

Synthesis and Anticancer Evaluation of O-Alkylated (E)-Chalcone Derivatives: A Focus on Estrogen Receptor Inhibition
 , , and
, , and
Abstract
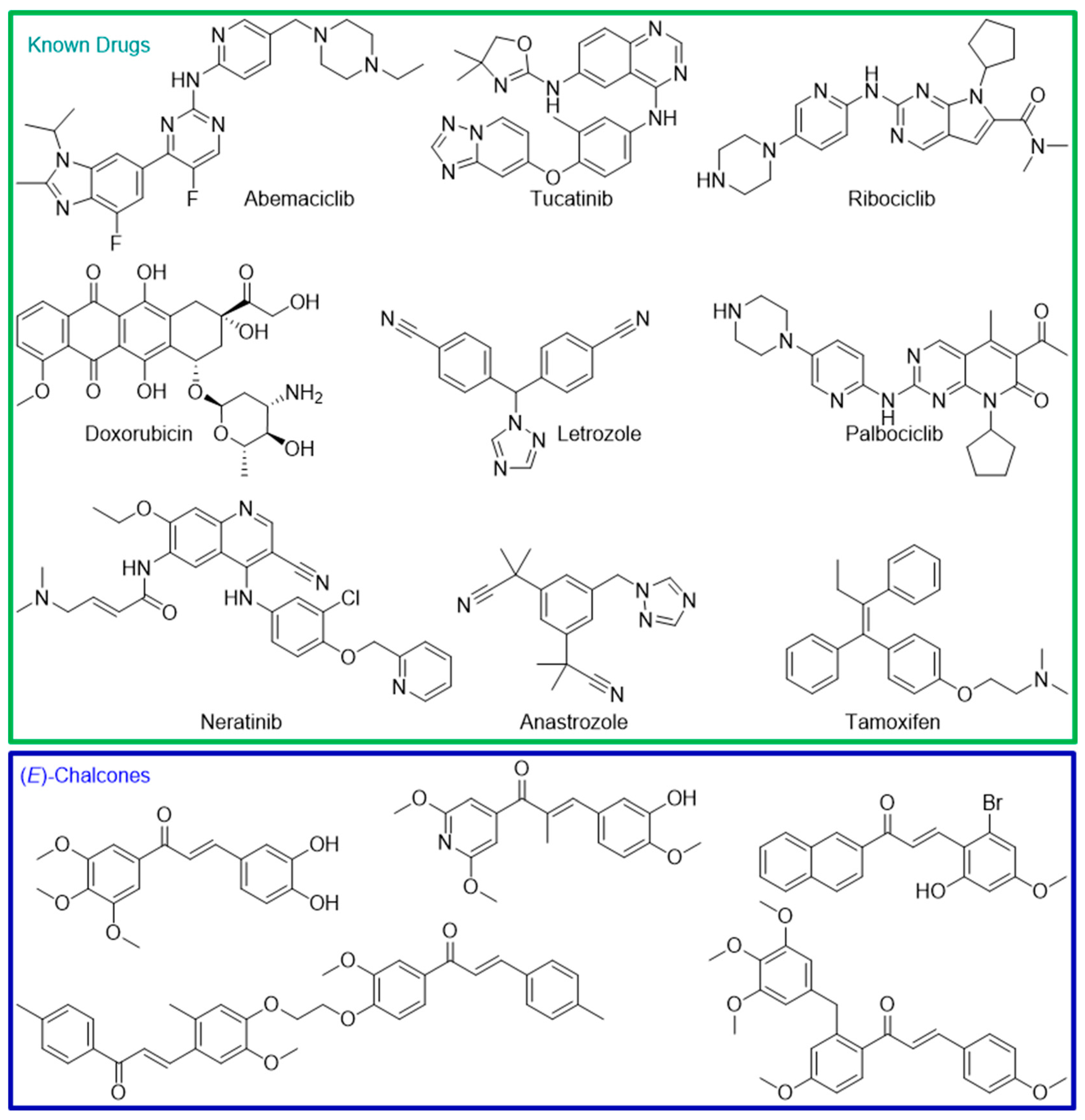
1. Introduction
2. Results and Discussion
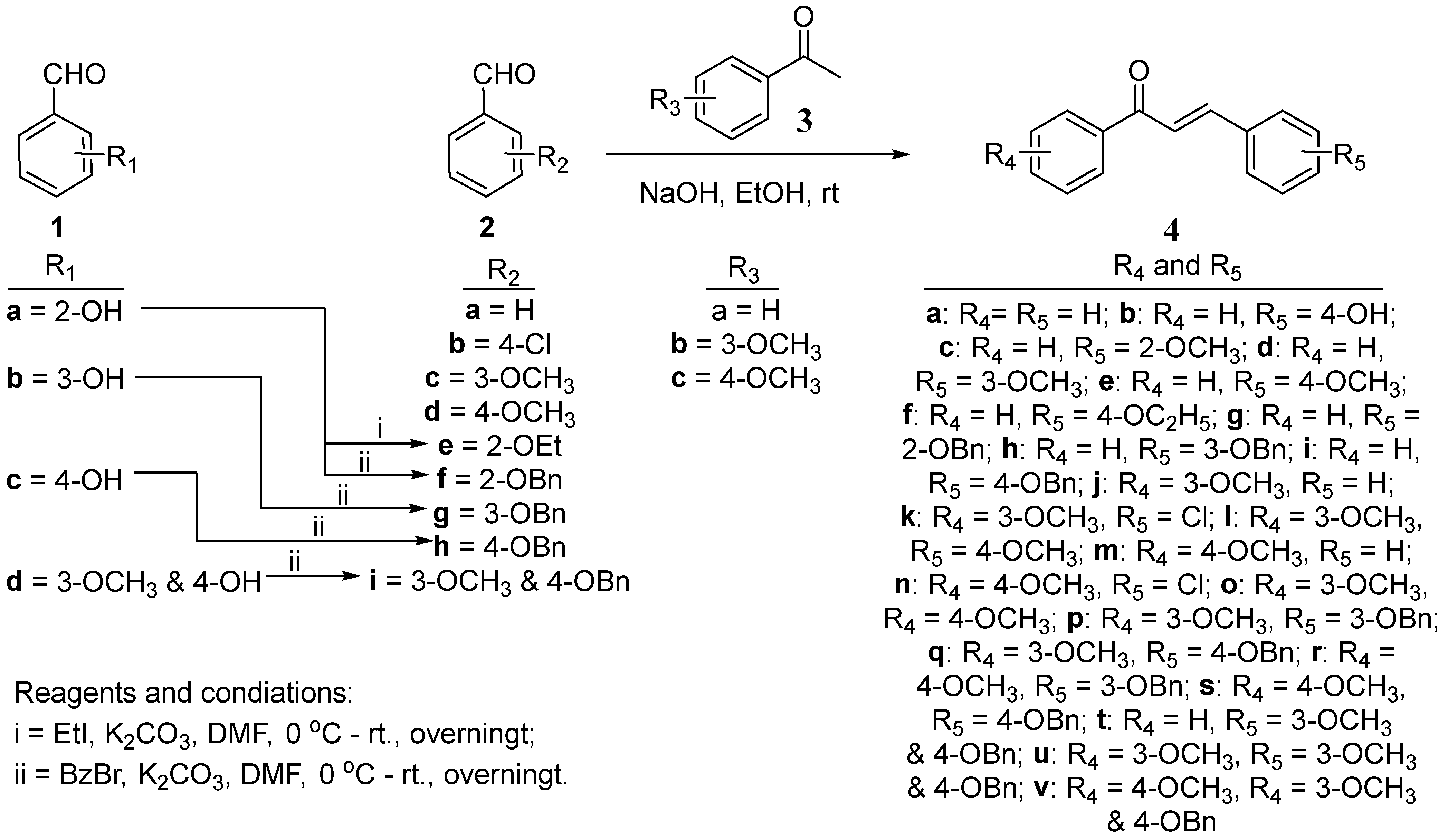
2.1. Synthesis of (E)-Chalcone Derivatives 4a–4v
2.2. Biological Evaluation of (E)-Chalcone Derivatives 4a–4v
2.2.1. In Vitro Cytotoxicity Evaluation of 4a–4v
2.2.2. In Vitro Protein Kinase Inhibition Assays of 4a, 4b, 4q and 4v
2.2.3. In Vitro Anti-Estrogenic Activity Assays of 4a, 4b, 4q, and 4v
2.2.4. In Vitro Aromatase Inhibition Assays of 4a, 4b, 4q, and 4v
2.2.5. In Silico Studies of the Synthesized Compounds 4a–4v
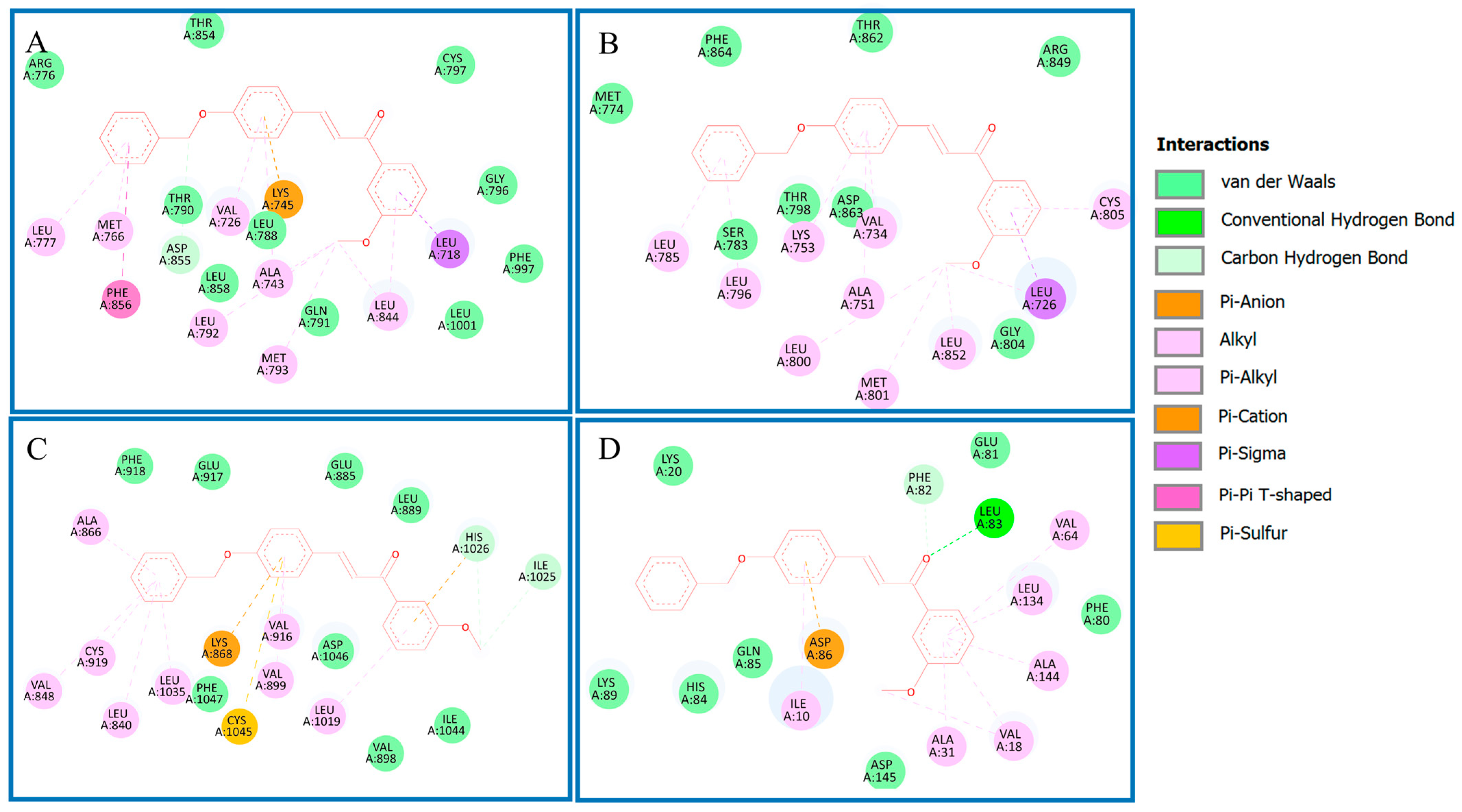
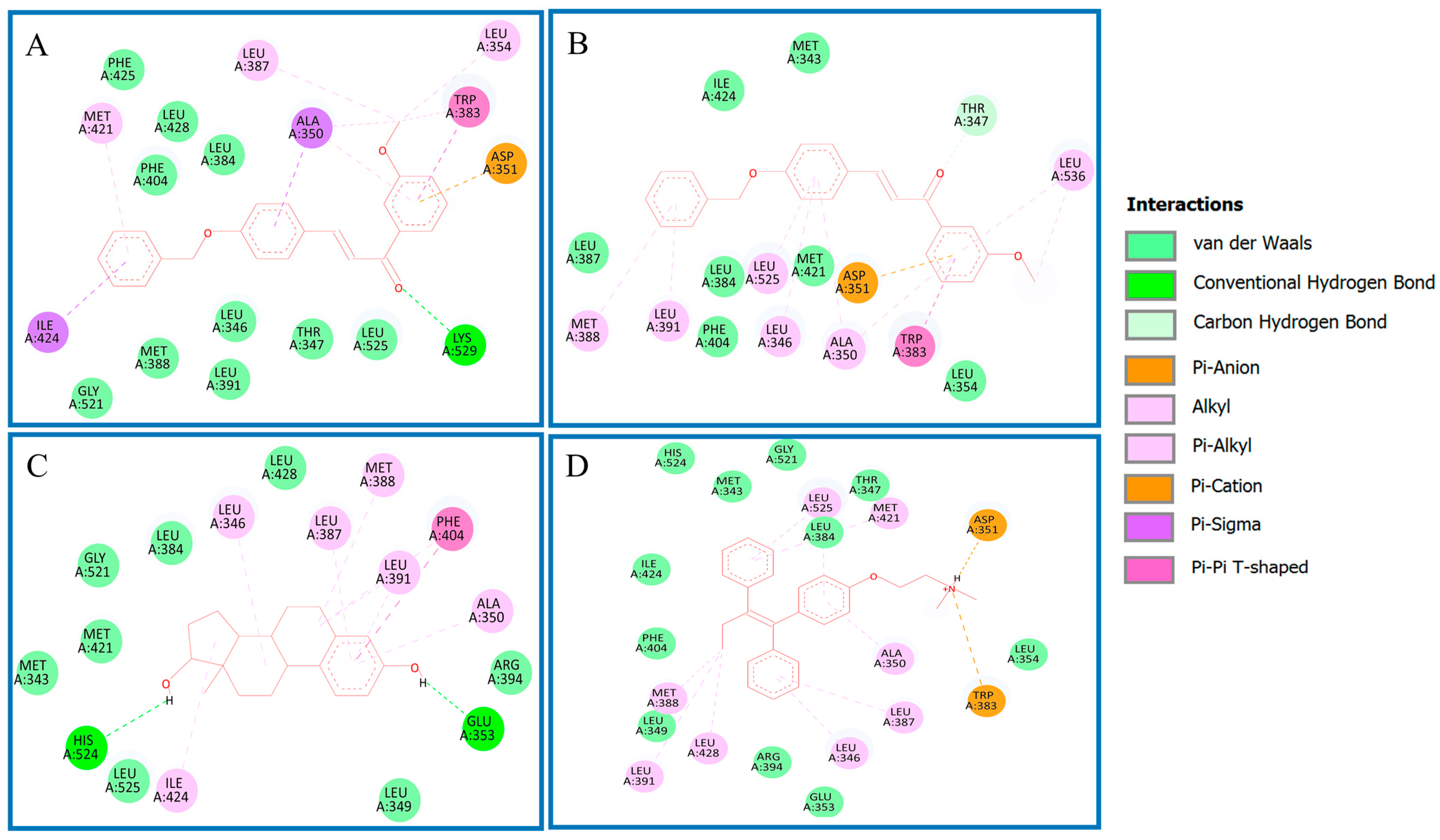
Analysis of Binding Mechanism of Compound 4q and Reference Compounds by Molecular Docking Study
In Silico Drug Likeness Property Analysis of 4a–4v
3. Materials and Methods
3.1. General
3.2. General Synthetic Method for the Synthesis of 2e–2i
3.3. General Synthetic Method for the Synthesis of 4a–4v
3.4. Biological Evaluation Assay
3.4.1. In Vitro Cytotoxicity Assay of 4a–4v
3.4.2. In Vitro Enzyme Assays of 4a, 4b, 4q, and 4v
3.4.3. In Vitro Anti-Estrogenic Activity Assay of 4a, 4b, 4q, and 4v
3.4.4. In Vitro Aromatase Assay of 4a, 4b, 4q, and 4v
3.5. In Silico Studies of 4q
3.5.1. Molecular Docking of 4q
3.5.2. In Silico ADME Studies of the Compounds 4a–4v
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Cancer Burden Growing, Amidst Mounting Need for Services. Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 15 September 2024).
- Basudan, A.M. Breast Cancer Incidence Patterns in the Saudi Female Population: A 17-Year Retrospective Analysis. Medicina 2022, 58, 1617. [Google Scholar] [CrossRef] [PubMed]
- Al Zomia, A.S.; Al Zehefa, I.A.M.; Lahiq, L.A.; Mirdad, M.T.; Alshahrani, A.S.; Alshahrani, T.; Almahfuth, N.N.; Mirdad, M.T.; Alqarni, A.A.; Alshareef, N.M.; et al. Tracking the epidemiological trends of female breast cancer in Saudi Arabia since 1990 and forecasting future statistics using global burden of disease data, time-series analysis. BMC Public Health 2024, 24, 1953. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, W.S.; Almufareh, N.A.; Domiaty, D.M.; Albasher, G.; Alduwish, M.A.; Alkhalaf, H.; Almuzzaini, B.; Al-Marshidy, S.S.; Alfraihi, R.; Elasbali, A.M.; et al. Epidemiology of cancer in Saudi Arabia thru 2010-2019: A systematic review with constrained meta-analysis. AIMS Public Health 2020, 7, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Sepucha, K.R.; Langford, A.T.; Belkora, J.K.; Chang, Y.; Moy, B.; Partridge, A.H.; Lee, C.N. Impact of Timing on Measurement of Decision Quality and Shared Decision Making: Longitudinal Cohort Study of Breast Cancer Patients. Med. Decis. Mak. 2019, 39, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Elkhalifa, D.; Alali, F.; Al Moustafa, A.E.; Khalil, A. Targeting triple negative breast cancer heterogeneity with chalcones: A molecular insight. J. Drug Target. 2019, 27, 830–838. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Wang, X. The Metabolic Mechanisms of Breast Cancer Metastasis. Front. Oncol. 2020, 10, 602416. [Google Scholar] [CrossRef]
- Gnant, M.; Harbeck, N.; Thomssen, C. St. Gallen 2011: Summary of the Consensus Discussion. Breast Care 2011, 6, 136–141. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Travis, R.C.; Key, T.J. Oestrogen exposure and breast cancer risk. Breast Cancer Res. 2003, 5, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Viale, P.H. The American Cancer Society’s Facts & Figures: 2020 Edition. J. Adv. Pract. Oncol. 2020, 11, 135–136. [Google Scholar] [CrossRef]
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Klein, P.; Tiersten, A.; Sparano, J.A. An emerging generation of endocrine therapies in breast cancer: A clinical perspective. npj Breast Cancer 2023, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Mechanisms of anticarcinogenesis and drug resistance. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2005, 591, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Torres-Guzmán, R.; Ganado, M.P.; Pérez, C.M.; Marugán, C.; Baquero, C.; Yang, Y.; Du, J.; Dios, A.d.; Puig, O.; Lallena, M.J. Abemaciclib, a CDK4 and 6 inhibitor with unique pharmacological properties for breast cancer therapy. J. Clin. Oncol. 2021, 39, e12506. [Google Scholar] [CrossRef]
- Chan, A.; Moy, B.; Mansi, J.; Ejlertsen, B.; Holmes, F.A.; Chia, S.; Iwata, H.; Gnant, M.; Loibl, S.; Barrios, C.H.; et al. Final Efficacy Results of Neratinib in HER2-positive Hormone Receptor-positive Early-stage Breast Cancer From the Phase III ExteNET Trial. Clin. Breast Cancer 2021, 21, 80–91.e87. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.; Azad, A.; Chan, P.Y.K.; Vigneswara, V.; Feldinger, K.; Nafi, S.N.M.; Laporte-Maguire, E.; De Santo, C.; Zuo, J.; Shaaban, A.M.; et al. Neratinib could be effective as monotherapy or in combination with trastuzumab in HER2-low breast cancer cells and organoid models. Br. J. Cancer 2024, 130, 1990–2002. [Google Scholar] [CrossRef]
- George, M.A.; Qureshi, S.; Omene, C.; Toppmeyer, D.L.; Ganesan, S. Clinical and Pharmacologic Differences of CDK4/6 Inhibitors in Breast Cancer. Front. Oncol. 2021, 11, 693104. [Google Scholar] [CrossRef] [PubMed]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Bustos, L.; Echiburú-Chau, C.; Castro-Alvarez, A.; Bradshaw, B.; Simirgiotis, M.J.; Mellado, M.; Parra, C.; Cuellar, M. Cytotoxic Effects on Breast Cancer Cell Lines of Chalcones Derived from a Natural Precursor and Their Molecular Docking Analysis. Molecules 2022, 27, 4387. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Gong, Z.; Huang, Y.; Li, Y.; Peng, Z. Design, synthesis, biological evaluation and molecular docking studies of new chalcone derivatives containing diaryl ether moiety as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 95, 103565. [Google Scholar] [CrossRef] [PubMed]
- Guruswamy, D.K.M.; Jayarama, S. Proapoptotic and anti-angiogenic activity of (2E)-3-(2-bromo-6-hydroxy-4-methoxyphenyl)-1-(naphthalene-2-yl) prop-2-en-1-one in MCF7 cell line. Chem. Pap. 2020, 74, 2229–2237. [Google Scholar] [CrossRef]
- Wilhelm, A.; Bonnet, S.L.; Twigge, L.; Rarova, L.; Stenclova, T.; Visser, H.G.; Schutte-Smith, M. Synthesis, characterization and cytotoxic evaluation of chalcone derivatives. J. Mol. Struct. 2022, 1251, 132001. [Google Scholar] [CrossRef]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Harshitha, K.R.; Sarojini, B.K.; Narayana, B.; Lobo, A.G.; Kalal, B.S. Molecular docking of 4-ehoxychalcones on oxidoreductase/pirin inhibitors and cytotoxic evaluation on breast/skin cancer cell lines. Lett. Drug Des. Discov. 2020, 17, 1245–1260. [Google Scholar] [CrossRef]
- Al-Kaabi, M.M.; Rady Al-Hazam, H.A.; Arwa. Microwave assisted synthesis, characterization and biochemical study of new chalcones. Egypt. J. Chem. 2021, 64, 4027–4035. [Google Scholar] [CrossRef]
- Mangoud, M.M.; Hussein, M.Z.; El-Bordany, E.A. Design and Synthesis of Novel Pyrazoles, Pyrazolines, and Pyridines from Chalcone Derivatives with Evaluation of Their In Vitro Anticancer Activity Against T-47D and UACC-257 Cell Lines. Egypt. J. Chem. 2020, 63, 5203–5218. [Google Scholar] [CrossRef]
- Ahn, S.; Truong, V.N.-P.; Kim, B.; Yoo, M.; Lim, Y.; Cho, S.K.; Koh, D. Design, synthesis, and biological evaluation of chalcones for anticancer properties targeting glycogen synthase kinase 3 beta. Appl. Biol. Chem. 2022, 65, 17. [Google Scholar] [CrossRef]
- Calliste, C.A.; Le Bail, J.C.; Trouillas, P.; Pouget, C.; Habrioux, G.; Chulia, A.J.; Duroux, J.L. Chalcones: Structural requirements for antioxidant, estrogenic and antiproliferative activities. Anticancer Res. 2001, 21, 3949–3956. [Google Scholar] [PubMed]
- Kohno, Y.; Kitamura, S.; Sanoh, S.; Sugihara, K.; Fujimoto, N.; Ohta, S. Metabolism of the α,β-unsaturated ketones, chalcone and trans-4-phenyl-3-buten-2-one, by rat liver microsomes and estrogenic activity of the metabolites. Drug Metab. Dispos. 2005, 33, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef] [PubMed]
- Williamson, A. XLV. Theory of ætherification. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1850, 37, 350–356. [Google Scholar] [CrossRef]
- Mirgany, T.O.; Asiri, H.H.; Rahman, A.F.M.M.; Alanazi, M.M. Discovery of 1H-benzo[d]imidazole-(halogenated) Benzylidenebenzohydrazide Hybrids as Potential Multi-Kinase Inhibitors. Pharmaceuticals 2024, 17, 839. [Google Scholar] [CrossRef]
- Scheiner, S. Weak H-bonds. Comparisons of CH···O to NH···O in proteins and PH···N to direct P···N interactions. Phys. Chem. Chem. Phys. 2011, 13, 13860–13872. [Google Scholar] [CrossRef]
- Cannizzaro, C.E.; Houk, K.N. Magnitudes and chemical consequences of R(3)N(+)-C-H...O[double bond]C hydrogen bonding. J. Am. Chem. Soc. 2002, 124, 7163–7169. [Google Scholar] [CrossRef]
- Callahan, R.; Hurvitz, S. Human epidermal growth factor receptor-2-positive breast cancer: Current management of early, advanced, and recurrent disease. Curr. Opin. Obstet. Gynecol. 2011, 23, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Tung, B.T.; Son, N.N.; Kim, N.B.; Khanh, D.T.H.; Minh, P.H. In silico screening of alkaloids as potential inhibitors of HER2 protein for breast cancer treatment. Vietnam J. Chem. 2023, 61, 308–317. [Google Scholar] [CrossRef]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Dougher, M.; Terman, B.I. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene 1999, 18, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gan, Y.; Li, H.; Yin, J.; He, X.; Lin, L.; Xu, S.; Fang, Z.; Kim, B.-W.; Gao, L.; et al. Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat. Commun. 2022, 13, 2835. [Google Scholar] [CrossRef] [PubMed]
- Talapati, S.R.; Nataraj, V.; Pothuganti, M.; Gore, S.; Ramachandra, M.; Antony, T.; More, S.S.; Krishnamurthy, N.R. Structure of cyclin-dependent kinase 2 (CDK2) in complex with the specific and potent inhibitor CVT-313. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 350–356. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Wang, J.P.; Li, Y.; Fan, P.; Liu, G.; Zhang, N.; Conaway, M.; Wang, H.; Korach, K.S.; Bocchinfuso, W.; et al. Effects of estrogen on breast cancer development: Role of estrogen receptor independent mechanisms. Int. J. Cancer 2010, 127, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Waller, C.L.; Swaan, P.W.; Cruciani, G.; Wrighton, S.A.; Wikel, J.H. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. Methods 2000, 44, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Tamuli, K.J.; Sahoo, R.K.; Bordoloi, M. Biocatalytic green alternative to existing hazardous reaction media: Synthesis of chalcone and flavone derivatives via the Claisen–Schmidt reaction at room temperature. New J. Chem. 2020, 44, 20956–20965. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, Y.; Zhang, Q.; Pan, W.; Li, T.; Yin, Y. Bi(OTf)3-Catalyzed Alkyl-Intercepted Meyer–Schuster Rearrangement of Propargylic Alcohols for the Synthesis of 1,2,3,5-Tetrasubstituted Pentane-1,5-diones. J. Org. Chem. 2022, 87, 3329–3340. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, A.P.; Souza, H.D.d.S.; Almeida-Júnior, A.; da Silva, M.F.R.; Cordeiro, L.V.; Lima, E.d.O.; Fiss, G.F.; de Athayde-Filho, P.F. Novel esters derived from 4-hydroxychalcones as potential sunscreens with antimicrobial action. Synth. Commun. 2024, 54, 973–991. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Rosaiah, J.N.; Kumar, A. Iodine-Catalyzed Mild and Efficient Method for the Synthesis of Chalcones. Synth. Commun. 2009, 39, 2288–2296. [Google Scholar] [CrossRef]
- Pereira de Oliveira Borlot, J.R.; Schlittler dos Santos, L.; Schwarzt Sampaio, G.J.; Santos Borges, A.; Rodrigues, R.P.; de Cássia Ribeiro Gonçalves, R.; Bezerra dos Santos, R.; Kitagawa, R.R. Synthesis, Docking Studies and Evaluation of Chalcones as Anti-Helicobacter pylori and antitumoral Agents. Chem. Biodivers. 2023, 20, e202301066. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, E.; Amawi, H.; Hussein, N.; Karthikeyan, C.; Fetcenko, A.; Narayana Moorthy, N.S.H.; Trivedi, P.; Tiwari, A.K. Design and discovery of silybin analogues as antiproliferative compounds using a ring disjunctive—Based, natural product lead optimization approach. Eur. J. Med. Chem. 2017, 133, 365–378. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Kamal, A.; Singh, H.K.; Maury, S.K.; Mondal, T.; Singh, S. Photoinduced, Metal-Free Hydroacylation of Aromatic Alkynes for Synthesis of alpha,beta-Unsaturated Ketones via C(sp)-H Functionalization. Org. Lett. 2024, 26, 1416–1420. [Google Scholar] [CrossRef]
- Shadakshari, U.; Nayak, S.K. Enantioselective conjugate addition of diethylzinc to chalcones catalysed by N-trityl aziridine-2-(S)-(diphenyl)methanol and Ni(acac)2. Tetrahedron 2001, 57, 8185–8188. [Google Scholar] [CrossRef]
- Bowman, M.D.; Jacobson, M.M.; Blackwell, H.E. Discovery of Fluorescent Cyanopyridine and Deazalumazine Dyes Using Small Molecule Macroarrays. Org. Lett. 2006, 8, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Toan, V.N.; Thanh, N.D.; Hai, D.S.; Tri, N.M. Synthesis, anticancer activity, and molecular simulation of N-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)thioureas containing a pyrimidine ring. New J. Chem. 2024, 48, 9208–9223. [Google Scholar] [CrossRef]
- Soleiman-Beigi, M.; Ghalavand, S.; Venovel, H.G.; Kohzadi, H. Synthesis of lithium/cesium-Zagronas from zagrosian natural asphalt and study of their activity as novel, green, heterogeneous and homogeneous nanocatalysts in the Claisen?Schmidt and Knoevenagel condensations. J. Iran. Chem. Soc. 2021, 18, 3267–3279. [Google Scholar] [CrossRef]
- Schwartz, M.A.; Rose, B.F.; Holton, R.A.; Scott, S.W.; Vishnuvajjala, B. Intramolecular oxidative coupling of diphenolic, monophenolic, and nonphenolic substrates. J. Am. Chem. Soc. 1977, 99, 2571–2578. [Google Scholar] [CrossRef]
- Tajudeen Bale, A.; Mohammed Khan, K.; Salar, U.; Chigurupati, S.; Fasina, T.; Ali, F.; Wadood, A.; Taha, M.; Sekhar Nanda, S.; Ghufran, M.; et al. Chalcones and bis-chalcones: As potential α-amylase inhibitors; synthesis, in vitro screening, and molecular modelling studies. Bioorg. Chem. 2018, 79, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yáñez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Chalcones: A Valid Scaffold for Monoamine Oxidases Inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, A.A.; Asiri, H.H.; Rahman, A.F.M.M.; Alanazi, M.M. Novel pyrrolo[2,3-d]pyrimidine derivatives as multi-kinase inhibitors with VEGFR-2 selectivity. J. Saudi Chem. Soc. 2023, 27, 101712. [Google Scholar] [CrossRef]
- Alotaibi, A.A.; Alanazi, M.M.; Rahman, A.F.M.M. Discovery of New Pyrrolo[2,3-d]pyrimidine Derivatives as Potential Multi-Targeted Kinase Inhibitors and Apoptosis Inducers. Pharmaceuticals 2023, 16, 1324. [Google Scholar] [CrossRef]
- Brander, S.M.; He, G.; Smalling, K.L.; Denison, M.S.; Cherr, G.N. The in vivo estrogenic and in vitro anti-estrogenic activity of permethrin and bifenthrin. Environ. Toxicol. Chem. 2012, 31, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.S.; Badshah, Y.; Shabbir, M.; Rafiq, M. Molecular Docking Using Chimera and Autodock Vina Software for Nonbioinformaticians. JMIR Bioinform. Biotechnol. 2020, 1, e14232. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.-H. Inhibition of Cyclin-Dependent Kinases by Purine Analogues. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cytotoxicity IC50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HCT-116 | SI | MDA-MB-231 | SI | HeLa | SI | MCF-7 | SI | WI-38 | |
| 4a | 9.72 ± 0.7 | 3.7 | 7.06 ± 0.5 | 5.1 | 15.38 ± 1.3 | 2.4 | 5.16 ± 0.4 | 7.0 | 36.28 ± 2.2 |
| 4b | 6.59 ± 0.3 | 8.3 | 4.93 ± 0.3 | 11.1 | 13.07 ± 1.1 | 4.2 | 2.08 ± 0.1 | 26.2 | 54.59 ± 3.1 |
| 4c | 28.35 ± 1.8 | 3.5 | 35.86 ± 2.1 | 2.8 | 42.46 ± 2.5 | 2.4 | 23.51 ± 1.6 | 4.3 | >100 |
| 4d | 36.01 ± 2.2 | 2.1 | 43.59 ± 2.3 | 1.8 | 39.43 ± 2.3 | 2.0 | 29.67 ± 1.9 | 2.6 | 77.26 ± 3.8 |
| 4e | 20.73 ± 1.5 | 4.8 | 24.30 ± 1.8 | 4.1 | 29.52 ± 2.0 | 3.4 | 16.90 ± 1.3 | 5.9 | >100 |
| 4f | 57.59 ± 3.3 | 1.1 | 46.12 ± 2.4 | 1.4 | 49.60 ± 2.8 | 1.3 | 35.28 ± 2.1 | 1.8 | 64.65 ± 3.6 |
| 4g | 53.96 ± 3.0 | 0.5 | 59.19 ± 3.5 | 0.5 | 36.47 ± 2.4 | 0.8 | 48.43 ± 2.6 | 0.6 | 27.81 ± 1.9 |
| 4h | 46.81 ± 2.7 | 1.8 | 53.80 ± 3.3 | 1.6 | 25.63 ± 1.8 | 3.3 | 42.07 ± 2.4 | 2.0 | 83.39 ± 4.2 |
| 4i | >100 | 1.0 | >100 | 1.0 | >100 | 1.0 | 85.54 ± 4.3 | 1.2 | >100 |
| 4j | 24.60 ± 1.7 | 3.2 | 29.34 ± 1.9 | 2.7 | 34.91 ± 2.2 | 2.2 | 18.83 ± 1.5 | 4.2 | 78.22 ± 3.9 |
| 4k | >100 | 1.0 | >100 | 1.0 | >100 | 1.0 | >100 | 1.0 | >100 |
| 4l | 83.96 ± 4.2 | 0.2 | 91.72 ± 4.5 | 0.2 | 88.83 ± 4.6 | 0.2 | 75.38 ± 3.8 | 0.2 | 16.24 ± 1.3 |
| 4m | 59.62 ± 3.5 | 1.5 | 45.93 ± 2.6 | 1.9 | 63.65 ± 3.7 | 1.4 | 33.04 ± 2.0 | 2.7 | 87.62 ± 4.6 |
| 4n | 66.08 ± 3.8 | 0.9 | 48.59 ± 2.8 | 1.3 | 74.51 ± 3.9 | 0.8 | 36.97 ± 2.3 | 1.7 | 61.53 ± 3.4 |
| 4o | 89.17 ± 4.5 | 0.3 | 73.48 ± 4.0 | 0.3 | >100 | 0.2 | 61.85 ± 3.4 | 0.4 | 24.18 ± 1.7 |
| 4p | 39.84 ± 2.4 | 2.3 | 68.42 ± 3.7 | 1.3 | 45.21 ± 2.4 | 2.0 | 57.11 ± 3.2 | 1.6 | 92.06 ± 4.9 |
| 4q | 17.06 ± 1.3 | 5.9 | 13.58 ± 1.1 | 7.4 | 22.64 ± 1.7 | 4.4 | 11.56 ± 0.9 | 8.7 | >100 |
| 4r | 62.60 ± 3.7 | 1.6 | 51.84 ± 2.9 | 1.9 | 57.03 ± 3.2 | 1.8 | 39.34 ± 2.4 | 2.5 | >100 |
| 4s | 73.53 ± 3.9 | 0.3 | 82.37 ± 4.1 | 0.3 | 86.96 ± 4.3 | 0.3 | 64.19 ± 3.5 | 0.3 | 22.34 ± 1.6 |
| 4t | 68.91 ± 3.7 | 0.6 | 64.09 ± 3.8 | 0.6 | 59.28 ± 3.4 | 0.7 | 55.62 ± 2.9 | 0.7 | 40.06 ± 2.5 |
| 4u | 95.27 ± 4.9 | 0.4 | >100 | 0.4 | >100 | 0.4 | 82.75 ± 4.1 | 0.5 | 38.95 ± 2.4 |
| 4v | 14.53 ± 1.1 | 5.2 | 10.24 ± 0.8 | 7.3 | 18.16 ± 1.5 | 4.1 | 8.67 ± 0.6 | 8.7 | 75.17 ± 3.9 |
| Doxorubicin | 5.23 ± 0.3 | 1.3 | 3.18 ± 0.1 | 2.1 | 5.57 ± 0.4 | 1.2 | 4.17 ± 0.2 | 1.6 | 6.72 ± 0.5 |
| Sorafenib | 5.47 ± 0.3 | 1.9 | 7.64 ± 0.4 | 1.4 | 8.04 ± 0.5 | 1.3 | 7.26 ± 0.3 | 1.5 | 10.65 ± 0.8 |
| Compound | Protein Kinase Inhibition [IC50 (µM) a/Inhibition % at 0.1 µM b] | |||
|---|---|---|---|---|
| EGFR | HER2 | VEGFR-2 | CDK2 | |
| 4a | 0.367 ± 0.012 | 0.691 ± 0.027 | 1.548 ± 0.053 | 0.84 ± 0.026 |
| 4b | 54 c | 0.387 ± 0.015 | 0.163 ± 0.006 | 0.288 ± 0.009 |
| 4q | 0.151 ± 0.005 | 48 c | 0.287 ± 0.01 | 41.9 c |
| 4v | 0.671 ± 0.022 | 1.568 ± 0.061 | 1.824 ± 0.062 | 1.032 ± 0.032 |
| Erlotinib | 0.056 ± 0.002 | 63.9 c | - | - |
| Sorafenib | - | - | 57 c | - |
| Dinaciclib | - | - | - | 60 c |
| Compound | Cell Proliferation | |||
|---|---|---|---|---|
| MCF-7 | MCF-7a | |||
| Cell Count | % | Cell Count | % | |
| 4a | 14,553 ± 518 | 79 | 21,155 ± 611 | 102 |
| 4b | 6213 ± 221 | 34 | 20,669 ± 597 | 99 |
| 4q | 6874 ± 245 | 37 | 20,630 ± 595 | 99 |
| 4v | 13,611 ± 485 | 73 | 20,397 ± 589 | 98 |
| Tamoxifen | 3185 ± 113 | 17 | 20,698 ± 597 | 100 |
| 17β-E2 (Control) | 18,524 ± 659 | 100 | 20,776 ± 600 | 100 |
| Compound | Aromatase Inhibition [IC50 (µM) a/Inhibition % at 0.1 µM b] |
|---|---|
| 4a | 0.434 ± 0.014 |
| 4b | 1.634 ± 0.054 |
| 4q | 0.801 ± 0.03 |
| 4v | 0.357 ± 0.012 |
| Letrozole | 55 c |
| Entry | Binding Affinity (kcal/mol) | Hydrogen Bonds | ||||||
|---|---|---|---|---|---|---|---|---|
| EGFR | HER2 | VEGFR2 | CDK2 | EGFR | HER2 | VEGFR2 | CDK2 | |
| 4q | −10.0 | −10.5 | −10.1 | −8.5 | - | - | - | Leu83 |
| Erlotinib | −7.3 | −8.3 | Met793, Lys745, Asp855 | - | - | - | ||
| Sorafenib | −10.7 | - | - | Glu885, Asp1046, Cys919 | - | |||
| Dinaciclib | −9.1 | - | - | - | Leu83, Lys89 | |||
| Entry | MW a | LogP | HBA b | HBD c | SA d | WS e | IA f | SP g | BBB h | CNS i | CYP j | CYP k | TC l | MTD m | ORAT n | ORCT o | SS p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 208.3 | 3.6 | 1.0 | 0.0 | 95.1 | −4.6 | 99.1 | −1.8 | 0.6 | −0.9 | No | No | 0.2 | 1.8 | 1.9 | 1.6 | Yes |
| 4b | 224.3 | 3.3 | 2.0 | 1.0 | 99.8 | −4.0 | 96.2 | −2.3 | 0.2 | −1.2 | No | No | 0.1 | 1.7 | 2.0 | 2.5 | Yes |
| 4c | 238.3 | 3.6 | 2.0 | 0.0 | 106.5 | −4.6 | 99.1 | −2.1 | 0.4 | −1.0 | No | No | 0.3 | 1.7 | 2.0 | 2.4 | Yes |
| 4d | 238.3 | 3.6 | 2.0 | 0.0 | 106.5 | −4.6 | 99.1 | −2.1 | 0.4 | −1.0 | No | No | 0.2 | 1.7 | 2.0 | 2.4 | Yes |
| 4e | 238.3 | 3.6 | 2.0 | 0.0 | 106.5 | −4.6 | 99.1 | −2.1 | 0.4 | −1.0 | No | No | 0.2 | 1.7 | 2.0 | 2.4 | Yes |
| 4f | 252.3 | 4.0 | 2.0 | 0.0 | 112.9 | −4.9 | 98.7 | −2.2 | 0.5 | −1.0 | No | No | 0.2 | 1.7 | 2.0 | 2.5 | Yes |
| 4g | 314.4 | 5.2 | 2.0 | 0.0 | 141.6 | −5.8 | 98.3 | −2.6 | 0.6 | −0.6 | No | Yes | 0.2 | 1.6 | 2.1 | 2.7 | No |
| 4h | 314.4 | 5.2 | 2.0 | 0.0 | 141.6 | −5.8 | 98.3 | −2.6 | 0.6 | −0.6 | No | Yes | 0.2 | 1.6 | 2.1 | 2.7 | No |
| 4i | 314.4 | 5.2 | 2.0 | 0.0 | 141.6 | −5.8 | 98.3 | −2.6 | 0.6 | −0.6 | No | Yes | 0.2 | 1.6 | 2.1 | 2.7 | No |
| 4j | 238.3 | 3.6 | 2.0 | 0.0 | 106.5 | −4.6 | 99.1 | −2.1 | 0.4 | −1.0 | No | No | 0.2 | 1.7 | 2.0 | 2.4 | Yes |
| 4k | 272.7 | 4.2 | 2.0 | 0.0 | 116.8 | −5.3 | 97.4 | −2.2 | 0.4 | −1.0 | No | No | 0.1 | 1.6 | 2.2 | 2.3 | Yes |
| 4l | 268.3 | 3.6 | 3.0 | 0.0 | 118.0 | −4.6 | 99.1 | −2.5 | 0.3 | −1.0 | No | No | 0.2 | 1.6 | 2.0 | 2.3 | No |
| 4m | 238.3 | 3.6 | 2.0 | 0.0 | 106.5 | −4.6 | 99.1 | −2.1 | 0.4 | −1.0 | No | No | 0.2 | 1.7 | 2.0 | 2.4 | Yes |
| 4n | 272.7 | 4.2 | 2.0 | 0.0 | 116.8 | −5.4 | 97.4 | −2.2 | 0.4 | −1.0 | No | No | 0.0 | 1.6 | 2.2 | 2.3 | No |
| 4o | 268.3 | 3.6 | 3.0 | 0.0 | 118.0 | −4.6 | 99.1 | −2.5 | 0.3 | −1.0 | No | No | 0.2 | 1.6 | 2.0 | 2.3 | No |
| 4p | 344.4 | 5.2 | 3.0 | 0.0 | 153.1 | −5.9 | 98.3 | −2.7 | 0.3 | −0.7 | No | Yes | 0.2 | 1.5 | 2.1 | 2.5 | No |
| 4q | 344.4 | 5.2 | 3.0 | 0.0 | 153.1 | −5.9 | 98.3 | −2.7 | 0.3 | −0.7 | No | Yes | 0.2 | 1.5 | 2.1 | 2.5 | No |
| 4r | 344.4 | 5.2 | 3.0 | 0.0 | 153.1 | −5.9 | 98.3 | −2.7 | 0.3 | −0.7 | No | Yes | 0.2 | 1.5 | 2.1 | 2.5 | No |
| 4s | 344.4 | 5.2 | 3.0 | 0.0 | 153.1 | −5.9 | 98.3 | −2.7 | 0.3 | −0.7 | No | Yes | 0.2 | 1.5 | 2.1 | 2.5 | No |
| 4t | 344.4 | 5.2 | 3.0 | 0.0 | 153.1 | −5.9 | 98.3 | −2.7 | 0.3 | −0.7 | No | Yes | 0.2 | 1.5 | 2.1 | 2.5 | No |
| 4u | 374.4 | 5.2 | 4.0 | 0.0 | 164.5 | −6.0 | 98.3 | −2.8 | 0.1 | −0.8 | No | Yes | 0.2 | 1.4 | 2.2 | 2.4 | No |
| 4v | 374.4 | 5.2 | 4.0 | 0.0 | 164.5 | −6.0 | 98.3 | −2.8 | 0.1 | −0.8 | No | Yes | 0.2 | 1.4 | 2.2 | 2.4 | No |
| So q | 464.8 | 5.5 | 4.0 | 3.0 | 185.1 | −6.6 | 88.5 | −3.0 | −1.4 | −1.9 | No | Yes | −0.2 | 0.7 | 3.2 | 1.1 | No |
| Do r | 543.5 | 0.0 | 12.0 | 6.0 | 222.1 | −3.9 | 55.8 | −2.8 | −1.6 | −4.1 | No | Yes | 1.0 | −0.1 | 1.8 | 2.1 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Ghamdi, A.R.; Ahmed, W.U.; Al-Wabli, R.I.; Al-Mutairi, M.S.; Rahman, A.F.M.M. Synthesis and Anticancer Evaluation of O-Alkylated (E)-Chalcone Derivatives: A Focus on Estrogen Receptor Inhibition. Int. J. Mol. Sci. 2025, 26, 833. https://doi.org/10.3390/ijms26020833
Al-Ghamdi AR, Ahmed WU, Al-Wabli RI, Al-Mutairi MS, Rahman AFMM. Synthesis and Anticancer Evaluation of O-Alkylated (E)-Chalcone Derivatives: A Focus on Estrogen Receptor Inhibition. International Journal of Molecular Sciences. 2025; 26(2):833. https://doi.org/10.3390/ijms26020833
Chicago/Turabian StyleAl-Ghamdi, Alwah R., Wahid U. Ahmed, Reem I. Al-Wabli, Maha S. Al-Mutairi, and A. F. M. Motiur Rahman. 2025. "Synthesis and Anticancer Evaluation of O-Alkylated (E)-Chalcone Derivatives: A Focus on Estrogen Receptor Inhibition" International Journal of Molecular Sciences 26, no. 2: 833. https://doi.org/10.3390/ijms26020833
APA StyleAl-Ghamdi, A. R., Ahmed, W. U., Al-Wabli, R. I., Al-Mutairi, M. S., & Rahman, A. F. M. M. (2025). Synthesis and Anticancer Evaluation of O-Alkylated (E)-Chalcone Derivatives: A Focus on Estrogen Receptor Inhibition. International Journal of Molecular Sciences, 26(2), 833. https://doi.org/10.3390/ijms26020833