
Protection Strategies Against Palmitic Acid-Induced Lipotoxicity in Metabolic Syndrome and Related Diseases

 ,
,  and
and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Palmitic Acid Biosynthesis in Metabolic Syndrome and Obesity
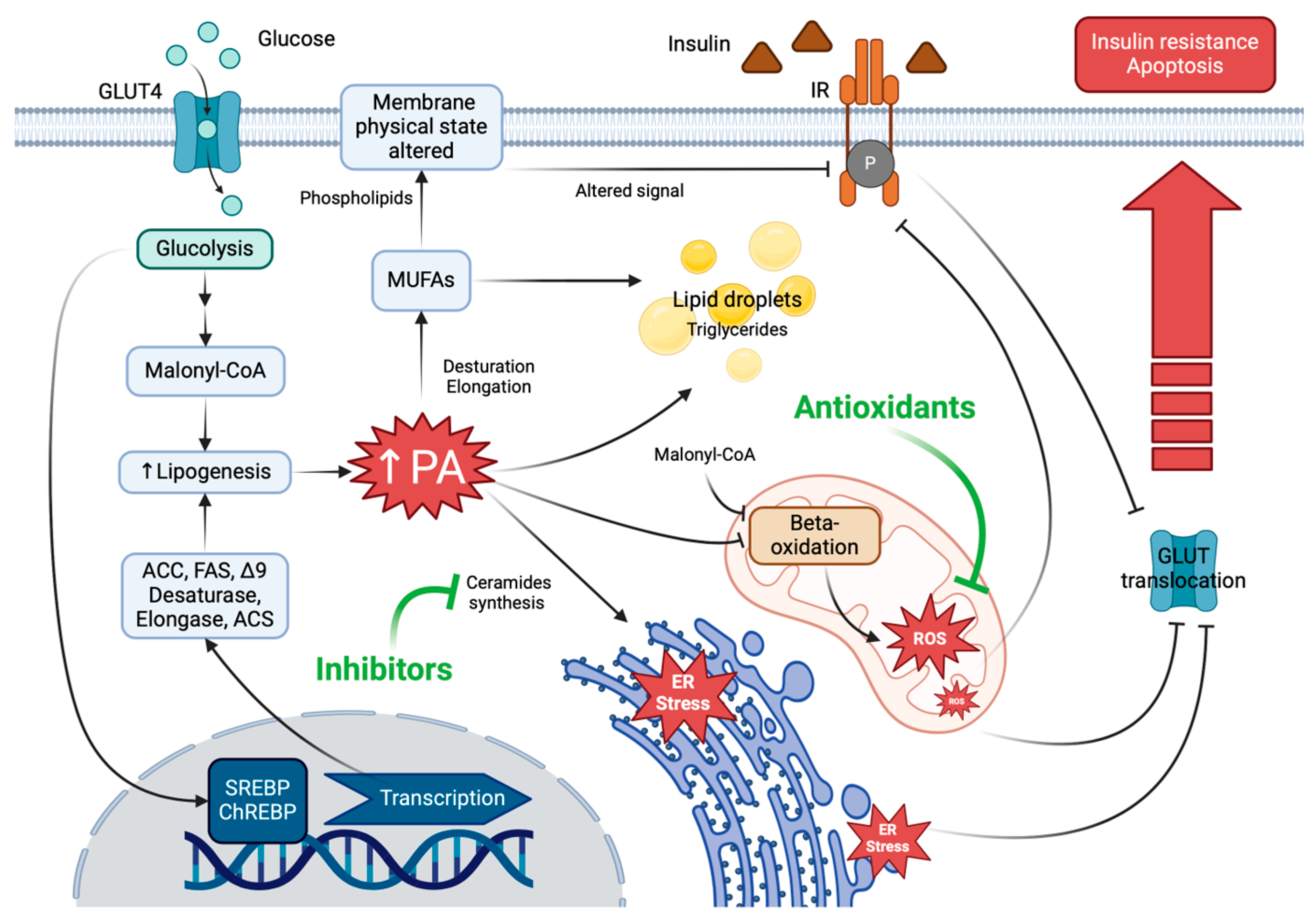
3. Palmitic Acid-Induced Lipotoxicity
3.1. Palmitic Acid-Induced Endoplasmic Reticulum (ER) Stress
3.2. Palmitic Acid-Induced Mitochondrial Dysfunction
3.3. Palmitic Acid-Induced Inflammation
4. PA-Induced Lipotoxicity and Free Fatty Acid Receptor Intervention
5. Palmitic Acid Reduced Insulin Sensitivity and Increased Vascular Dysfunction
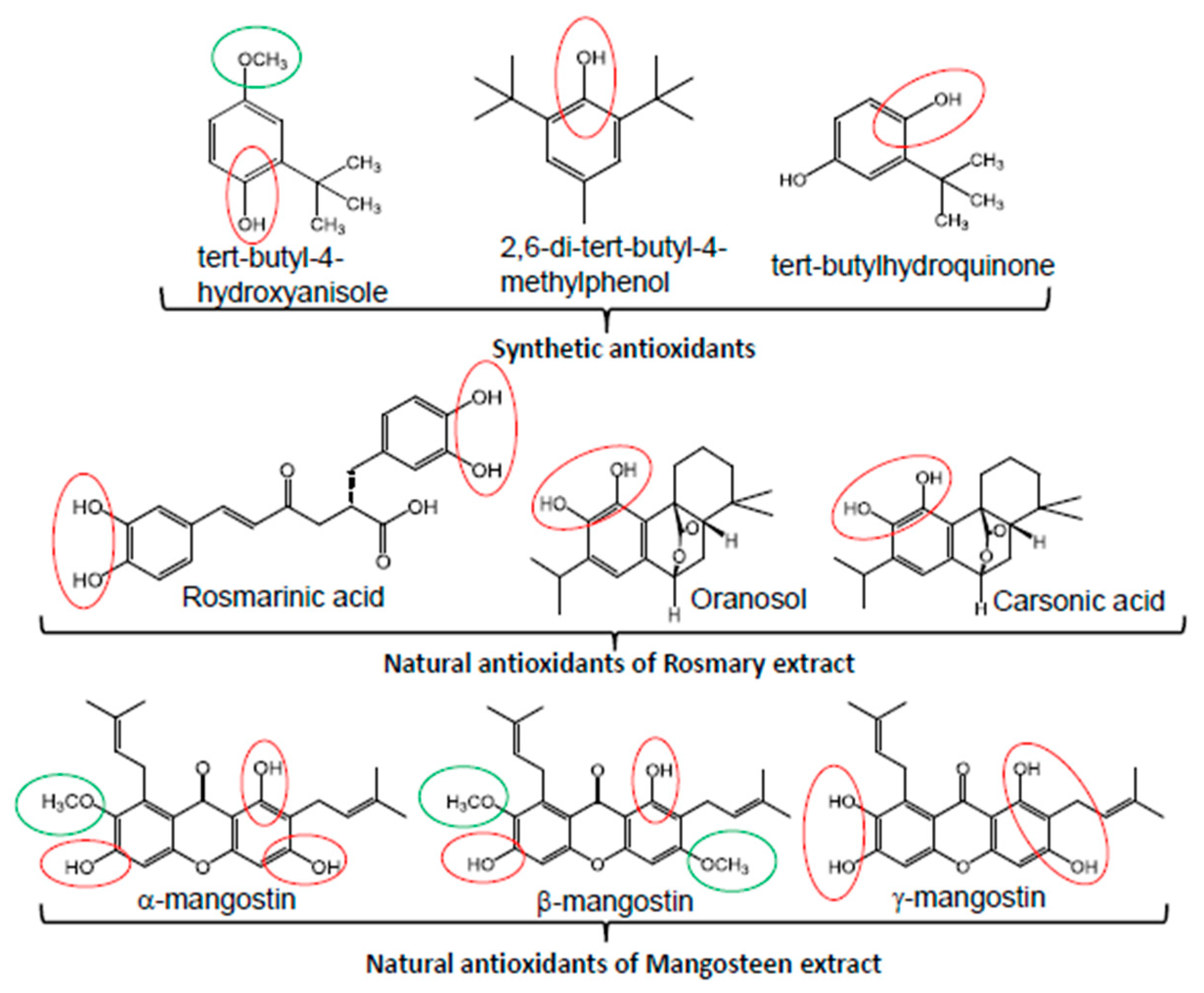
6. Palmitic Acid-Induced Lipotoxicity Is Attenuated by Natural Antioxidants
7. PA-Induced Lipotoxicity Is Attenuated by Lipid Droplet (LD) Functionality
8. Natural Antioxidants and PA-Induced Lipid Droplets
9. Antioxidant Strategies to Prevent Lipid Peroxidation
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apekey, T.A.; Maynard, M.J.; Kittana, M.; Kunutsor, S.K. Comparison of the Effectiveness of Low Carbohydrate Versus Low Fat Diets, in Type 2 Diabetes: Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2022, 14, 4391. [Google Scholar] [CrossRef] [PubMed]
- Pour Abbasi, M.S.; Shojaei, N.; Farhangi, M.A. Low-carbohydrate diet score is associated with improved blood pressure and cardio-metabolic risk factors among obese adults. Physiol. Rep. 2022, 10, e15375. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; Iqbal, N.; Seshadri, P.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Williams, T.; Williams, M.; Gracely, E.J.; Stern, L. A Low-Carbohydrate as Compared with a Low-Fat Diet in Severe Obesity. N. Engl. J. Med. 2003, 348, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Hyde, P.N.; Sapper, T.N.; Crabtree, C.D.; LaFountain, R.A.; Bowling, M.L.; Buga, A.; Fell, B.; McSwiney, F.T.; Dickerson, R.M.; Miller, V.J.; et al. Dietary carbohydrate restriction improves metabolic syndrome independent of weight loss. JCI Insight 2019, 4, e128308. [Google Scholar] [CrossRef]
- Hall, K.D.; Chung, S.T. Low-carbohydrate diets for the treatment of obesity and type 2 diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 308–312. [Google Scholar] [CrossRef]
- Ismael, S.A. Effects of low carbohydrate diet compared to low fat diet on reversing the metabolic syndrome, using NCEP ATP III criteria: A randomized clinical trial. BMC Nutr. 2021, 7, 62. [Google Scholar] [CrossRef]
- Murru, E.; Manca, C.; Carta, G.; Banni, S. Impact of Dietary Palmitic Acid on Lipid Metabolism. Front. Nutr. 2022, 9, 861664. [Google Scholar] [CrossRef]
- Mthembu, S.X.H.; Mazibuko-Mbeje, S.E.; Silvestri, S.; Orlando, P.; Marcheggiani, F.; Cirilli, I.; Nkambule, B.B.; Muller, C.J.F.; Tiano, L.; Dludla, P.V. Low levels and partial exposure to palmitic acid improves mitochondrial function and the oxidative status of cultured cardiomyoblasts. Toxicol. Rep. 2024, 12, 234–243. [Google Scholar] [CrossRef]
- Rocca, C.; De Bartolo, A.; Guzzi, R.; Crocco, M.C.; Rago, V.; Romeo, N.; Perrotta, I.; De Francesco, E.M.; Muoio, M.G.; Granieri, M.C.; et al. Palmitate-Induced Cardiac Lipotoxicity Is Relieved by the Redox-Active Motif of SELENOT through Improving Mitochondrial Function and Regulating Metabolic State. Cells 2023, 12, 1042. [Google Scholar] [CrossRef]
- Engin, A.B. What Is Lipotoxicity? In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2017; pp. 197–220. [Google Scholar]
- Chong, M.F.-F.; Hodson, L.; Bickerton, A.S.; Roberts, R.; Neville, M.; Karpe, F.; Frayn, K.N.; Fielding, B.A. Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3 d of high-carbohydrate feeding. Am. J. Clin. Nutr. 2008, 87, 817–823. [Google Scholar] [CrossRef]
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2017, 19, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Ngandiri, D.A.; Llerins Perez, M.; Wolf, A.; Wang, Y. The Molecular Brakes of Adipose Tissue Lipolysis. Front. Physiol. 2022, 13, 826314. [Google Scholar] [CrossRef] [PubMed]
- Alvheim, A.R.; Malde, M.K.; Osei-Hyiaman, D.; Hong, Y.H.; Pawlosky, R.J.; Madsen, L.; Kristiansen, K.; Frøyland, L.; Hibbeln, J.R. Dietary Linoleic Acid Elevates Endogenous 2-AG and Anandamide and Induces Obesity. Obesity 2012, 20, 1984–1994. [Google Scholar] [CrossRef]
- Miklankova, D.; Markova, I.; Hüttl, M.; Stankova, B.; Malinska, H. The Different Insulin-Sensitising and Anti-Inflammatory Effects of Palmitoleic Acid and Oleic Acid in a Prediabetes Model. J. Diabetes Res. 2022, 2022, 4587907. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Liu, X.; Luo, R.; Liao, G.; Li, L.; Liu, J.; Cheng, J.; Lu, Y.; Chen, Y. Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis. Life Sci. 2018, 203, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Cho, Y. Protection of palmitic acid-mediated lipotoxicity by arachidonic acid via channeling of palmitic acid into triglycerides in C2C12. J. Biomed. Sci. 2014, 21, 13. [Google Scholar] [CrossRef]
- Cho, Y.S.; Kim, C.H.; Kim, K.Y.; Cheon, H.G. Protective effects of arachidonic acid against palmitic acid-mediated lipotoxicity in HIT-T15 cells. Mol. Cell. Biochem. 2012, 364, 19–28. [Google Scholar] [CrossRef]
- Qiu, T.; Yang, X.; Wang, J.; Pan, C.; Chu, X.; Xiong, J.; Xie, J.; Chang, Y.; Wang, C.; Zhang, J. Obesity-induced elevated palmitic acid promotes inflammation and glucose metabolism disorders through GPRs/NF-κB/KLF7 pathway. Nutr. Diabetes 2022, 12, 23. [Google Scholar] [CrossRef]
- Schwarz, J.-M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef]
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose Triglyceride Lipase and Hormone-sensitive Lipase Are the Major Enzymes in Adipose Tissue Triacylglycerol Catabolism. J. Biol. Chem. 2006, 281, 40236–40241. [Google Scholar] [CrossRef]
- Kennedy, A.; Martinez, K.; Chuang, C.-C.; LaPoint, K.; McIntosh, M. Saturated Fatty Acid-Mediated Inflammation and Insulin Resistance in Adipose Tissue: Mechanisms of Action and Implications. J. Nutr. 2009, 139, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Phan, F.; Foufelle, F. SREBP-1c and lipogenesis in the liver: An update. Biochem. J. 2021, 478, 3723–3739. [Google Scholar] [CrossRef] [PubMed]
- Régnier, M.; Carbinatti, T.; Parlati, L.; Benhamed, F.; Postic, C. The role of ChREBP in carbohydrate sensing and NAFLD development. Nat. Rev. Endocrinol. 2023, 19, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Botolin, D.; Xu, J.; Christian, B.; Mitchell, E.; Jayaprakasam, B.; Nair, M.; Peters, J.M.; Busik, J.; Olson, L.K.; et al. Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J. Lipid Res. 2006, 47, 2028–2041. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.-D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Moon, Y.A.; Ochoa, C.R.; Mitsche, M.A.; Hammer, R.E.; Horton, J.D. Deletion of ELOVL6 blocks the synthesis of oleic acid but does not prevent the development of fatty liver or insulin resistance. J. Lipid Res. 2015, 55, 2597–2605. [Google Scholar] [CrossRef]
- Poudyal, H.; Brown, L. Stearoyl-CoA Desaturase: A Vital Checkpoint in the Development and Progression of Obesity. Endocr. Metab. Immune Disord.-Drug Targets 2011, 11, 217–231. [Google Scholar] [CrossRef]
- El Hafidi, M.; Cuéllar, A.; Ramırez, J.; Baños, G. Effect of sucrose addition to drinking water, that induces hypertension in the rats, on liver microsomal Δ9 and Δ5-desaturase activities. J. Nutr. Biochem. 2001, 12, 396–403. [Google Scholar] [CrossRef]
- Ruiz-Ramírez, A.; Chávez-Salgado, M.; Peñeda-Flores, J.A.; Zapata, E.; Masso, F.; El-Hafidi, M. High-sucrose diet increases ROS generation, FFA accumulation, UCP2 level, and proton leak in liver mitochondria. Am. J. Physiol.-Endocrinol. Metab. 2011, 301, E1198–E1207. [Google Scholar] [CrossRef]
- Ravaut, G.; Légiot, A.; Bergeron, K.-F.; Mounier, C. Monounsaturated Fatty Acids in Obesity-Related Inflammation. Int. J. Mol. Sci. 2020, 22, 330. [Google Scholar] [CrossRef]
- Lenighan, Y.M.; McNulty, B.A.; Roche, H.M. Dietary fat composition: Replacement of saturated fatty acids with PUFA as a public health strategy, with an emphasis on α-linolenic acid. Proc. Nutr. Soc. 2019, 78, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Panov, A.; Dikalova, A. Critical Role of Mitochondrial Fatty Acid Metabolism in Normal Cell Function and Pathological Conditions. Int. J. Mol. Sci. 2024, 25, 6498. [Google Scholar] [CrossRef]
- Sharifi, S.; Yamamoto, T.; Zeug, A.; Elsner, M.; Avezov, E.; Mehmeti, I. Non-esterified fatty acid palmitate facilitates oxidative endoplasmic reticulum stress and apoptosis of β-cells by upregulating ERO-1α expression. Redox Biol. 2024, 73, 103170. [Google Scholar] [CrossRef]
- Yang, H.-Y.; Chen, J.-Y.; Huo, Y.-N.; Yu, P.-L.; Lin, P.-Z.; Hsu, S.-C.; Huang, S.-M.; Tsai, C.-S.; Lin, C.-Y. The Role of Sirtuin 1 in Palmitic Acid-Induced Endoplasmic Reticulum Stress in Cardiac Myoblasts. Life 2022, 12, 182. [Google Scholar] [CrossRef]
- Ljubkovic, M.; Gressette, M.; Bulat, C.; Cavar, M.; Bakovic, D.; Fabijanic, D.; Grkovic, I.; Lemaire, C.; Marinovic, J. Disturbed Fatty Acid Oxidation, Endoplasmic Reticulum Stress, and Apoptosis in Left Ventricle of Patients With Type 2 Diabetes. Diabetes 2019, 68, 1924–1933. [Google Scholar] [CrossRef]
- Baldwin, A.C.; Green, C.D.; Olson, L.K.; Moxley, M.A.; Corbett, J.A. A role for aberrant protein palmitoylation in FFA-induced ER stress and β-cell death. Am. J. Physiol. Metab. 2012, 302, E1390–E1398. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, P.; Li, J.; Zhao, Q.; Chang, Y.; He, X. Protein lipidation in health and disease: Molecular basis, physiological function and pathological implication. Signal Transduct. Target. Ther. 2024, 9, 60. [Google Scholar] [CrossRef]
- Martínez, L.; Torres, S.; Baulies, A.; Alarcón-Vila, C.; Elena, M.; Fabriàs, G.; Casas, J.; Caballeria, J.; Fernandez-Checa, J.C.; García-Ruiz, C. Myristic acid potentiates palmitic acid-induced lipotoxicity and steatohepatitis associated with lipodystrophy by sustaning de novo ceramide synthesis. Oncotarget 2015, 6, 41479–41496. [Google Scholar] [CrossRef]
- Imierska, M.; Zabielski, P.; Roszczyc-Owsiejczuk, K.; Sokołowska, E.; Pogodzińska, K.; Kojta, I.; Błachnio-Zabielska, A. Serine Palmitoyltransferase Gene Silencing Prevents Ceramide Accumulation and Insulin Resistance in Muscles in Mice Fed a High-Fat Diet. Cells 2022, 11, 1123. [Google Scholar] [CrossRef]
- Lallement, J.; Raho, I.; Merlen, G.; Rainteau, D.; Croyal, M.; Schiffano, M.; Kassis, N.; Doignon, I.; Soty, M.; Lachkar, F.; et al. Deletion of serine palmitoyl transferase 2 in hepatocytes impairs ceramide/sphingomyelin balance, prevents obesity and leads to liver damage in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ruiz, M.; Henricsson, M.; Borén, J.; Pilon, M. Palmitic acid causes increased dihydroceramide levels when desaturase expression is directly silenced or indirectly lowered by silencing AdipoR2. Lipids Health Dis. 2021, 20, 173. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Huang, X.; Zhang, J.; Huang, Y.; Tang, Y.; Wen, H.; Xu, Y.; Zhang, S.; Wei, X.; Sun, S.; et al. Palmitic acid induces β-cell ferroptosis by activating ceramide signaling pathway. Exp. Cell Res. 2024, 440, 114134. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Luo, H.; Zhang, N.; Wang, Y.; Li, Y.; Huang, H.; Liu, Y.; Hu, Y.; Liu, H.; Zhang, J.; et al. Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation. Int. J. Mol. Sci. 2019, 20, 6268. [Google Scholar] [CrossRef]
- Wei, C.-D.; Li, Y.; Zheng, H.-Y.; Tong, Y.-Q.; Dai, W. Palmitate induces H9c2 cell apoptosis by increasing reactive oxygen species generation and activation of the ERK1/2 signaling pathway. Mol. Med. Rep. 2013, 7, 855–861. [Google Scholar] [CrossRef]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Müller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Alizadeh, J.; da Silva Rosa, S.C.; Weng, X.; Jacobs, J.; Lorzadeh, S.; Ravandi, A.; Vitorino, R.; Pecic, S.; Zivkovic, A.; Stark, H.; et al. Ceramides and ceramide synthases in cancer: Focus on apoptosis and autophagy. Eur. J. Cell Biol. 2023, 102, 151337. [Google Scholar] [CrossRef]
- Chang, Y.C.; Fong, Y.; Tsai, E.-M.; Chang, Y.-G.; Chou, H.L.; Wu, C.-Y.; Teng, Y.-N.; Liu, T.-C.; Yuan, S.-S.; Chiu, C.-C. Exogenous C8-Ceramide Induces Apoptosis by Overproduction of ROS and the Switch of Superoxide Dismutases SOD1 to SOD2 in Human Lung Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3010. [Google Scholar] [CrossRef]
- Kuang, H.; Sun, X.; Liu, Y.; Tang, M.; Wei, Y.; Shi, Y.; Li, R.; Xiao, G.; Kang, J.; Wang, F.; et al. Palmitic acid-induced ferroptosis via CD36 activates ER stress to break calcium-iron balance in colon cancer cells. FEBS J. 2023, 290, 3664–3687. [Google Scholar] [CrossRef]
- Chen, X.; Chen, K.; Hu, J.; Dong, Y.; Zheng, M.; Jiang, J.; Hu, Q.; Zhang, W. Palmitic acid induces lipid droplet accumulation and senescence in nucleus pulposus cells via ER-stress pathway. Commun. Biol. 2024, 7, 539. [Google Scholar] [CrossRef]
- Alnahdi, A.; John, A.; Raza, H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients 2019, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mao, M.; Zuo, Z. Palmitate Induces Mitochondrial Energy Metabolism Disorder and Cellular Damage via the PPAR Signaling Pathway in Diabetic Cardiomyopathy. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Raja, A.A.; Dandare, A.; Khan, M.J.; Khan, M.J. Free Fatty Acid Overload Targets Mitochondria: Gene Expression Analysis of Palmitic Acid-Treated Endothelial Cells. Genes 2022, 13, 1704. [Google Scholar] [CrossRef]
- Song, Y.; Wang, L.; Luo, Z.; Hogstrand, C.; Lai, X.-H.; Zheng, F. Moderate replacement of fish oil with palmitic acid-stimulated mitochondrial fusion promotes β-oxidation by Mfn2 interacting with Cpt1α via its GTPase-domain. J. Nutr. Biochem. 2024, 126, 109559. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.-M.; Xu, A.-M.; Dai, R.-P.; Zhou, Z.-G. Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef]
- Haffar, T.; Akoumi, A.; Bousette, N. Lipotoxic Palmitate Impairs the Rate of β-Oxidation and Citric Acid Cycle Flux in Rat Neonatal Cardiomyocytes. Cell. Physiol. Biochem. 2016, 40, 969–981. [Google Scholar] [CrossRef]
- Barrios-Maya, M.-A.; Ruiz-Ramírez, A.; Quezada, H.; Céspedes Acuña, C.L.; El-Hafidi, M. Palmitoyl-CoA effect on cytochrome c release, a key process of apoptosis, from liver mitochondria of rat with sucrose diet-induced obesity. Food Chem. Toxicol. 2021, 154, 112351. [Google Scholar] [CrossRef]
- Ma, X.M.; Geng, K.; Law, B.Y.-K.; Wang, P.; Pu, Y.L.; Chen, Q.; Xu, H.W.; Tan, X.Z.; Jiang, Z.Z.; Xu, Y. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol. Toxicol. 2023, 39, 277–299. [Google Scholar] [CrossRef]
- Heilig, R.; Lee, J.; Tait, S.W.G. Mitochondrial DNA in cell death and inflammation. Biochem. Soc. Trans. 2023, 51, 457–472. [Google Scholar] [CrossRef]
- Mansuri, M.L.; Sharma, G.; Parihar, P.; Dube, K.T.; Sharma, T.; Parihar, A.; Parihar, M.S. Increased oxidative stress and mitochondrial impairments associated with increased expression of TNF-α and caspase-3 in palmitic acid-induced lipotoxicity in myoblasts. J. Biochem. Mol. Toxicol. 2021, 35, e22744. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Lee, A.Y.; Park, S.; Kim, J.-H.; Cho, M.-H. Multiple pathways are involved in palmitic acid-induced toxicity. Food Chem. Toxicol. 2014, 67, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramírez, A.; Barrios-Maya, M.-A.; López-Acosta, O.; Molina-Ortiz, D.; El-Hafidi, M. Cytochrome c release from rat liver mitochondria is compromised by increased saturated cardiolipin species induced by sucrose feeding. Am. J. Physiol.-Endocrinol. Metab. 2015, 309, E777–E786. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, K.E.; Abriata, L.A.; Meireles, F.T.P.; Moss, F.R.; Frost, A.; Dal Peraro, M.; Aydin, H. Cardiolipin clustering promotes mitochondrial membrane dynamics. bioRxiv 2024. [Google Scholar] [CrossRef]
- Oemer, G.; Edenhofer, M.-L.; Lackner, K.; Leman, G.; Koch, J.; Lindner, H.H.; Dubrac, S.; Zschocke, J.; Keller, M.A. Fatty Acyl Availability Modulates Cardiolipin Composition and Alters Mitochondrial Function in HeLa Cells. J. Lipid Res. 2020, 62, 100111. [Google Scholar] [CrossRef]
- El-Hafidi, M.; Correa, F.; Zazueta, C. Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165744. [Google Scholar] [CrossRef]
- Venkatraman, K.; Budin, I. Cardiolipin remodeling maintains the inner mitochondrial membrane in cells with saturated lipidomes. J. Lipid Res. 2024, 65, 100601. [Google Scholar] [CrossRef]
- Sapandowski, A.; Stope, M.; Evert, K.; Evert, M.; Zimmermann, U.; Peter, D.; Päge, I.; Burchardt, M.; Schild, L. Cardiolipin composition correlates with prostate cancer cell proliferation. Mol. Cell. Biochem. 2015, 410, 175–185. [Google Scholar] [CrossRef]
- Bi, C.; Zhang, T.; Li, Y.; Zhao, H.; Zhang, P.; Wang, Y.; Xu, Y.; Gu, K.; Liu, Y.; Yu, J.; et al. A Proteomics- and Metabolomics-Based Study Revealed That Disorder of Palmitic Acid Metabolism by Aconitine Induces Cardiac Injury. Chem. Res. Toxicol. 2020, 33, 3031–3040. [Google Scholar] [CrossRef]
- Yang, L.; Guan, G.; Lei, L.; Liu, J.; Cao, L.; Wang, X. Oxidative and endoplasmic reticulum stresses are involved in palmitic acid-induced H9c2 cell apoptosis. Biosci. Rep. 2019, 39, BSR20190225. [Google Scholar] [CrossRef]
- Shilovsky, G.A.; Putyatina, T.S.; Ashapkin, V.V.; Yamskova, O.V.; Lyubetsky, V.A.; Sorokina, E.V.; Shram, S.I.; Markov, A.V.; Vyssokikh, M.Y. Biological Diversity and Remodeling of Cardiolipin in Oxidative Stress and Age-Related Pathologies. Biochem. 2019, 84, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, R.H.; Leandro, C.G.; Vinolo, M.A.; Nachbar, R.T.; dos Reis Silveira, L.; Hirabara, S.M.; Curi, R.; Pithon-Curi, T.C. The Effects of Palmitic Acid on Nitric Oxide Production by Rat Skeletal Muscle: Mechanism via Superoxide and iNOS Activation. Cell. Physiol. Biochem. 2012, 30, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Shimazaki, S.; Kaneko, Y.; Karasawa, T.; Takahashi, M.; Ohkuchi, A.; Takahashi, H.; Kurosawa, A.; Torii, Y.; Iwata, H.; et al. Palmitic acid activates NLRP3 inflammasome and induces placental inflammation during pregnancy in mice. J. Reprod. Dev. 2020, 66, 241–248. [Google Scholar] [CrossRef]
- Liu, B.; Deng, X.; Jiang, Q.; Li, G.; Zhang, J.; Zhang, N.; Xin, S.; Xu, K. Scoparone improves hepatic inflammation and autophagy in mice with nonalcoholic steatohepatitis by regulating the ROS/P38/Nrf2 axis and PI3K/AKT/mTOR pathway in macrophages. Biomed. Pharmacother. 2020, 125, 109895. [Google Scholar] [CrossRef]
- Amine, H.; Benomar, Y.; Taouis, M. Palmitic acid promotes resistin-induced insulin resistance and inflammation in SH-SY5Y human neuroblastoma. Sci. Rep. 2021, 11, 5427. [Google Scholar] [CrossRef]
- Sergi, D.; Luscombe-Marsh, N.; Naumovski, N.; Abeywardena, M.; O’Callaghan, N. Palmitic Acid, but Not Lauric Acid, Induces Metabolic Inflammation, Mitochondrial Fragmentation, and a Drop in Mitochondrial Membrane Potential in Human Primary Myotubes. Front. Nutr. 2021, 8, 663838. [Google Scholar] [CrossRef]
- Tanwar, V.S.; Reddy, M.A.; Das, S.; Samara, V.A.; Abdollahi, M.; Dey, S.; Malek, V.; Ganguly, R.; Stapleton, K.; Lanting, L.; et al. Palmitic Acid–Induced Long Noncoding RNA PARAIL Regulates Inflammation via Interaction With RNA-Binding Protein ELAVL1 in Monocytes and Macrophages. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1157–1175. [Google Scholar] [CrossRef]
- Al Mahri, S.; Malik, S.S.; Al Ibrahim, M.; Haji, E.; Dairi, G.; Mohammad, S. Free Fatty Acid Receptors (FFARs) in Adipose: Physiological Role and Therapeutic Outlook. Cells 2022, 11, 750. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Latour, M.G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T.L.; Luo, J.; Lin, D.C.-H.; Poitout, V. GPR40 Is Necessary but Not Sufficient for Fatty Acid Stimulation of Insulin Secretion In Vivo. Diabetes 2007, 56, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, H.; Shachar, S.; Sekler, I.; Hershfinkel, M.; Walker, M.D. Role of GPR40 in fatty acid action on the β cell line INS-1E. Biochem. Biophys. Res. Commun. 2005, 335, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, K.; Yabuki, C.; Maruyama, M.; Abiru, A.; Komatsu, H.; Negoro, N.; Tsujihata, Y.; Takeuchi, K.; Habata, Y.; Mori, M. Fasiglifam (TAK-875) has dual potentiating mechanisms via G α q-GPR40/FFAR1 signaling branches on glucose-dependent insulin secretion. Pharmacol. Res. Perspect. 2016, 4, e00237. [Google Scholar] [CrossRef]
- Trexler, A.J.; Taraska, J.W. Regulation of insulin exocytosis by calcium-dependent protein kinase C in beta cells. Cell Calcium 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Kim, H.-S.; Hwang, Y.-C.; Koo, S.-H.; Park, K.S.; Lee, M.-S.; Kim, K.-W.; Lee, M.-K. PPAR-γ Activation Increases Insulin Secretion through the Up-regulation of the Free Fatty Acid Receptor GPR40 in Pancreatic β-Cells. PLoS ONE 2013, 8, e50128. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, H.; Bergsten, P.; Sargsyan, E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 3248–3257. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, H.; Smith, D.M.; Bergsten, P.; Sargsyan, E. FFAR1 Is Involved in Both the Acute and Chronic Effects of Palmitate on Insulin Secretion. Endocrinology 2013, 154, 4078–4088. [Google Scholar] [CrossRef]
- Marafie, S.K.; Al-Shawaf, E.M.; Abubaker, J.; Arefanian, H. Palmitic acid-induced lipotoxicity promotes a novel interplay between Akt-mTOR, IRS-1, and FFAR1 signaling in pancreatic β-cells. Biol. Res. 2019, 52, 44. [Google Scholar] [CrossRef]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The Orphan G Protein-coupled Receptor GPR40 Is Activated by Medium and Long Chain Fatty Acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Song, Q.; Griffiths, A.; Song, Z. mTORC1-IRE1α pathway activation contributes to palmitate-elicited triglyceride secretion and cell death in hepatocytes. Exp. Biol. Med. 2020, 245, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Gosis, B.S.; Wada, S.; Thorsheim, C.; Li, K.; Jung, S.; Rhoades, J.H.; Yang, Y.; Brandimarto, J.; Li, L.; Uehara, K.; et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 2022, 376, eabf8271. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Pavlíková, Z.; Kučera, T.; Kozlík, P.; Šopin, T.; Vacík, T.; Ľupták, M.; Duda, M.; Slanař, O.; Kutinová Canová, N. Pharmacological effects of mTORC1/C2 inhibitor in a preclinical model of NASH progression. Biomed. Pharmacother. 2023, 167, 115447. [Google Scholar] [CrossRef]
- Gehrmann, W.; Elsner, M.; Lenzen, S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic β -cells. Diabetes Obes. Metab. 2010, 12, 149–158. [Google Scholar] [CrossRef]
- Graciano, M.F.; Valle, M.M.; Curi, R.; Carpinelli, A.R. Evidence for the involvement of GPR40 and NADPH oxidase in palmitic acid-induced superoxide production and insulin secretion. Islets 2013, 5, 139–148. [Google Scholar] [CrossRef]
- Leifke, E.; Naik, H.; Wu, J.; Viswanathan, P.; DeManno, D.; Kipnes, M.; Vakilynejad, M. A multiple-ascending-dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK-875, in subjects with type 2 diabetes. Clin. Pharmacol. Ther. 2012, 92, 29–39. [Google Scholar] [CrossRef]
- Araki, T.; Hirayama, M.; Hiroi, S.; Kaku, K. GPR40-induced insulin secretion by the novel agonist TAK-875: First clinical findings in patients with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 271–278. [Google Scholar] [CrossRef]
- Dragano, N.R.V.; Milbank, E.; Haddad-Tóvolli, R.; Garrido-Gil, P.; Nóvoa, E.; Fondevilla, M.F.; Capelli, V.; Zanesco, A.M.; Solon, C.; Morari, J.; et al. Hypothalamic free fatty acid receptor-1 regulates whole-body energy balance. Mol. Metab. 2024, 79, 101840. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chen, H.-W.; Lo, C.-W.; Wang, Y.-R.; Li, C.-C.; Liu, K.-L.; Lii, C.-K. Luteolin ameliorates palmitate-induced lipotoxicity in hepatocytes by mediating endoplasmic reticulum stress and autophagy. Food Chem. Toxicol. 2023, 171, 113554. [Google Scholar] [CrossRef]
- Kim, C.; Joo, S.; Kim, I.; Choi, H.-I.; Bae, E.; Kim, S.; Ma, S. Anti-Apoptotic Effect of G-Protein-Coupled Receptor 40 Activation on Tumor Necrosis Factor-α-Induced Injury of Rat Proximal Tubular Cells. Int. J. Mol. Sci. 2019, 20, 3386. [Google Scholar] [CrossRef]
- Shen, X.; Yang, L.; Yan, S.; Wei, W.; Liang, L.; Zheng, H.; Cai, X. The effect of FFAR1 on pioglitazone-mediated attenuation of palmitic acid-induced oxidative stress and apoptosis in βTC6 cells. Metabolism 2014, 63, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Y.; Feng, D.D.; Wang, D.; Sun, J.; Li, S.; Chen, C. Long-term in vitro treatment of INS -1 rat pancreatic β-cells by unsaturated free fatty acids protects cells against gluco- and lipotoxicities via activation of GPR 40 receptors. Clin. Exp. Pharmacol. Physiol. 2012, 39, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yang, L.; Shen, X. The relationship between GPR40 and lipotoxicity of the pancreatic β-cells as well as the effect of pioglitazone. Biochem. Biophys. Res. Commun. 2010, 403, 36–39. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, T.; Zhang, D.; Leung, P.S. GPR120 protects lipotoxicity-induced pancreatic β-cell dysfunction through regulation of PDX1 expression and inhibition of islet inflammation. Clin. Sci. 2019, 133, 101–116. [Google Scholar] [CrossRef]
- Martínez-García, C.; Izquierdo-Lahuerta, A.; Vivas, Y.; Velasco, I.; Yeo, T.-K.; Chen, S.; Medina-Gomez, G. Renal Lipotoxicity-Associated Inflammation and Insulin Resistance Affects Actin Cytoskeleton Organization in Podocytes. PLoS ONE 2015, 10, e0142291. [Google Scholar] [CrossRef]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Alen, R.; Pereira, L.; Povo-Retana, A.; Astudillo, A.M.; Hitos, A.B.; Gomez-Hurtado, I.; Lopez-Collazo, E.; Boscá, L.; Francés, R.; et al. Saturated fatty acid-enriched small extracellular vesicles mediate a crosstalk inducing liver inflammation and hepatocyte insulin resistance. JHEP Rep. 2023, 5, 100756. [Google Scholar] [CrossRef]
- Ferrara, P.J.; Rong, X.; Maschek, J.A.; Verkerke, A.R.P.; Siripoksup, P.; Song, H.; Green, T.D.; Krishnan, K.C.; Johnson, J.M.; Turk, J.; et al. Lysophospholipid acylation modulates plasma membrane lipid organization and insulin sensitivity in skeletal muscle. J. Clin. Investig. 2021, 131, e135963. [Google Scholar] [CrossRef]
- Tokarz, V.L.; Mylvaganam, S.; Klip, A. Palmitate-induced insulin resistance causes actin filament stiffness and GLUT4 mis-sorting without altered Akt signalling. J. Cell Sci. 2023, 136, jcs261300. [Google Scholar] [CrossRef]
- Den Hartogh, D.J.; Vlavcheski, F.; Tsiani, E. Muscle Cell Insulin Resistance is Attenuated by Rosmarinic Acid: Elucidating the Mechanisms Involved. Int. J. Mol. Sci. 2023, 24, 5094. [Google Scholar] [CrossRef]
- Tang, C.; Wang, Y.; Xu, Z.; Chen, D.; Xu, J.; Yang, D.; Zhang, L.; Liu, J.; Kan, J. The relationships between high-fat diet and metabolic syndrome: Potential mechanisms. Food Biosci. 2024, 59, 104261. [Google Scholar] [CrossRef]
- Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; Ortis, F.; Carpinelli, A.R. Lipotoxicity and β-cell failure in type 2 diabetes: Oxidative stress linked to NADPH oxidase and ER stress. Cells 2021, 10, 3328. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Adak, S.; Spyropoulos, G.; Zhang, Q.; Feng, C.; Yin, L.; Speck, S.L.; Shyr, Z.; Morikawa, S.; Kitamura, R.A.; et al. Palmitoylation couples insulin hypersecretion with β cell failure in diabetes. Cell Metab. 2023, 35, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier, J.; Andersson, D.C.; Zhang, S.-J.; Lanner, J.T.; Wibom, R.; Katz, A.; Bruton, J.D.; Westerblad, H. Effects of Palmitate on Ca2+ Handling in Adult Control and ob/ob Cardiomyocytes. Diabetes 2007, 56, 1136–1142. [Google Scholar] [CrossRef]
- Ren, G.; Bhatnagar, S.; Hahn, D.J.; Kim, J. Long-chain acyl-CoA synthetase-1 mediates the palmitic acid-induced inflammatory response in human aortic endothelial cells. Am. J. Physiol. Metab. 2020, 319, E893–E903. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 2017, 8, 13997. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, L.; Liu, J.; Ma, Y.; Qiu, C.; Liu, C.; Gong, Y.; Yuwen, Y.; Guan, G.; Zhang, Y.; et al. Palmitic acid in type 2 diabetes mellitus promotes atherosclerotic plaque vulnerability via macrophage Dll4 signaling. Nat. Commun. 2024, 15, 1281. [Google Scholar] [CrossRef]
- Trombetta, A.; Togliatto, G.; Rosso, A.; Dentelli, P.; Olgasi, C.; Cotogni, P.; Brizzi, M.F. Increase of Palmitic Acid Concentration Impairs Endothelial Progenitor Cell and Bone Marrow–Derived Progenitor Cell Bioavailability. Diabetes 2013, 62, 1245–1257. [Google Scholar] [CrossRef]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef]
- Longhitano, L.; Distefano, A.; Amorini, A.M.; Orlando, L.; Giallongo, S.; Tibullo, D.; Lazzarino, G.; Nicolosi, A.; Alanazi, A.M.; Saoca, C.; et al. (+)-Lipoic Acid Reduces Lipotoxicity and Regulates Mitochondrial Homeostasis and Energy Balance in an In Vitro Model of Liver Steatosis. Int. J. Mol. Sci. 2023, 24, 14491. [Google Scholar] [CrossRef]
- Wong, K.-L.; Wu, Y.-R.; Cheng, K.-S.; Chan, P.; Cheung, C.-W.; Lu, D.-Y.; Su, T.-H.; Liu, Z.-M.; Leung, Y.-M. Palmitic acid-induced lipotoxicity and protection by (+)-catechin in rat cortical astrocytes. Pharmacol. Rep. 2014, 66, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wei, J.; Sheng, F.; Li, P. Attenuation of Palmitic Acid—Induced Lipotoxicity by Chlorogenic Acid through Activation of SIRT1 in Hepatocytes. Mol. Nutr. Food Res. 2019, 63, 1801432. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liu, L.; Zhao, Y.; Yang, L.; Cheng, J.; Hua, R.; Zhang, Z.; Li, Q. Melatonin protects mouse testes from palmitic acid-induced lipotoxicity by attenuating oxidative stress and DNA damage in a SIRT1-dependent manner. J. Pineal Res. 2020, 69, e12690. [Google Scholar] [CrossRef]
- Wu, Z.; Geng, Y.; Buist-Homan, M.; Moshage, H. Scopoletin and umbelliferone protect hepatocytes against palmitate- and bile acid-induced cell death by reducing endoplasmic reticulum stress and oxidative stress. Toxicol. Appl. Pharmacol. 2022, 436, 115858. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Li, Y.; Guan, Y.; Zhang, K.; Liu, J.; Fan, M.; Qian, H.; Wang, L. Highland barley tea represses palmitic acid-induced apoptosis and mitochondrial dysfunction via regulating AMPK/SIRT3/FoxO3a in myocytes. Food Biosci. 2021, 40, 100893. [Google Scholar] [CrossRef]
- Yoon, H.J.; Bang, M.-H.; Kim, H.; Imm, J.-Y. Improvement of palmitate-induced insulin resistance in C2C12 skeletal muscle cells using Platycodon grandiflorum seed extracts. Food Biosci. 2018, 25, 61–67. [Google Scholar] [CrossRef]
- Xia, M.; Wu, Z.; Wang, J.; Buist-Homan, M.; Moshage, H. The Coumarin-Derivative Esculetin Protects against Lipotoxicity in Primary Rat Hepatocytes via Attenuating JNK-Mediated Oxidative Stress and Attenuates Free Fatty Acid-Induced Lipid Accumulation. Antioxidants 2023, 12, 1922. [Google Scholar] [CrossRef]
- Kang, B.-B.; Chiang, B.-H. EGCG regulation of non-insulin-responsive endosomal compartments in insulin-resistant skeletal muscle. Food Biosci. 2019, 28, 689. [Google Scholar] [CrossRef]
- Pan, X.; Liu, C.; Wang, X.; Zhao, M.; Zhang, Z.; Zhang, X.; Wang, C.; Song, G. Resveratrol improves palmitic acid-induced insulin resistance via the DDIT4/mTOR pathway in C2C12 cells. Mol. Med. Rep. 2023, 28, 181. [Google Scholar] [CrossRef]
- Tong, T.; Ren, N.; Soomi, P.; Wu, J.; Guo, N.; Kang, H.; Kim, E.; Wu, Y.; He, P.; Tu, Y.; et al. Theaflavins Improve Insulin Sensitivity through Regulating Mitochondrial Biosynthesis in Palmitic Acid-Induced HepG2 Cells. Molecules 2018, 23, 3382. [Google Scholar] [CrossRef]
- Turpaev, K.T. Keap1-Nrf2 signaling pathway: Mechanisms of regulation and role in protection of cells against toxicity caused by xenobiotics and electrophiles. Biochemistry 2013, 78, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Majewska, M.; Leß, B.; Mehmeti, I.; Wollnitzke, P.; Semleit, N.; Levkau, B.; Saba, J.D.; van Echten-Deckert, G.; Gurgul-Convey, E. The fate of intracellular S1P regulates lipid droplet turnover and lipotoxicity in pancreatic beta-cells. J. Lipid Res. 2024, 65, 100587. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar] [PubMed]
- Cnop, M.; Hannaert, J.C.; Hoorens, A.; Eizirik, D.L.; Pipeleers, D.G. Inverse Relationship Between Cytotoxicity of Free Fatty Acids in Pancreatic Islet Cells and Cellular Triglyceride Accumulation. Diabetes 2001, 50, 1771–1777. [Google Scholar] [CrossRef]
- Obaseki, E.; Adebayo, D.; Bandyopadhyay, S.; Hariri, H. Lipid droplets and fatty acid-induced lipotoxicity: In a nutshell. FEBS Lett. 2024, 598, 1207–1214. [Google Scholar] [CrossRef]
- Lin, P.; Zhou, D.H. Subcellular organelles: Lipid droplets and the multifunctional roles. In The Molecular Nutrition of Fats; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- de Souza, C.O.; Vannice, G.K.; Rosa Neto, J.C.; Calder, P.C. Is Palmitoleic Acid a Plausible Nonpharmacological Strategy to Prevent or Control Chronic Metabolic and Inflammatory Disorders? Mol. Nutr. Food Res. 2018, 62, 1700504. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Li, N.; Sancak, Y.; Frasor, J.; Atilla-Gokcumen, G.E. A Protective Role for Triacylglycerols during Apoptosis. Biochemistry 2018, 57, 72–80. [Google Scholar] [CrossRef]
- Cabodevilla, A.G.; Son, N.; Goldberg, I.J. Intracellular lipase and regulation of the lipid droplet. Curr. Opin. Lipidol. 2024, 35, 85–92. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [PubMed]
- Drizyte-Miller, K.; Schott, M.B.; McNiven, M.A. Lipid Droplet Contacts With Autophagosomes, Lysosomes, and Other Degradative Vesicles. Contact 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Danielli, M.; Perne, L.; Jarc Jovičić, E.; Petan, T. Lipid droplets and polyunsaturated fatty acid trafficking: Balancing life and death. Front. Cell Dev. Biol. 2023, 11, 1104725. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shrestha, R.; Yang, X.; Wu, X.; Jia, J.; Chiba, H.; Hui, S.-P. Oxidative Stress and Lipid Dysregulation in Lipid Droplets: A Connection to Chronic Kidney Disease Revealed in Human Kidney Cells. Antioxidants 2022, 11, 1387. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Dibwe, D.F.; Kitayama, E.; Oba, S.; Takeishi, N.; Chiba, H.; Hui, S.-P. Inhibition of Lipid Accumulation and Oxidation in Hepatocytes by Bioactive Bean Extracts. Antioxidants 2024, 13, 513. [Google Scholar] [CrossRef]
- Granieri, M.C.; Rocca, C.; De Bartolo, A.; Nettore, I.C.; Rago, V.; Romeo, N.; Ceramella, J.; Mariconda, A.; Macchia, P.E.; Ungaro, P.; et al. Quercetin and Its Derivative Counteract Palmitate-Dependent Lipotoxicity by Inhibiting Oxidative Stress and Inflammation in Cardiomyocytes. Int. J. Environ. Res. Public Health 2023, 20, 3492. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhang, T.; Liang, Y.; Zhang, J. Baicalein ameliorates lipid accumulation in HepG2 cells via the pregnane X receptor signaling pathway. Food Biosci. 2024, 60, 104482. [Google Scholar] [CrossRef]
- Chen, Q.; Tang, L.; Xin, G.; Li, S.; Ma, L.; Xu, Y.; Zhuang, M.; Xiong, Q.; Wei, Z.; Xing, Z.; et al. Oxidative stress mediated by lipid metabolism contributes to high glucose-induced senescence in retinal pigment epithelium. Free Radic. Biol. Med. 2019, 130, 48–58. [Google Scholar] [CrossRef]
- Zacharewicz, E.; Hesselink, M.K.C.; Schrauwen, P. Exercise counteracts lipotoxicity by improving lipid turnover and lipid droplet quality. J. Intern. Med. 2018, 284, 505–518. [Google Scholar] [CrossRef]
- Zhao, P.; Jin, Y.; Wu, X.; Huang, J.; Chen, L.; Tan, Y.; Yuan, H.; Wu, J.; Ren, Z. Artificial Lipid Droplets: Novel Effective Biomaterials to Protect Cells against Oxidative Stress and Lipotoxicity. Nanomaterials 2022, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Zadoorian, A.; Du, X.; Yang, H. Lipid droplet biogenesis and functions in health and disease. Nat. Rev. Endocrinol. 2023, 19, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Panov, A. Perhydroxyl Radical (HO2•) as Inducer of the Isoprostane Lipid Peroxidation in Mitochondria. Mol. Biol. 2018, 52, 295–305. [Google Scholar] [CrossRef]
- Panov, A.; Mayorov, V.I.; Dikalov, S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022, 23, 4047. [Google Scholar] [CrossRef] [PubMed]
- Rahmani-Manglano, N.E.; García-Moreno, P.J.; Pérez-Gálvez, R.; Guadix, E.M. Antioxidants location affects the oxidative stability of spray-dried microcapsules loaded with fish oil. Food Biosci. 2023, 56, 103074. [Google Scholar] [CrossRef]
- Xi, B.-N.; Zhang, J.-J.; Li, C.; Xu, X.; Zeng, Q.; Zhang, Y.; Chen, B.; Shen, Y. Effects of natural and synthetic antioxidants addition on the characteristic flavor and metabolites of walnut oil during oxidation. Food Biosci. 2024, 61, 104788. [Google Scholar] [CrossRef]
- Nyanhongo, G.S.; Sygmund, C.; Ludwig, R.; Prasetyo, E.N.; Guebitz, G.M. An antioxidant regenerating system for continuous quenching of free radicals in chronic wounds. Eur. J. Pharm. Biopharm. 2013, 83, 396–404. [Google Scholar] [CrossRef]
- Bayram, I.; Decker, E.A. Underlying mechanisms of synergistic antioxidant interactions during lipid oxidation. Trends Food Sci. Technol. 2023, 133, 219–230. [Google Scholar] [CrossRef]
- Pérez, M.; Dominguez-López, I.; Lamuela-Raventós, R.M. The Chemistry Behind the Folin—Ciocalteu Method for the Estimation of (Poly)phenol Content in Food: Total Phenolic Intake in a Mediterranean Dietary Pattern. J. Agric. Food Chem. 2023, 71, 17543–17553. [Google Scholar] [CrossRef]
- Platzer, M.; Kiese, S.; Tybussek, T.; Herfellner, T.; Schneider, F.; Schweiggert-Weisz, U.; Eisner, P. Radical Scavenging Mechanisms of Phenolic Compounds: A Quantitative Structure-Property Relationship (QSPR) Study. Front. Nutr. 2022, 9, 882458. [Google Scholar] [CrossRef]
- Fernández Sosa, E.I.; Ehman, N.; Felissia, F.E.; Chaves, M.G.; Area, M.C. Progress and potentialities in wood extractives-based materials for active food packaging applications. Food Biosci. 2024, 60, 104489. [Google Scholar] [CrossRef]
- da Costa Monção, É.; Grisi, C.V.B.; de Moura Fernandes, J.; Souza, P.S.; de Souza, A.L. Active packaging for lipid foods and development challenges for marketing. Food Biosci. 2022, 45, 101370. [Google Scholar] [CrossRef]
- Liu, R.; Mabury, S.A. Synthetic Phenolic Antioxidants: A Review of Environmental Occurrence, Fate, Human Exposure, and Toxicity. Environ. Sci. Technol. 2020, 54, 11706–11719. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, A.; Hu, S.; Ares, I.; Martínez-Larrañaga, M.-R.; Wang, X.; Martínez, M.; Anadón, A.; Martínez, M.-A. Synthetic phenolic antioxidants: Metabolism, hazards and mechanism of action. Food Chem. 2021, 353, 129488. [Google Scholar] [CrossRef]
- Chong, Y.M.; Chang, S.K.; Sia, W.C.M.; Yim, H.S. Antioxidant efficacy of mangosteen (Garcinia mangostana Linn.) peel extracts in sunflower oil during accelerated storage. Food Biosci. 2015, 12, 18–25. [Google Scholar] [CrossRef]
- Shahid, M.S.; Zhou, S.; Nie, W.; Wang, L.; Lv, H.; Yuan, J. Phytogenic Antioxidants Prolong n-3 Fatty Acid-Enriched Eggs’ Shelf Life by Activating the Nrf-2 Pathway through Phosphorylation of MAPK. Foods 2022, 11, 3158. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Perez, T.I.; Zuidhof, M.J.; Renema, R.A.; Wu, J. Oxidative Stability of Omega-3 Polyunsaturated Fatty Acids Enriched Eggs. J. Agric. Food Chem. 2013, 61, 11595–11602. [Google Scholar] [CrossRef] [PubMed]
- Larsson, K.; Harrysson, H.; Havenaar, R.; Alminger, M.; Undeland, I. Formation of malondialdehyde (MDA), 4-hydroxy-2-hexenal (HHE) and 4-hydroxy-2-nonenal (HNE) in fish and fish oil during dynamic gastrointestinal in vitro digestion. Food Funct. 2016, 7, 1176–1187. [Google Scholar] [CrossRef]
- Guichardant, M.; Bacot, S.; Molière, P.; Lagarde, M. Hydroxy-alkenals from the peroxidation of n-3 and n-6 fatty acids and urinary metabolites. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 179–182. [Google Scholar] [CrossRef]
- Feng, R.; Zhang, H.; Ding, N.; Ma, H.; Luo, Y.; Tan, Y.; Wangtueai, S.; Hong, H. Effect of lipoxygenase-catalyzed linoleic acid oxidation and 4-hydroxy-2-nonenal on digestibility and gel properties of myofibrillar protein. Food Biosci. 2024, 61, 104882. [Google Scholar] [CrossRef]
- Moumtaz, S.; Percival, B.C.; Parmar, D.; Grootveld, K.L.; Jansson, P.; Grootveld, M. Toxic aldehyde generation in and food uptake from culinary oils during frying practices: Peroxidative resistance of a monounsaturate-rich algae oil. Sci. Rep. 2019, 9, 4125. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Richards, M.P.; Undeland, I. Lipid oxidation and antioxidant delivery systems in muscle food. Compr. Rev. Food Sci. Food Saf. 2022, 21, 1275–1299. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceja-Galicia, Z.A.; Cespedes-Acuña, C.L.A.; El-Hafidi, M. Protection Strategies Against Palmitic Acid-Induced Lipotoxicity in Metabolic Syndrome and Related Diseases. Int. J. Mol. Sci. 2025, 26, 788. https://doi.org/10.3390/ijms26020788
Ceja-Galicia ZA, Cespedes-Acuña CLA, El-Hafidi M. Protection Strategies Against Palmitic Acid-Induced Lipotoxicity in Metabolic Syndrome and Related Diseases. International Journal of Molecular Sciences. 2025; 26(2):788. https://doi.org/10.3390/ijms26020788
Chicago/Turabian StyleCeja-Galicia, Zeltzin Alejandra, Carlos Leonardo Armando Cespedes-Acuña, and Mohammed El-Hafidi. 2025. "Protection Strategies Against Palmitic Acid-Induced Lipotoxicity in Metabolic Syndrome and Related Diseases" International Journal of Molecular Sciences 26, no. 2: 788. https://doi.org/10.3390/ijms26020788
APA StyleCeja-Galicia, Z. A., Cespedes-Acuña, C. L. A., & El-Hafidi, M. (2025). Protection Strategies Against Palmitic Acid-Induced Lipotoxicity in Metabolic Syndrome and Related Diseases. International Journal of Molecular Sciences, 26(2), 788. https://doi.org/10.3390/ijms26020788