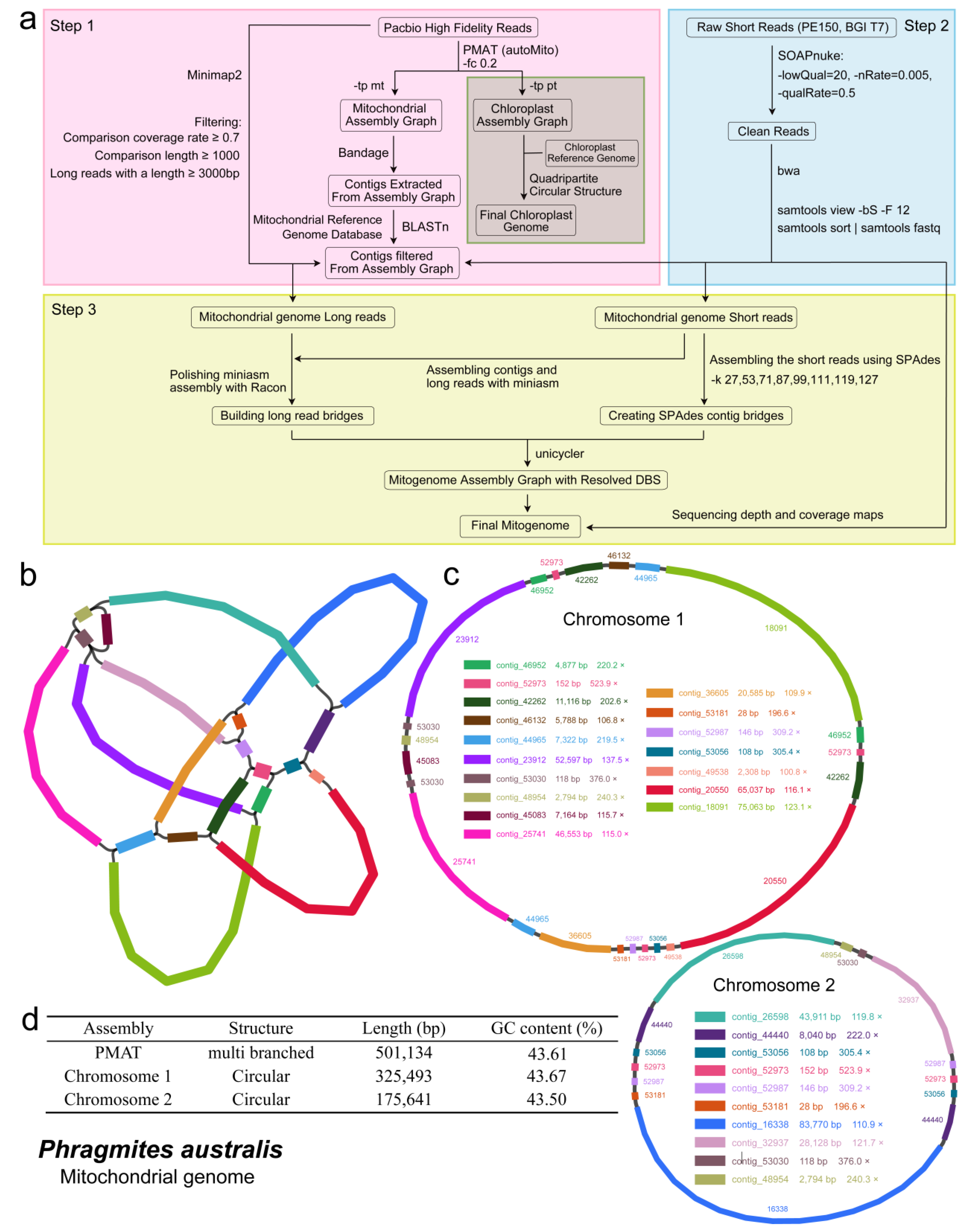
2.1. Assembly and Annotation of P. australis Mitogenome
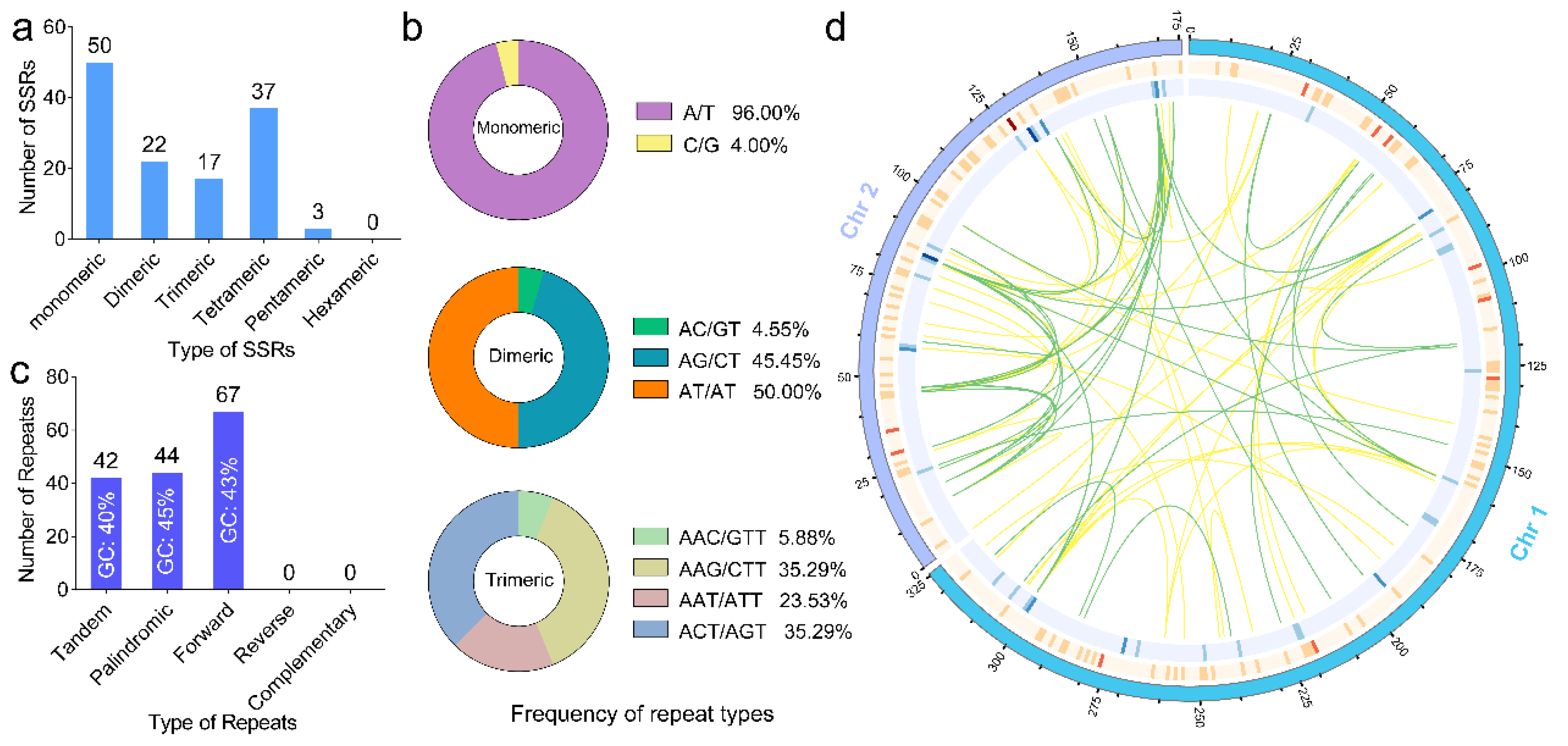
2.2. Analysis of Repetitive Sequences in the P. australis Mitogenome
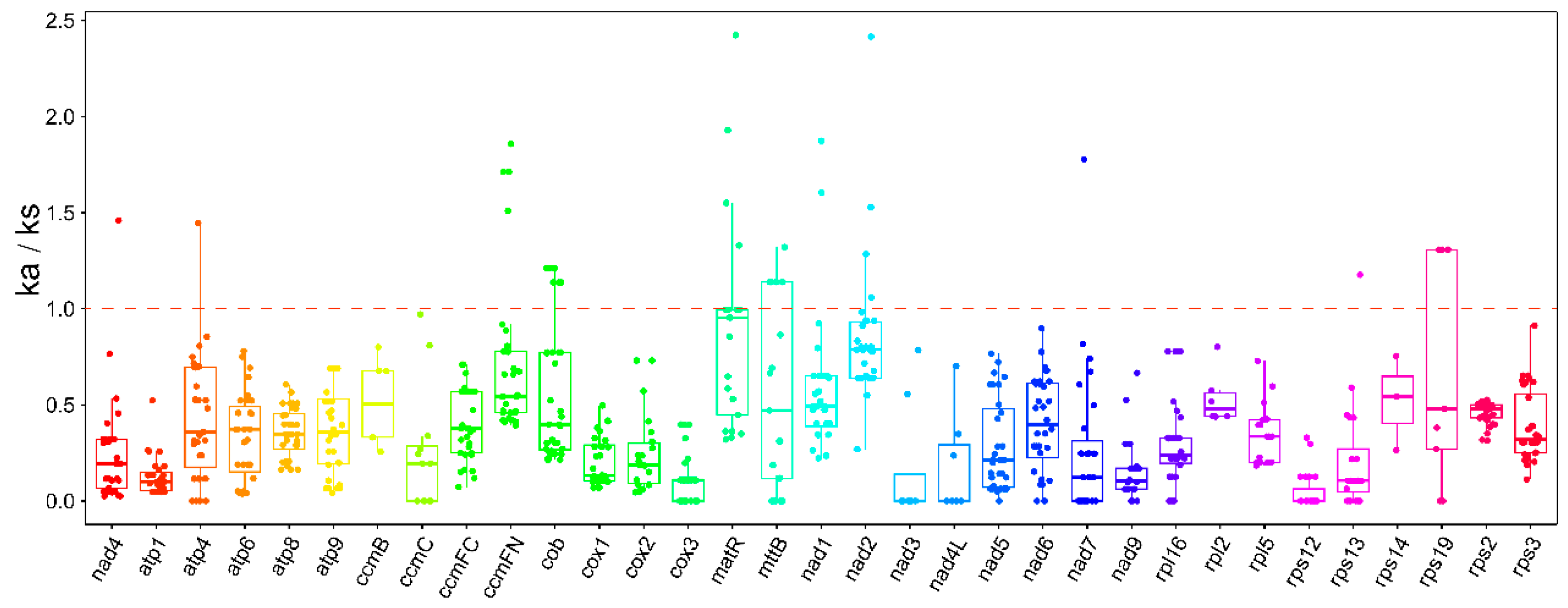
2.3. Ka/Ks Analysis
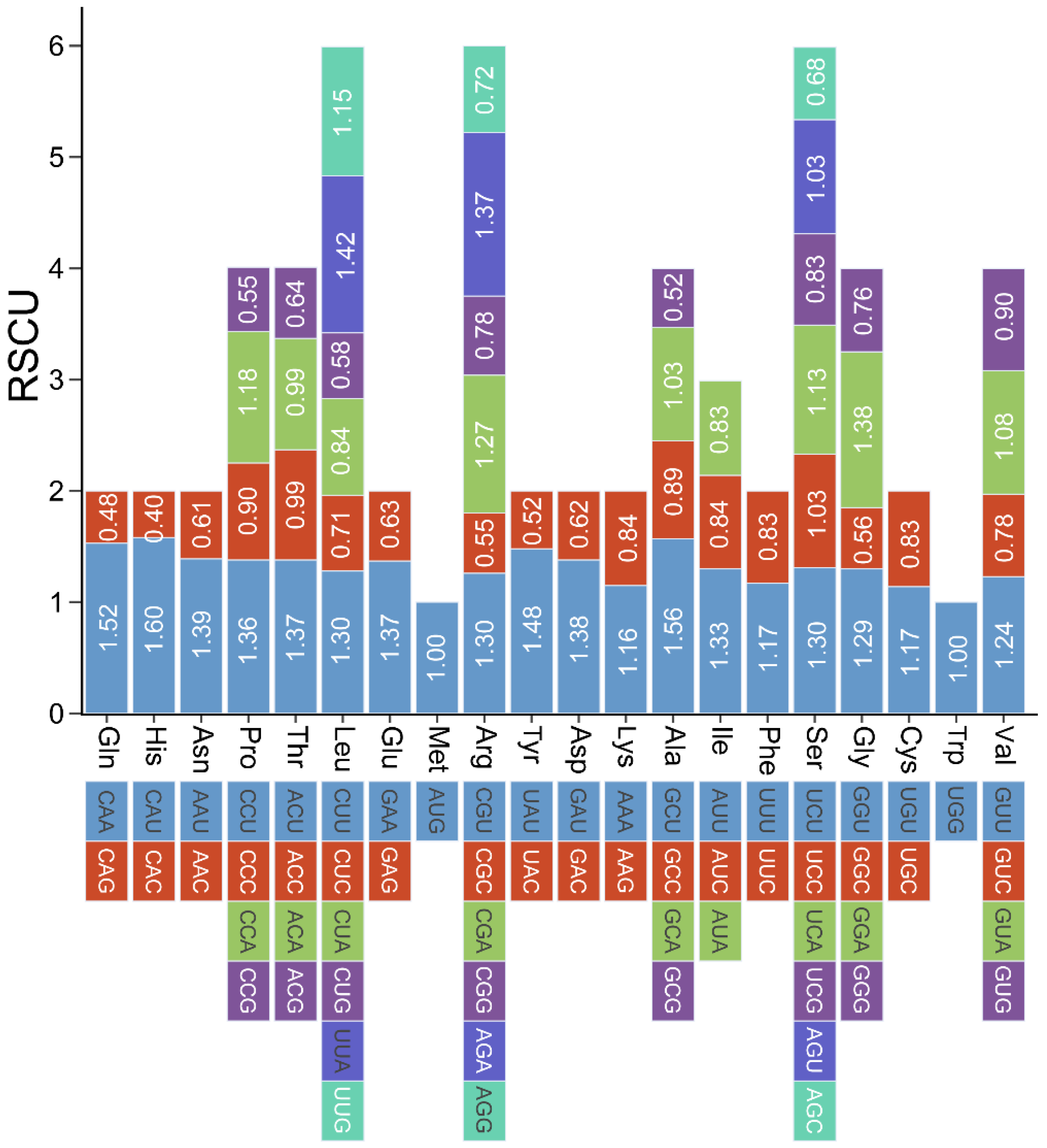
2.4. Analysis of Codon Usage in PCGs
2.5. Intracellular Gene Transfer (IGT) of P. australis Mitogenome
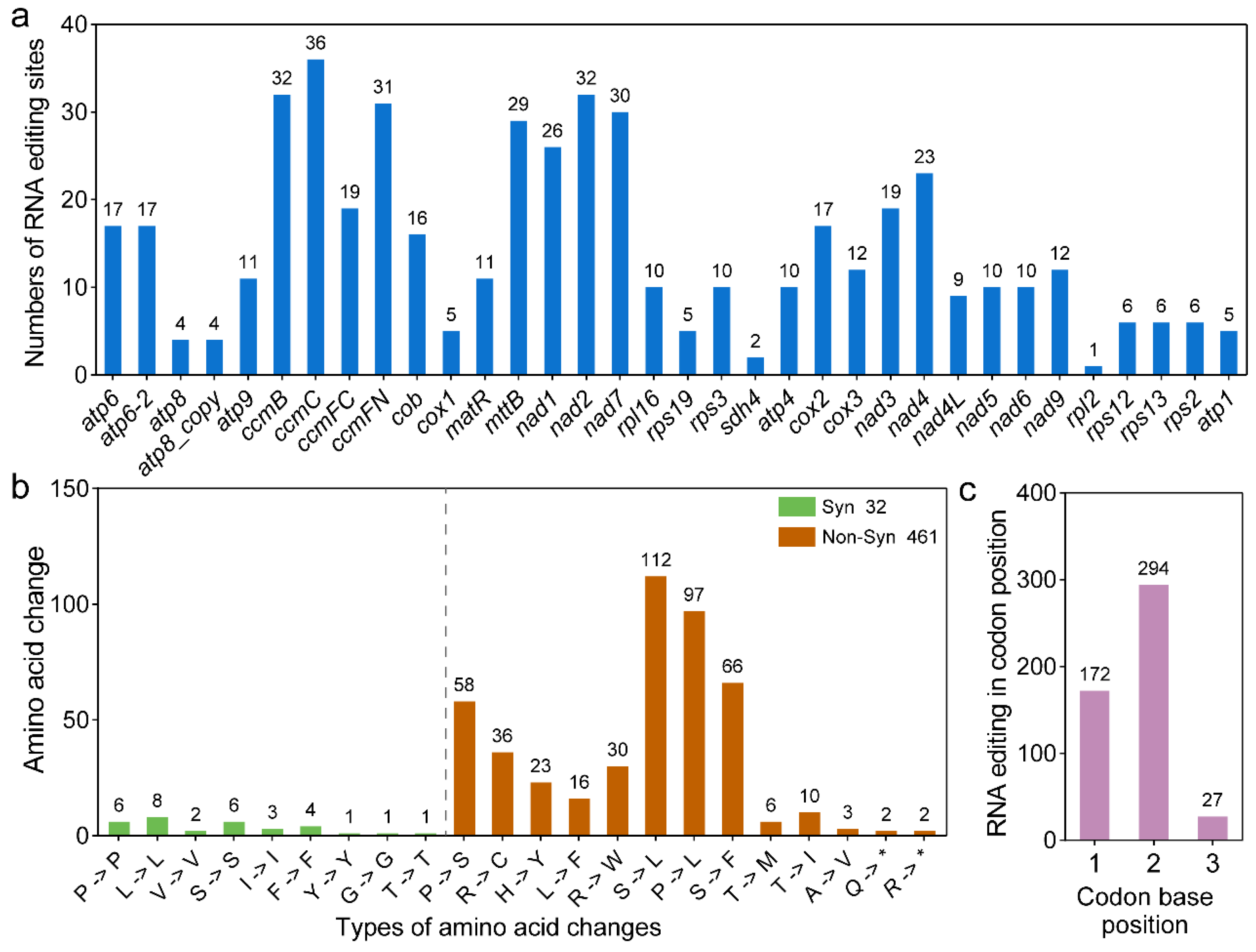
2.6. Prediction and Validation of RNA Editing
2.7. Analysis of Mitochondria-Related Differentially Expressed Genes (mtDEG)
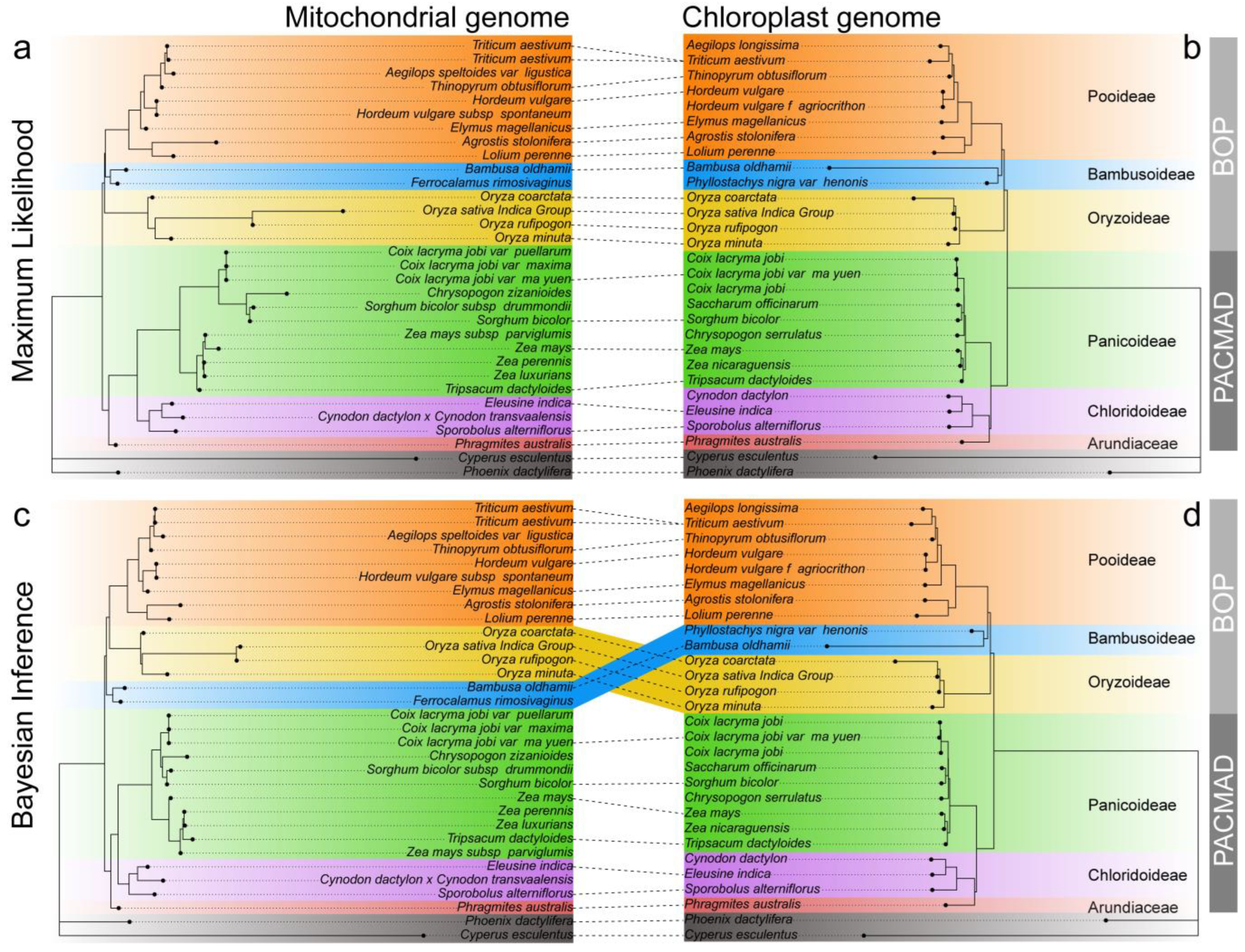
2.8. Phylogenetic Analysis
3. Discussion
3.1. Genome Assembly and Annotation of the Mitogenome of P. australis
3.2. Repetitive Sequences Impact Mitogenome GC Content
3.3. Analysis of P. australis Mitochondrial PCGs Ka/Ks and Codon Usage Preferences
3.4. Extensive and Frequent Intracellular Gene Transfer (IGT) Events Occur in P. australis
3.5. Prediction and Validation of RNA Editing
3.6. Analysis of Tissue-Specific Differential Expression Gene of P. australis Mitogenomes
3.7. Phylogenetic Analyses
4. Materials and Methods
4.1. Sample Collection and Sequencing Data
4.2. Genome Assembly and Annotation
4.3. Identification of the Succinate Dehydrogenase Gene Family
4.4. Identification of Repeat Sequences
4.5. Analysis of Synonymous and Nonsynonymous Substitution Rates
4.6. Codon Usage Analysis
4.7. Identification of Mitochondrial Plastid DNA Segments (MTPTs) and Nuclear Mitochondrial DNA Segments (NUMTs)
4.8. RNA Editing Site Prediction and Mitochondrial Gene Expression Analysis
4.9. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cahoon, D.R.; McKee, K.L. How Plants Influence Resilience of Salt Marsh and Mangrove Wetlands to Sea-Level Rise. Estuaries Coasts 2021, 44, 883–898. [Google Scholar] [CrossRef]
- Cui, J.P.; Qiu, T.H. De novo full-length transcriptome analysis of two ecotypes of Phragmites australis (swamp reed and dune reed) provides new insights into the transcriptomic complexity of dune reed and its long-term adaptation to desert environments. BMC Genom. 2023, 24, 180. [Google Scholar] [CrossRef]
- Hua, W.T.; Hu, W.Q. Identification of microbial consortia for sustainable disposal of constructed wetland reed litter wastes. Environ. Sci. Pollut. Res. 2023, 30, 58019–58029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Cui, X.J. Cyclic utilization of reed litters to enhance nitrogen removal efficiency in simulated estuarine wetland. Environ. Sci. Pollut. Res. 2021, 28, 39071–39081. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.X.; Fang, Z. Phragmites rhizoma polysaccharide-based nanocarriers for synergistic treatment of ulcerative colitis. Int. J. Biol. Macromol. 2022, 220, 22–32. [Google Scholar] [CrossRef]
- Ren, Y.; Cui, G.D. Traditional Uses, Phytochemistry, Pharmacology and Toxicology of Rhizoma phragmitis: A Narrative Review. Chin. J. Integr. Med. 2022, 28, 1127–1136. [Google Scholar] [CrossRef]
- Zhou, R.M.; Cui, M.X.; Wang, Y.; Zhang, M.; Li, F.; Liu, K. Isolation, structure identification and anti-inflammatory activity of a polysaccharide from Phragmites rhizoma. Int. J. Biol. Macromol. 2020, 161, 810–817. [Google Scholar] [CrossRef]
- Brownfield, L.; Yi, J. Organelles maintain spindle position in plant meiosis. Nat. Commun. 2015, 6, 6492. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Osiewacz, H.D.; Brust, D. Mitochondrial pathways governing stress resistance, life, and death in the fungal aging model Podospora anserina. Ann. N. Y. Acad. Sci. 2010, 1197, 54–66. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Cheng, N.; Lo, Y.S. Correlation between mtDNA complexity and mtDNA replication mode in developing cotyledon mitochondria during mung bean seed germination. New Phytol. 2017, 213, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Cuthbert, J.M. The massive mitochondrial genome of the angiosperm is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 10185–10191. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Yamamoto, A. Comprehensive Analysis of Mitochondria in Roots and Hypocotyls of Soybean under Flooding Stress using Proteomics and Metabolomics Techniques. J. Proteome Res. 2011, 10, 3993–4004. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.Z.; Meng, X.H. Oxidative Damage of Mitochondrial Proteins Contributes to Fruit Senescence: A Redox Proteomics Analysis. J. Proteome Res. 2009, 8, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.F.; Millar, A.H. Components of Mitochondrial Oxidative Phosphorylation Vary in Abundance Following Exposure to Cold and Chemical Stresses. J. Proteome Res. 2012, 11, 3860–3879. [Google Scholar] [CrossRef] [PubMed]
- Colombatti, F.; Mencia, R. The mitochondrial oxidation resistance protein AtOXR2 increases plant biomass and tolerance to oxidative stress. J. Exp. Bot. 2019, 70, 3177–3195. [Google Scholar] [CrossRef]
- Wang, J.Y.; Xu, G.J. Mitochondrial functions in plant immunity. Trends Plant Sci. 2022, 27, 1063–1076. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y. A mitochondrial RNA processing protein mediates plant immunity to a broad spectrum of pathogens by modulating the mitochondrial oxidative burst. Plant Cell 2022, 34, 2343–2363. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Qu, Y. Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 2022, 13, 1031769. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Q.; Shang, L.G. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, R. Comparative analysis of mitochondrial genomes of invasive weed Mikania micrantha and its indigenous congener Mikania cordata. Int. J. Biol. Macromol. 2024, 281, 136357. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Schon, K.R. Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes. Nature 2022, 611, 105–114. [Google Scholar] [CrossRef]
- Raven, J.A. Implications of mutation of organelle genomes for organelle function and evolution. J. Exp. Bot. 2015, 66, 5639–5650. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.H.; Cui, S.X. Evolutionary analysis for Phragmites ecotypes based on full-length plastomes. Aquat. Bot. 2021, 170, 103349. [Google Scholar] [CrossRef]
- Wang, C.; Liu, L. Chromosome-level genome assemblies reveal genome evolution of an invasive plant Phragmites australis. Commun. Biol. 2024, 7, 1007. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M.; Fay, M.F. The genome sequence of common reed, Phragmites australis (Cav.) Steud. (Poaceae). Wellcome Open Res. 2024, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, X.X. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2011, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhang, Q. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ran, Z.; Xiao, X.; Yan, C.; Xu, J.; Tang, M.; An, M. Comparative analysis of the whole mitochondrial genomes of four species in sect. Chrysantha (Camellia L.), endemic taxa in China. BMC Plant Biol. 2024, 24, 955. [Google Scholar]
- Wang, R.; Luo, Y.; Lan, Z.; Qiu, D. Insights into structure, codon usage, repeats, and RNA editing of the complete mitochondrial genome of Perilla frutescens (Lamiaceae). Sci. Rep. 2024, 14, 13940. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Luo, X.; Huang, Y.; Ke, S.-J.; Liu, Z.-J. The Complete Mitogenome of Apostasia fujianica Y.Li & S.Lan and Comparative Analysis of Mitogenomes across Orchidaceae. Int. J. Mol. Sci. 2024, 25, 8151. [Google Scholar] [CrossRef]
- Xie, P.; Wu, J.; Lu, M.; Tian, T.; Wang, D.; Luo, Z.; Yang, D.; Li, L.; Yang, X.; Liu, D.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Fritillaria ussuriensis Maxim. (Liliales: Liliaceae), an endangered medicinal plant. BMC Genom. 2024, 25, 773. [Google Scholar] [CrossRef]
- Adams, K.L.; Palmer, J.D. Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenet Evol. 2003, 29, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Archibald, J.M. Origin of eukaryotic cells: 40 years on. Symbiosis 2011, 54, 69–86. [Google Scholar] [CrossRef]
- Chen, Y.M.; Guo, Y.W. Pangenome-based trajectories of intracellular gene transfers in Poaceae unveil high cumulation in Triticeae. Plant Physiol. 2023, 193, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Liu, Z. Evolutionary transfers of mitochondrial genes to the nucleus in the Populus lineage and coexpression of nuclear and mitochondrial Sdh4 genes. New Phytol. 2006, 172, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.B.; Millar, A.H. Succinate dehydrogenase: The complex roles of a simple enzyme. Curr. Opin. Plant Biol. 2013, 16, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Hall, N.D.; Zhang, H.; Mower, J.P.; McElroy, J.S.; Goertzen, L.R. The Mitochondrial Genome of Eleusine indica and Characterization of Gene Content within Poaceae. Genome Biol. Evol. 2020, 12, 3684–3697. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Li, X.Y. Mitochondrial-to-nuclear communication in aging: An epigenetic perspective. Trends Biochem. Sci. 2022, 47, 645–659. [Google Scholar] [CrossRef]
- Guo, W.H.; Grewe, F. Ginkgo and Welwitschia Mitogenomes Reveal Extreme Contrasts in Gymnosperm Mitochondrial Evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Du, X.R. Assembly and analysis of the complete mitochondrial genome of Forsythia suspensa (Thunb.) Vahl. BMC Genom. 2023, 24, 708. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Chen, Y.P.P. Bacterial genomic G+C composition-eliciting environmental adaptation. Genomics 2010, 95, 7–15. [Google Scholar] [CrossRef]
- Smarda, P.; Bures, P. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc. Natl. Acad. Sci. USA 2014, 111, E4096–E4102. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.C. Plant Mitochondria are a Riddle Wrapped in a Mystery Inside an Enigma. J. Mol. Evol. 2021, 89, 151–156. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.G.; Zhang, X.R. Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genom. 2022, 23, 664. [Google Scholar] [CrossRef]
- Yang, J.; Wariss, H.M. De novo genome assembly of the endangered Acer yangbiense, a plant species with extremely small populations endemic to Yunnan, China. Gigascience 2019, 8, 085. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Liao, X.Z. Genomic architectural variation of plant mitochondria-A review of multichromosomal structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Chen, J.H.; Chen, S.T.; He, N.Y.; Wang, Q.L.; Zhao, Y.; Gao, W.; Guo, F.Q. Nuclear-encoded synthesis of the D1 subunit of photosystem II increases photosynthetic efficiency and crop yield. Nat. Plants 2020, 6, 570–580. [Google Scholar] [CrossRef]
- Ma, J.C.; Wang, S.H.; Zhu, X.J.; Sun, G.L.; Chang, G.X.; Li, L.H.; Hu, X.Y.; Zhang, S.Z.; Zhou, Y.; Song, C.P.; et al. Major episodes of horizontal gene transfer drove the evolution of land plants. Mol. Plant. 2022, 15, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Yamazaki, Y.; Murai, K.; Kanno, A.; Terachi, T.; Shiina, T.; Miyashita, N.; Nasuda, S.; Nakamura, C.; Mori, N.; et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33, 6235–6250. [Google Scholar] [CrossRef]
- Clifton, S.W.; Minx, P.; Fauron, C.M.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C.; et al. Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef]
- Li, J.X.; Chen, Y.L.; Liu, Y.L.; Wang, C.; Li, L.; Chao, Y.H. Complete mitochondrial genome of Agrostis stolonifera: Insights into structure, Codon usage, repeats, and RNA editing. BMC Genom. 2023, 24, 466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yuan, H.; Xu, J.; Cui, D.; Xiong, G.; Schwarzacher, T.; Heslop-Harrison, J.S. The mitochondrial genome of the diploid oat Avena longiglumis. BMC Plant Biol. 2023, 23, 218. [Google Scholar] [CrossRef]
- Anderson, B.M.; Krause, K.; Petersen, G. Mitochondrial genomes of two parasitic Cuscuta species lack clear evidence of horizontal gene transfer and retain unusually fragmented ccmF(C) genes. BMC Genom. 2021, 22, 816. [Google Scholar] [CrossRef] [PubMed]
- Bergthorsson, U.; Adams, K.L.; Thomason, B.; Palmer, J.D. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 2003, 424, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Joyce, P.B.; Gray, M.W. Chloroplast-like transfer RNA genes expressed in wheat mitochondria. Nucleic Acids Res. 1989, 17, 5461–5476. [Google Scholar] [CrossRef]
- Gray, M.W. RNA editing in plant mitochondria: 20 years later. IUBMB Life 2009, 61, 1101–1104. [Google Scholar] [CrossRef]
- Miyata, Y.; Sugita, M. Tissue- and stage-specific RNA editing of rps 14 transcripts in moss (Physcomitrella patens) chloroplasts. J. Plant Physiol. 2004, 161, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.E.; Ma, L.; Liu, J.Y.; Liu, X.Z.; Song, F.; Tian, L.; Cai, W.Z.; Li, H. The first A-to-I RNA editome of hemipteran species Coridius chinensis reveals overrepresented recoding and prevalent intron editing in early-diverging insects. Cell Mol. Life Sci. 2024, 81, 136. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Jiang, X.H.; Wang, T.F.; Zhang, X.J.; Zhang, A.D. Tissue-specificity of RNA editing in plant: Analysis of transcripts from three tobacco (Nicotiana tabacum) varieties. Plant Biotechnol. Rep. 2021, 15, 471–482. [Google Scholar] [CrossRef]
- Knoop, V. C-to-U and U-to-C: RNA editing in plant organelles and beyond. J. Exp. Bot. 2023, 74, 2273–2294. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, Y.; Cheng, W.; Wu, K.; Gao, H.; Liu, L.; Xu, L.; Gong, B. Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Diospyros oleifera, the first representative from the family Ebenaceae. Heliyon 2022, 8, e09870. [Google Scholar] [CrossRef] [PubMed]
- Zavrtanik, U.; Medved, T.; Puric, S.; Vranken, W.; Lah, J.; Hadzi, S. Leucine Motifs Stabilize Residual Helical Structure in Disordered Proteins. J. Mol. Biol. 2024, 436, 168444. [Google Scholar] [CrossRef]
- Zhu, B.Y.; Zhou, N.E.; Kay, C.M.; Hodges, R.S. Packing and hydrophobicity effects on protein folding and stability: Effects of beta-branched amino acids, valine and isoleucine, on the formation and stability of two-stranded alpha-helical coiled coils/leucine zippers. Protein Sci. 1993, 2, 383–394. [Google Scholar] [CrossRef]
- Hu, Y.X.; Huang, A.; Li, Y.; Molloy, D.P.; Huang, C. Emerging roles of the C-to-U RNA editing in plant stress responses. Plant Sci. 2024, 349, 112263. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wang, H.; Zhao, X.; Obel, H.O.; Yu, X.; Lou, Q.; Chen, J.; Cheng, C. Chloroplast Pan-Genomes and Comparative Transcriptomics Reveal Genetic Variation and Temperature Adaptation in the Cucumber. Int. J. Mol. Sci. 2023, 24, 8943. [Google Scholar] [CrossRef]
- Ramadan, A.M. Salinity effects on nad3 gene RNA editing of wild barley mitochondria. Mol. Biol. Rep. 2020, 47, 3857–3865. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, A.M.; Said, O.A.M.; Abushady, A.M. Salinity stress reveals three types of RNA editing sites in mitochondrial Nad7 gene of wild barley both in silico and in qRT-PCR experiments. Theor. Exp. Plant Physiol. 2022, 34, 13–22. [Google Scholar] [CrossRef]
- Ramadan, A.; Alnufaei, A.A.; Fiaz, S.; Khan, T.K.; Hassan, S.M. Effect of salinity on ccmfn gene RNA editing of mitochondria in wild barley and uncommon types of RNA editing. Funct. Integr. Genom. 2023, 23, 50. [Google Scholar] [CrossRef]
- Gao, J.; Guan, B.; Ge, M.; Eller, F.; Yu, J.; Wang, X.; Zuo, J. Can allelopathy of Phragmites australis extracts aggravate the effects of salt stress on the seed germination of Suaeda salsa? Front. Plant Sci. 2022, 13, 990541. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Kim, M.J.; Nelson, W.; Balbuena, T.S.; Kim, R.; Kramer, R.; Crow, J.A.; May, G.D.; Thelen, J.J.; Soderlund, C.A.; et al. Next-generation sequencing-based transcriptomic and proteomic analysis of the common reed, Phragmites australis (Poaceae), reveals genes involved in invasiveness and rhizome specificity. Am. J. Bot. 2012, 99, 232–247. [Google Scholar] [CrossRef]
- Williams, J.; Lambert, A.M.; Long, R.; Saltonstall, K. Does hybrid Phragmites australis differ from native and introduced lineages in reproductive, genetic, and morphological traits? Am. J. Bot. 2019, 106, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.Y.; Du, Z.X.; Li, X.Y.; Chen, J.Z. Physiological and ecological characteristics and reproductive responses of Phragmites australis to dry-wet conditions in inland saline marshes of Northeast China. PeerJ 2022, 10, e14269. [Google Scholar]
- Wu, J.; Gao, T.; Zhao, L.; Bao, H.; Yu, C.; Hu, J.; Ma, F. Investigating Phragmites australis response to copper exposure using physiologic, Fourier Transform Infrared and metabolomic approaches. Funct. Plant Biol. 2022, 49, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, L.; Yang, R.; Jiang, Y.; Liao, J.; Chai, S.; Deng, X.; Wang, L.; Pu, X.; Zhang, Y.; et al. Integrated Multiomics and Synergistic Functional Network Revealed the Mechanism in the Tolerance of Different Ecotypes of Salvia miltiorrhiza Bge. to Doxycycline Pollution. Environ. Sci. Technol. 2023, 57, 9603–9614. [Google Scholar] [CrossRef]
- Zhang, X.; Gu, C.; Zhang, T.; Tong, B.; Zhang, H.; Wu, Y.; Yang, C. Chloroplast (Cp) Transcriptome of P. davidiana Dode x P. bolleana Lauch provides insight into the Cp drought response and Populus Cp phylogeny. BMC Evol. Biol. 2020, 20, 51. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, H.; Zhou, J.M.; Smith, S.M.; Li, J. Malate Circulation: Linking Chloroplast Metabolism to Mitochondrial ROS. Trends Plant Sci. 2020, 25, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.M.; Zhang, Y. Plant Immunity: Danger Perception and Signaling. Cell 2020, 181, 978–989. [Google Scholar] [CrossRef]
- He, W.; Chen, C.; Adedze, Y.M.N.; Dong, X.; Xi, K.; Sun, Y.; Dang, T.; Jin, D. Multicentric origin and diversification of atp6-orf79-like structures reveal mitochondrial gene flows in Oryza rufipogon and Oryza sativa. Evol. Appl. 2020, 13, 2284–2299. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, C.J.; Wang, W.; Goffinet, B. Mitochondrial phylogenomics of early land plants: Mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst. Biol. 2014, 63, 862–878. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.P.; Wang, R.; Gu, R.Q.; Chen, M.H.; Wang, Z.Y.; Li, L.; Hong, J.M.; Cui, S.X. Telomere-to-telomere Phragmites australis reference genome assembly with a B chromosome provides new insights into its evolution and polysaccharide biosynthesis. Res. Sq. 2024, preprint. [Google Scholar] [CrossRef]
- Oh, D.H.; Kowalski, K.P.; Quach, Q.N.; Wijesinghege, C.; Tanford, P.; Dassanayake, M.; Clay, K. Novel genome characteristics contribute to the invasiveness of Phragmites australis (common reed). Mol. Ecol. 2022, 31, 1142–1159. [Google Scholar] [CrossRef]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Chen, Y.S.; Shi, C.M.; Huang, Z.B.; Zhang, Y.; Li, S.K.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2017, 7, gix120. [Google Scholar] [CrossRef]
- Bi, C.; Shen, F.; Han, F.; Qu, Y.; Hou, J.; Xu, K.; Xu, L.A.; He, W.; Wu, Z.; Yin, T. PMAT: An efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 2024, 11, uhae023. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.C.; Chen, H.M.; Jiang, M.; Wang, L.Q.; Wu, X.; Huang, L.F.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Wang, D.P.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.Y.; Gao, F.L.; Jakovlic, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. Imeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Chen, C.J.; Wu, Y.; Li, J.W.; Wang, X.; Zeng, Z.H.; Xu, J.; Liu, Y.L.; Feng, J.T.; Chen, H.; He, Y.H.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Edera, A.A.; Small, I.; Milone, D.H.; Sanchez-Puerta, M.V. Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 2021, 136, 104682. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, U357–U359. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Sébastien Lê, J.J.; François, H. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Name of Genes | |
|---|---|---|
| Protein coding genes (PCGs) | ATP synthase | atp1, atp4, atp6 (×2), atp8 (×2), atp9 |
| NADH dehydrogenase | nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 | |
| Cytochrome c biogenesis | cob | |
| Ubiquinol cytochrome c reductase | ccmB, ccmC, ccmFC, ccmFN | |
| Cytochrome c oxidase | cox1, cox2, cox3 | |
| Maturases | matR | |
| Transport membrane protein | mttB | |
| Large subunit of the ribosome | rpl5, rpl16, rpl2 | |
| Small subunit of ribosome | rps2, rps3, rps12, rps13, rps14, rps19 | |
| Succinate dehydrogenase | sdh4 | |
| Ribosomal RNA | Ribosomal RNAs | rrn5 (×2), rrn18 (×2), rrn26 (×2) |
| Transfer RNA | Transfer RNAs | trnC-GCA, trnD-GUC (×2), trnE-UUC, trnfM-CAU (×2), trnH-GUG, trnI-CAU (×2), trnL-CAA, trnN-GUU, trnP-UGG (×2), trnQ-UUG, trnR-UCU, trnS-GGA (×2), trnS-UGA, trnV-UAC, trnF-GAA (×2), trnK-UUU (×2), trnM-CAU, trnS-GCU, trnW-CCA, trnY-GUA |
| Repeats Type | Number of Repeats | Total | Proportion (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |||
| Monomeric | - | - | - | - | - | - | - | 37 | 7 | 2 | 3 | 1 | 50 | 38.76 |
| Dimeric | - | - | 17 | 4 | 1 | - | - | - | - | - | - | - | 22 | 17.05 |
| Trimeric | 13 | 1 | 1 | 1 | - | - | - | - | - | - | - | 17 | 13.18 | |
| Tetrameric | 35 | 1 | - | - | - | - | - | - | - | - | - | - | 37 | 28.68 |
| Pentameric | 2 | 1 | - | - | - | - | - | - | - | - | - | - | 3 | 2.33 |
| total | 38 | 16 | 18 | 5 | 2 | 0 | 0 | 37 | 7 | 2 | 3 | 1 | 129 | 100.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Yang, Q.; Zhang, J.; Ju, C.; Cui, S. Mitochondrial Genome Insights into Evolution and Gene Regulation in Phragmites australis. Int. J. Mol. Sci. 2025, 26, 546. https://doi.org/10.3390/ijms26020546
Cui J, Yang Q, Zhang J, Ju C, Cui S. Mitochondrial Genome Insights into Evolution and Gene Regulation in Phragmites australis. International Journal of Molecular Sciences. 2025; 26(2):546. https://doi.org/10.3390/ijms26020546
Chicago/Turabian StyleCui, Jipeng, Qianhui Yang, Jiyue Zhang, Chuanli Ju, and Suxia Cui. 2025. "Mitochondrial Genome Insights into Evolution and Gene Regulation in Phragmites australis" International Journal of Molecular Sciences 26, no. 2: 546. https://doi.org/10.3390/ijms26020546
APA StyleCui, J., Yang, Q., Zhang, J., Ju, C., & Cui, S. (2025). Mitochondrial Genome Insights into Evolution and Gene Regulation in Phragmites australis. International Journal of Molecular Sciences, 26(2), 546. https://doi.org/10.3390/ijms26020546