


Nicking Activity of M13 Bacteriophage Protein 2


Abstract
1. Introduction
2. Results
2.1. Cloning of P2 Protein Gene and Protein Expression
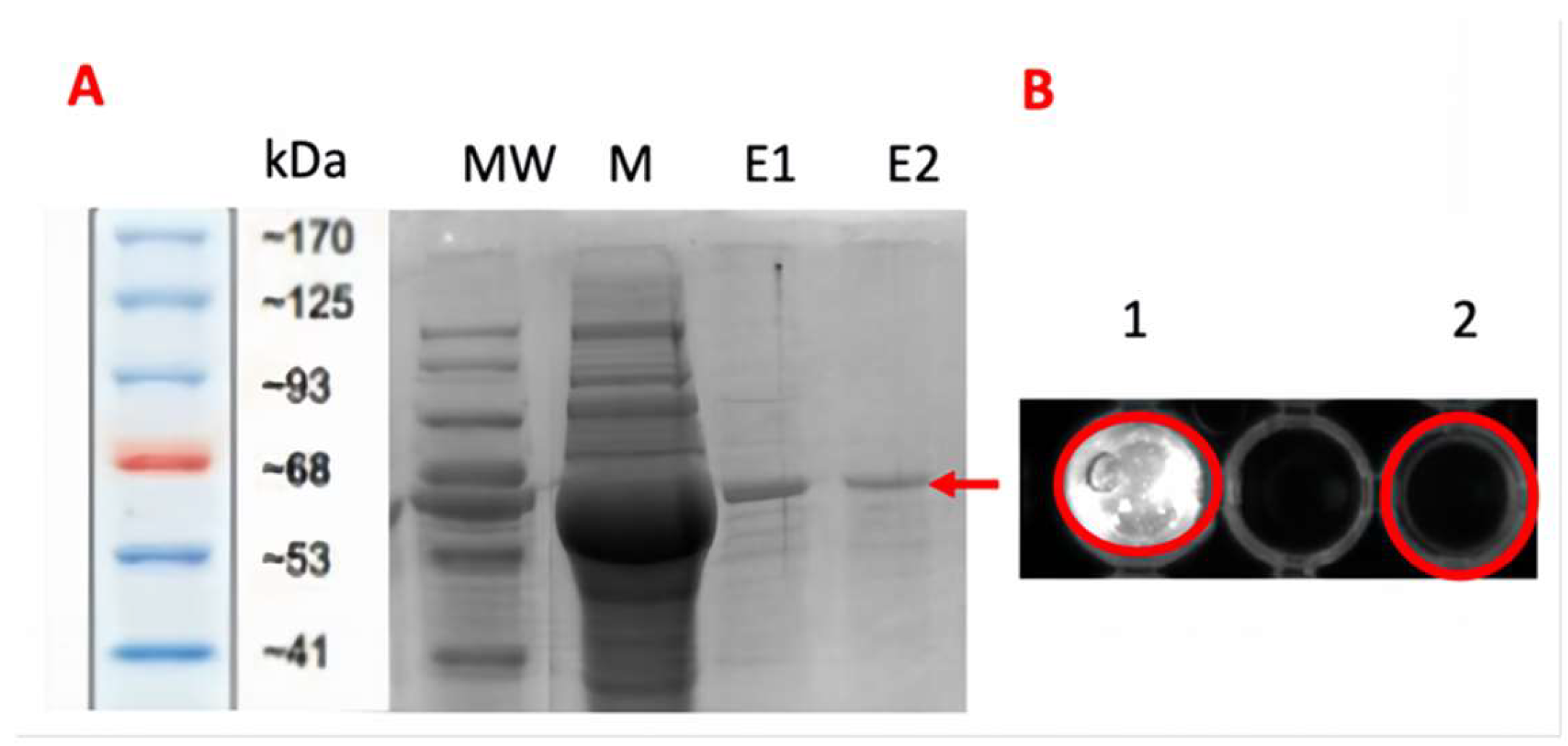
2.2. Protein Purification and Validation
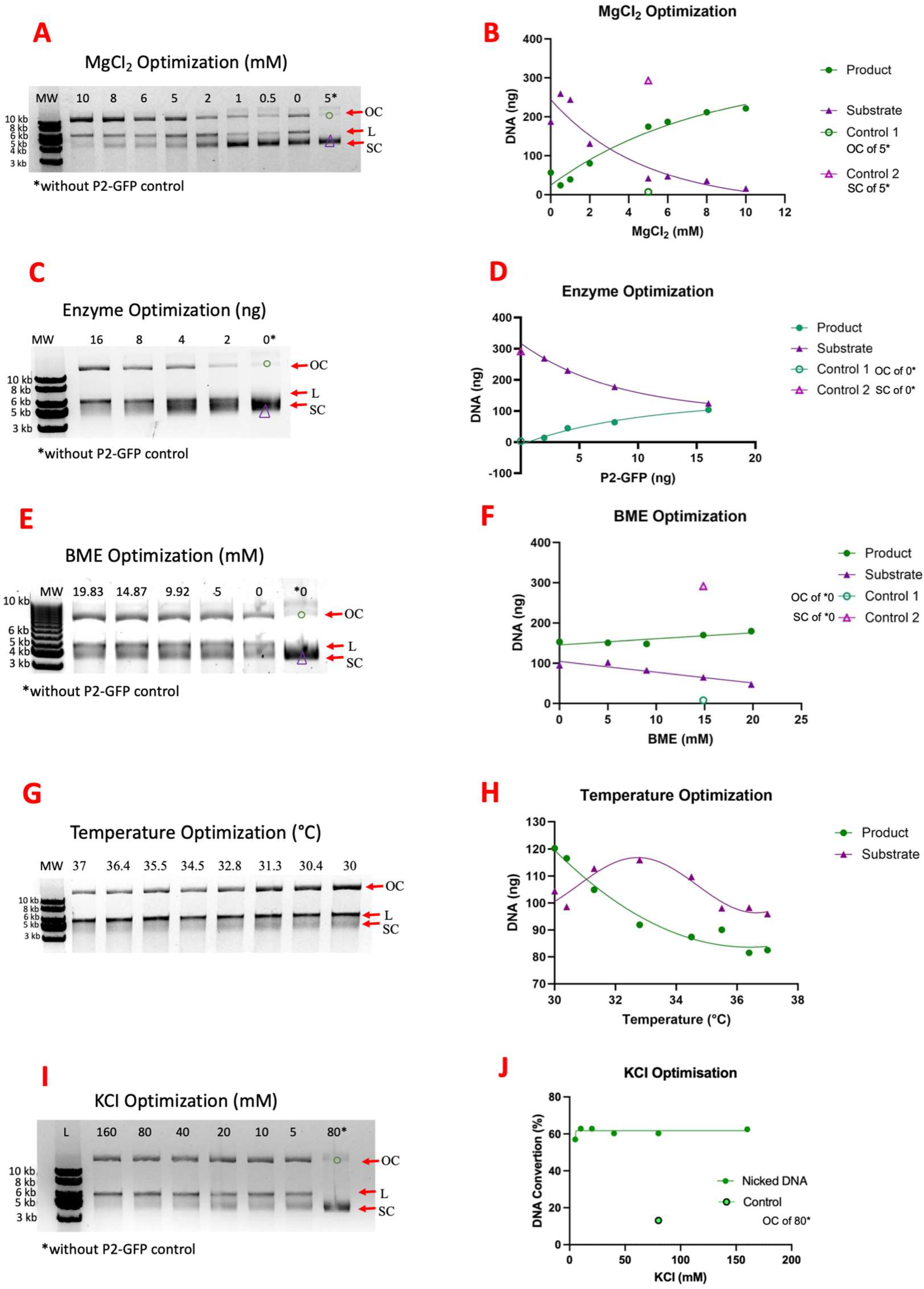
2.3. P2 Activity Test
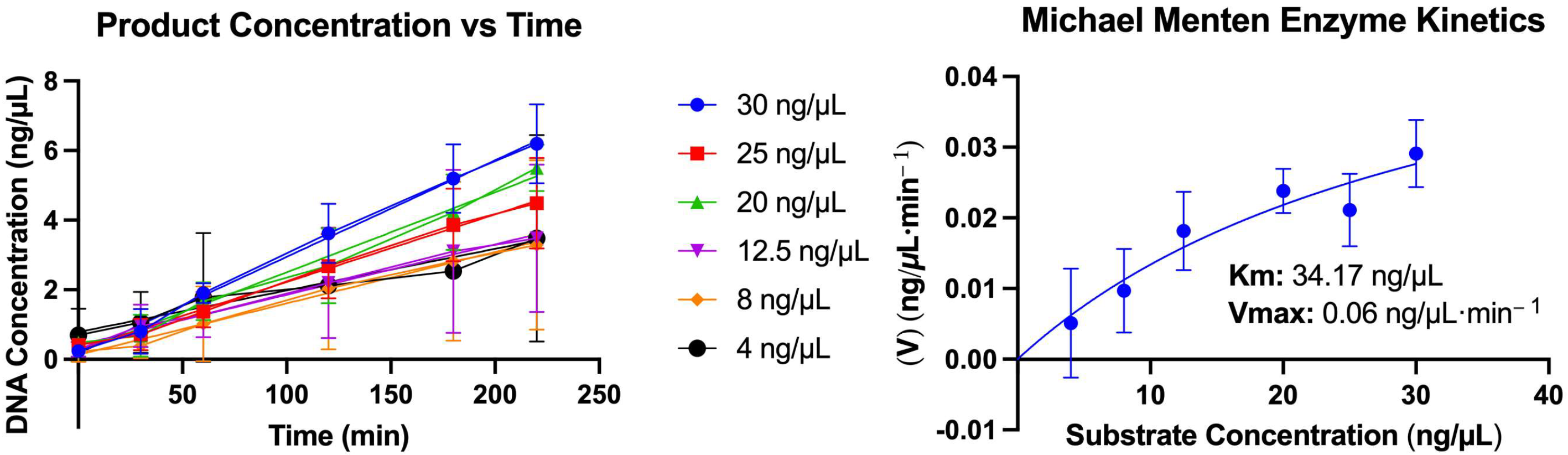
2.4. Enzyme Kinetics
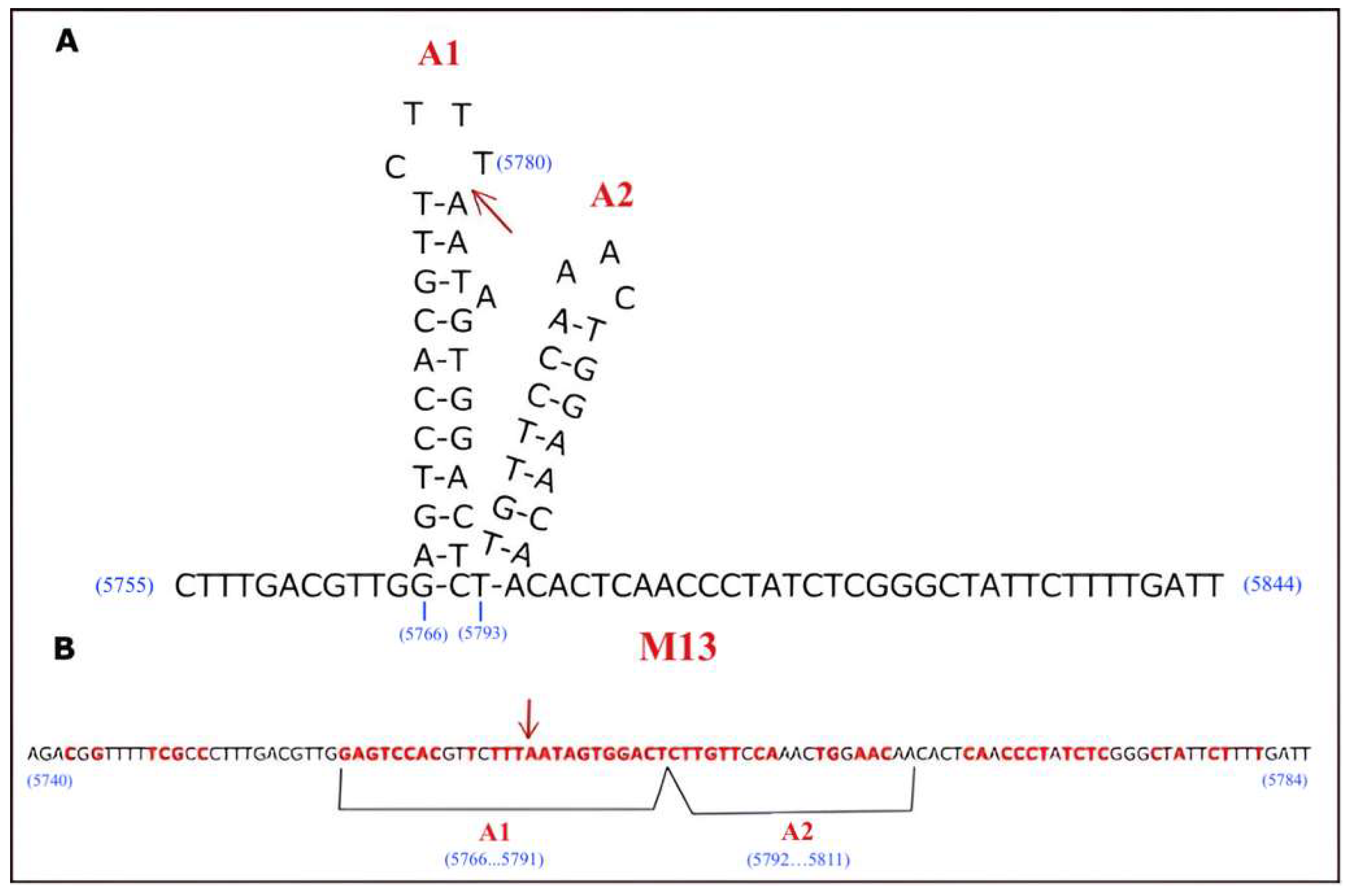
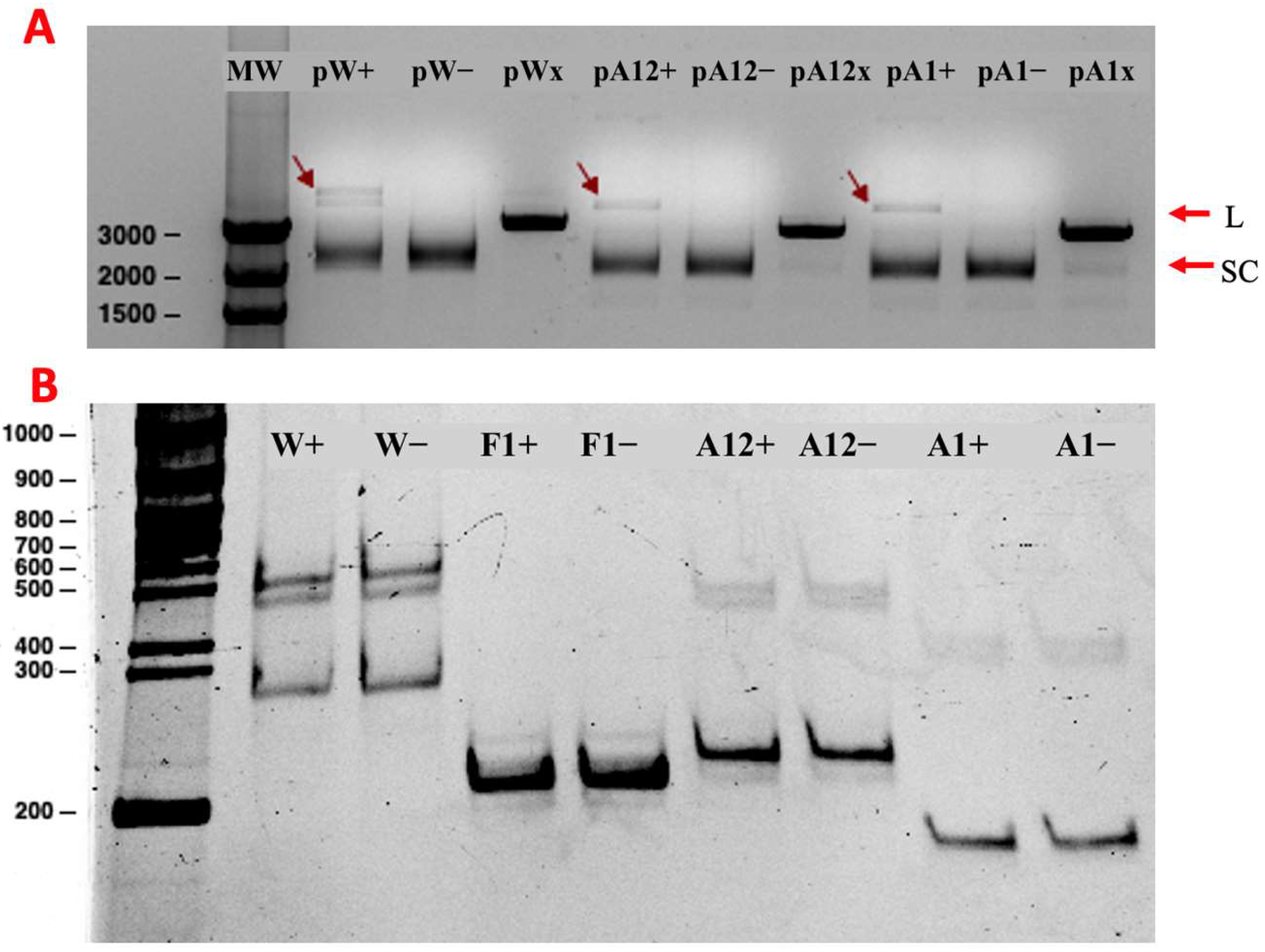
2.5. Determination of Minimal Nicking Site
3. Discussion
4. Materials and Methods
4.1. Cloning of the P2 Gene
4.2. Protein Expression
4.3. Solubilization of Inclusion Bodies
4.4. Protein Purification
4.5. Analysis of Purified P2-GFP
4.6. P2-GFP Activity Test
4.7. Determination of Minimal Recognition Sequence for P2 Nicking
4.7.1. Cloning of PCR Products Containing Different Segments of the P2 Nicking Site
4.7.2. Nicking Reactions of Different Segments of the P2 Recognition Sequence
4.8. Data Analysis
4.9. Enzyme Kinetics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suggs, S.V.; Ray, D.S. Nucleotide Sequence of the Origin for Bacteriophage M13 DNA Replication. Cold Spring Harb. Symp. Quant. Biol. 1979, 43, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.F.; Russell, D. M13 Bacteriophages. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Smeal, S.W.; Schmitt, M.A.; Pereira, R.R.; Prasad, A.; Fisk, J.D. Simulation of the M13 Life Cycle II: Investigation of the Control Mechanisms of M13 Infection and Establishment of the Carrier State. Virology 2017, 500, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Brasino, M. Engineering of Filamentous Bacteriophage for Protein Sensing. Ph.D. Thesis, University of Colorado at Boulder, Boulder, CO, USA, 2016. [Google Scholar]
- Russel, M. Moving through the Membrane with Filamentous Phages. Trends Microbiol. 1995, 3, 223–228. [Google Scholar] [CrossRef]
- Dotto, G.; Horiuchi, K.; Zinder, N. The Functional Origin of Bacteriophage F1 DNA Replication. Its Signals and Domains. J. Mol. Biol. 1984, 172, 507–521. [Google Scholar] [CrossRef]
- Smeal, S.W.; Schmitt, M.A.; Rodrigues, R.; Prasad, A.; Fisk, J.D. Simulation of the M13 Life Cycle I: Assembly of a Genetically-Structured Deterministic Chemical Kinetic Simulation. Virology 2017, 500, 259–274. [Google Scholar] [CrossRef]
- Rakonjac, J.; Russel, M.; Khanum, S.; Brooke, S.J.; Raji, M. Filamentous Phage: Structure and Biology. In Recombinant Antibodies for Infectious Diseases. Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar] [CrossRef]
- Slonczewski, J.; Foster, J. Viral Molecular Biology. In Microbiology an Evolving Science; W. W. Norton and Norton Company, Inc.: New York, NY, USA, 2017. [Google Scholar]
- Dotto, G.P.; Enea, V.; Zinder, N.D. Gene II of Phage F1: Its Functions and Its Products. Proc. Natl. Acad. Sci. USA 1981, 78, 5421–5424. [Google Scholar] [CrossRef]
- Greenstein, D.; Horiuchi, K. Interaction between the Replication Origin and the Initiator Protein of the Filamentous Phage F1. Binding Occurs in Two Steps. J. Mol. Biol. 1987, 197, 157–174. [Google Scholar] [CrossRef]
- Geider, K.; Meyer, T. Gene-II Protein of Bacteriophage Fd in Enzymatic Replication of Viral Duplex DNA. Cold Spring Harb. Symp. Quant. Biol. 1979, 43 Pt 1, 59–62. [Google Scholar] [CrossRef]
- Morag, O.; Sgourakis, G.N.; Abramov, G.; Goldbourt, A. Filamentous Bacteriophage Viruses: Preparation, Magic-Angle Spinning Solid-State NMR Experiments, and Structure Determination. In Protein NMR; Ghose, R., Ed.; Humana Press: New York, NY, USA, 2018; pp. 67–97. ISBN 978-1-4939-7386-6. [Google Scholar]
- Azzazy, H.M.E.; Highsmith, W.E. Phage Display Technology: Clinical Applications and Recent Innovations. Clin. Biochem. 2002, 35, 425–445. [Google Scholar] [CrossRef]
- Harth, G.; Bäumel, I.; Meyer, T.F.; Geider, K. Bacteriophage Fd Gene-2 Protein: Processing of Phage Fd Viral Straands Replicated by Phage T7 Enzymes. Eur. J. Biochem 1981, 119, 663–668. [Google Scholar] [CrossRef]
- Meyer, T.; Geider, K.; Kurz, C.; Schaller, H. Cleavage Site of Bacteriophage Fd Gene II-Protein in the Origin of Viral Strand Replication. Nature 1979, 278, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Peeters, B.P.H.; Schoenmakers, J.G.G.; Konings, R.N.H. Comparison of the DNA Sequences Involved in Replication and Packaging of the Filamentous Phages IKe and Ff (M13, Fd, and Fl). DNA 1987, 6, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.D.; Jansz, H. Single-Stranded DNA Phage Origins. Curr. Top. Microbiol. Immunol. 1988, 136, 31–70. [Google Scholar] [CrossRef] [PubMed]
- Higashitani, N.; Higashitani, A.; Guan, Z.W.; Horiuchi, K. Recognition Mechanisms of the Minus-Strand Origin of Phage F1 by Escherichia Coli RNA Polymerase. Genes Cells 1996, 1, 829–841. [Google Scholar] [CrossRef]
- Horiuchi, K. Initiation Mechanisms in Replication of Filamentous Phage DNA. Genes Cells 1997, 2, 425–432. [Google Scholar] [CrossRef]
- Dennehy, J.J. Bacteriophages as Model Organisms for Virus Emergence Research. Trends Microbiol. 2009, 17, 450–457. [Google Scholar] [CrossRef]
- Geider, K.; Bäumel, I.; Meyer, T. Intermediate Stages in Enzymatic Replication of Bacteriophage Fd Duplex DNA. J. Biol. Chem. 1982, 257, 6488–6493. [Google Scholar] [CrossRef]
- Horiuchi, K.; Zinder, N. Origin and Direction of Synthesis of Bacteriophage Fl DNA. Proc. Natl. Acad. Sci. USA 1976, 73, 2341–2345. [Google Scholar] [CrossRef]
- Cleary, J.M.; Ray, D. Deletion Analysis of the Cloned Replication Origin Region from Bacteriophage M13. J. Virol. 1981, 40, 197–203. [Google Scholar] [CrossRef]
- Johnston, S.; Ray, D. Interference between M13 and OriM13 Plasmids Is Mediated by a Replication Enhancer Sequence near the Viral Strand Origin. J. Mol. Biol. 1984, 177 4, 685–700. [Google Scholar] [CrossRef]
- Meyer, T.F.; Geider, K. Bacteriophage Fd Gene II-Protein. II. Specific Cleavage and Relaxation of Supercoiled RF from Filamentous Phages. J. Biol. Chem. 1979, 254, 12642–12646. [Google Scholar] [CrossRef]
- Meyer, T.; Geider, K. Enzymatic Synthesis of Bacteriophage Fd Viral DNA. Nature 1982, 296, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Pratt, D. Bacteriophage M 13 Gene 2 Protein: Increasing Its Yield in Infected Cells, and Identification and Localization. Virology 1974, 61, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Schaller, H.; Uhlmann, A.; Geider, K. A DNA Fragment from the Origin of Single-Strand to Double-Strand DNA Replication of Bacteriophage Fd. Proc. Natl. Acad. Sci. USA 1976, 73, 49–53. [Google Scholar] [CrossRef]
- Dotto, G.; Enea, V.; Zinder, N. Functional Analysis of Bacteriophage F1 Intergenic Region. Virology 1981, 114, 463–473. [Google Scholar] [CrossRef]
- Meyer, T.F.; Geider, K. Cloning of the Bacteriophage Fd Gene 2 and Construction of a Plasmid Dependent on Fd Gene 2 Protein. Proc. Natl. Acad. Sci. USA 1981, 78, 5416–5420. [Google Scholar] [CrossRef]
- Dotto, G.; Horiuchi, K.; Jakes, K.; Zinder, N. Replication Origin of Bacteriophage F1. Two Signals Required for Its Function. J. Mol. Biol. 1982, 162, 335–343. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Evolution of Eukaryotic Single-Stranded DNA Viruses of the Bidnaviridae Family from Genes of Four Other Groups of Widely Different Viruses. Sci. Rep. 2014, 4, 5347. [Google Scholar] [CrossRef]
- Delwart, E.; Li, L. Rapidly Expanding Genetic Diversity and Host Range of the Circoviridae Viral Family and Other Rep Encoding Small Circular SsDNA Genomes. Virus Res. 2012, 164, 114–121. [Google Scholar] [CrossRef]
- Steinfeldt, T.; Finsterbusch, T.; Mankertz, A. Rep and Rep’ Protein of Porcine Circovirus Type 1 Bind to the Origin of Replication in Vitro. Virology 2001, 291, 152–160. [Google Scholar] [CrossRef]
- Faurez, F.; Dory, D.; Grasland, B.; Jestin, A. Replication of Porcine Circoviruses. Virol. J. 2009, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Spurgers, K.B.; Sharkey, C.M.; Warfield, K.L.; Bavari, S. Oligonucleotide Antiviral Therapeutics: Antisense and RNA Interference for Highly Pathogenic RNA Viruses. Antiviral. Res. 2008, 78, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, S.S.; Qiu, J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front. Cell. Infect. Microbiol. 2018, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Heo, J.; Jang, J.W.; Yoon, J.H.; Kweon, Y.O.; Park, S.J.; Tami, Y.; You, S.; Yates, P.; Tao, Y.; et al. Safety, Tolerability and Antiviral Activity of the Antisense Oligonucleotide Bepirovirsen in Patients with Chronic Hepatitis B: A Phase 2 Randomized Controlled Trial. Nat. Med. 2021, 27, 1725–1734. [Google Scholar] [CrossRef]
- Quemener, A.M.; Galibert, M.D. Antisense Oligonucleotide: A Promising Therapeutic Option to Beat COVID-19. Wiley Interdiscip. Rev. RNA 2022, 13, 1–12. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ishimoto, T.; Fujita, S.; Kiryu, S.; Wada, M.; Akatsuka, T.; Saito, M.; Kawano, M. Antimicrobial Antisense RNA Delivery to F-Pili Producing Multidrug-Resistant Bacteria via a Genetically Engineered Bacteriophage. Biochem. Biophys. Res. Commun. 2020, 530, 533–540. [Google Scholar] [CrossRef]
- Guan, W.; Huang, Q.; Cheng, F.; Qiu, J. Internal Polyadenylation of the Parvovirus B19 Precursor MRNA Is Regulated by Alternative Splicing. J. Biol. Chem. 2011, 286, 24793–24805. [Google Scholar] [CrossRef]
- Ly, C.V.; Miller, T.M. Emerging Antisense Oligonucleotide and Viral Therapies for Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 2018, 31, 648–654. [Google Scholar] [CrossRef]
- Chung, C.T.; Niemela, S.L.; Miller, R.H. One-Step Preparation of Competent Escherichia Coli: Transformation and Storage of Bacterial Cells in the Same Solution. Proc. Natl. Acad. Sci. USA 1989, 86, 2172–2175. [Google Scholar] [CrossRef]
- Arndt, C.; Koristka, S.; Bartsch, H.; Bachmann, M. Native Polyacrylamide Gels. In Methods in Molecular Biology; Walker, J.M., Kurien, B.T., Scofield, R.H., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 49–53. ISBN 9781617798207. [Google Scholar]
- Green, M.R.; Sambrook, J. Polyacrylamide Gel Electrophoresis. Cold Spring Harb. Protoc. 2020, 2020, 525–532. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Stock Concentration | Final Concentration | Volume (µL) |
|---|---|---|---|
| Reaction Buffer (20 mM Tris pH: 8.5, 9.10% sorbitol, 5 µg BSA) | 3× | 1× | 7 |
| BME (1/100) | 143 mM | 9.9 mM | 1.45 |
| P2-GFP/P2 Enzyme | 2 µM | 2 µM | 1 |
| Substrate DNA in 1× KCI Buffer (300 ng in 20 mM Tris-HCl pH: 8.5, 80 mM KCl) | - | 300 ng | - |
| ddH2O | - | - | Up to 21-µL |
| Preparation of Substrate DNA | |||
| DNA | 300 ng | ||
| KCI Buffer (20 mM Tris-HCl pH: 8.5, 80 mM KCl) | 5× | 1× | |
| Reaction Conditions | |||
| Incubation at 30 °C for 30 min | |||
| Insert Name | Description | Primer Pairs (Set) | Product Size (bp) |
|---|---|---|---|
| Insert W | dsDNA: SARS_CoV19 N1 site amplification with “whole” part recognition site included primers | N1P2 Fw/Std1 P2 Rv | 264 |
| Insert A12 | dsDNA: SARS_CoV19 N1 site amplification with “A1 and A2 loop” part recognition site included primers | N1P2 A12 Fw/Std 1 P2 A12 Rv | 208 |
| Insert A1 | dsDNA: SARS_CoV19 N1 site amplification with “A1 loop” part recognition site included primers | N1P2 A1 Fw/Std 1 P2 A1 Rv | 158 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aybakan, E.; Kocagoz, T.; Can, O. Nicking Activity of M13 Bacteriophage Protein 2. Int. J. Mol. Sci. 2025, 26, 789. https://doi.org/10.3390/ijms26020789
Aybakan E, Kocagoz T, Can O. Nicking Activity of M13 Bacteriophage Protein 2. International Journal of Molecular Sciences. 2025; 26(2):789. https://doi.org/10.3390/ijms26020789
Chicago/Turabian StyleAybakan, Esma, Tanil Kocagoz, and Ozge Can. 2025. "Nicking Activity of M13 Bacteriophage Protein 2" International Journal of Molecular Sciences 26, no. 2: 789. https://doi.org/10.3390/ijms26020789
APA StyleAybakan, E., Kocagoz, T., & Can, O. (2025). Nicking Activity of M13 Bacteriophage Protein 2. International Journal of Molecular Sciences, 26(2), 789. https://doi.org/10.3390/ijms26020789