

Investigating the Inhibitory Effects of Paliperidone on RAGEs: Docking, DFT, MD Simulations, MMPBSA, MTT, Apoptosis, and Immunoblotting Studies


Abstract
:1. Introduction
2. Results
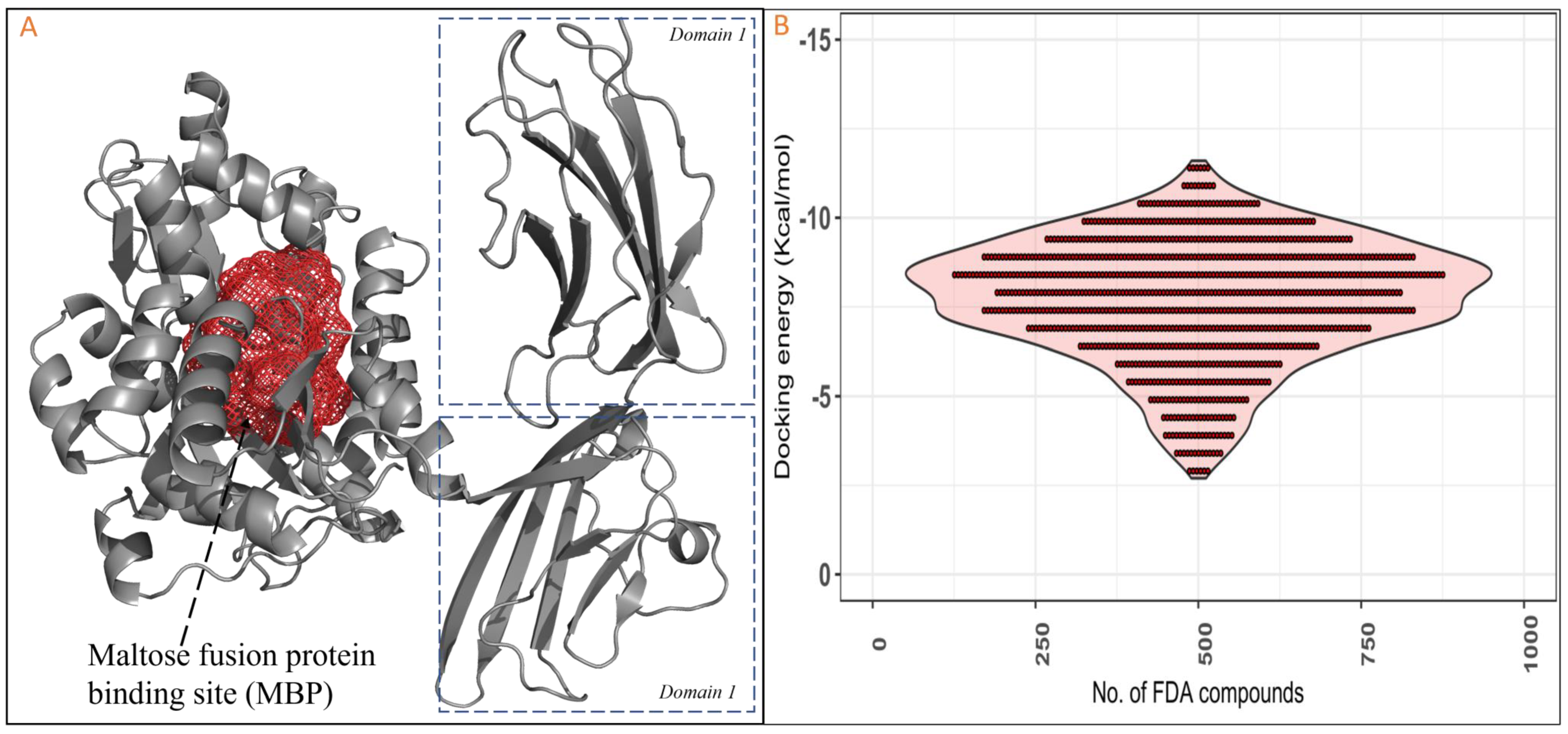
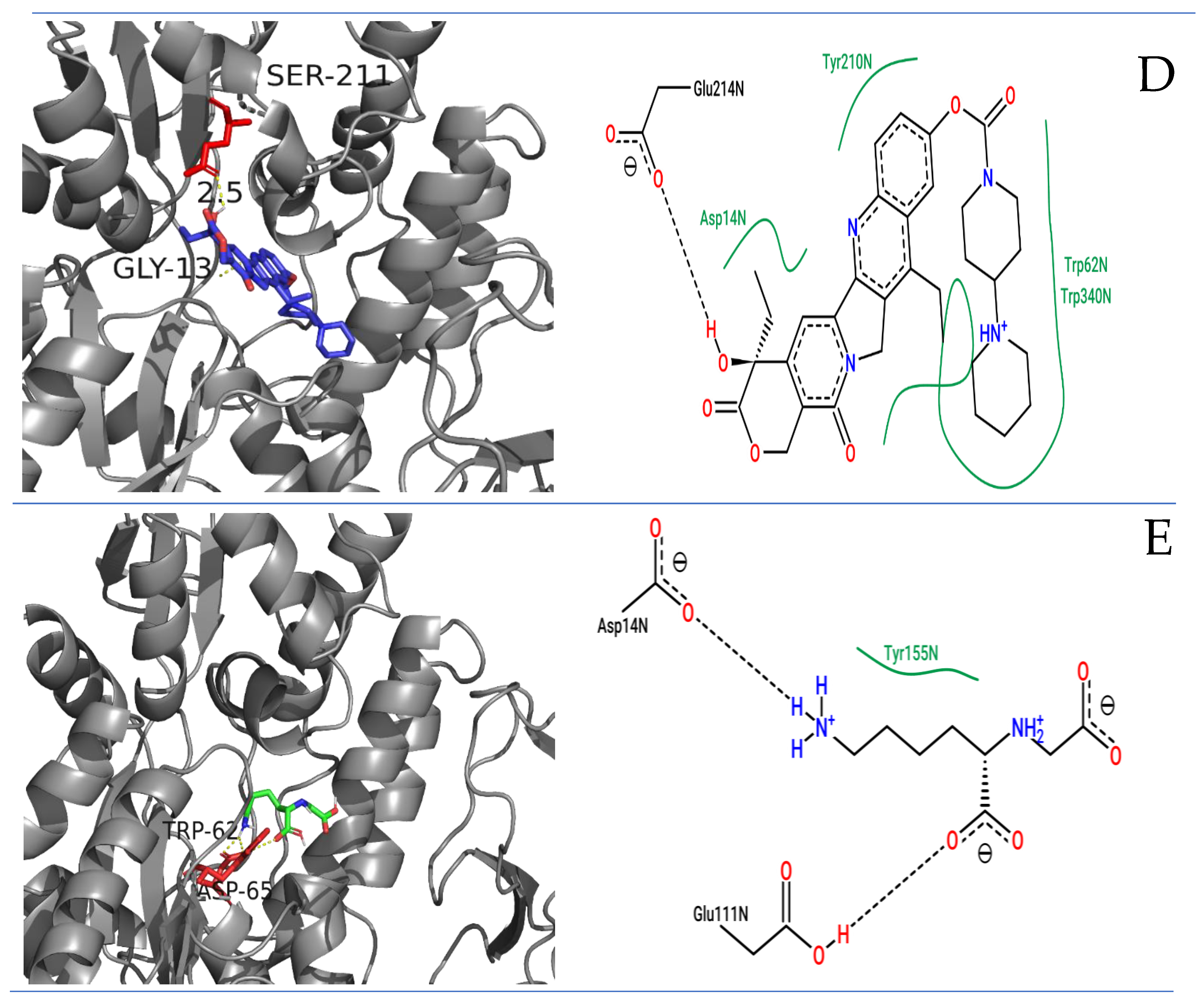
2.1. Molecular Docking Studies of RAGE with FDA Compounds
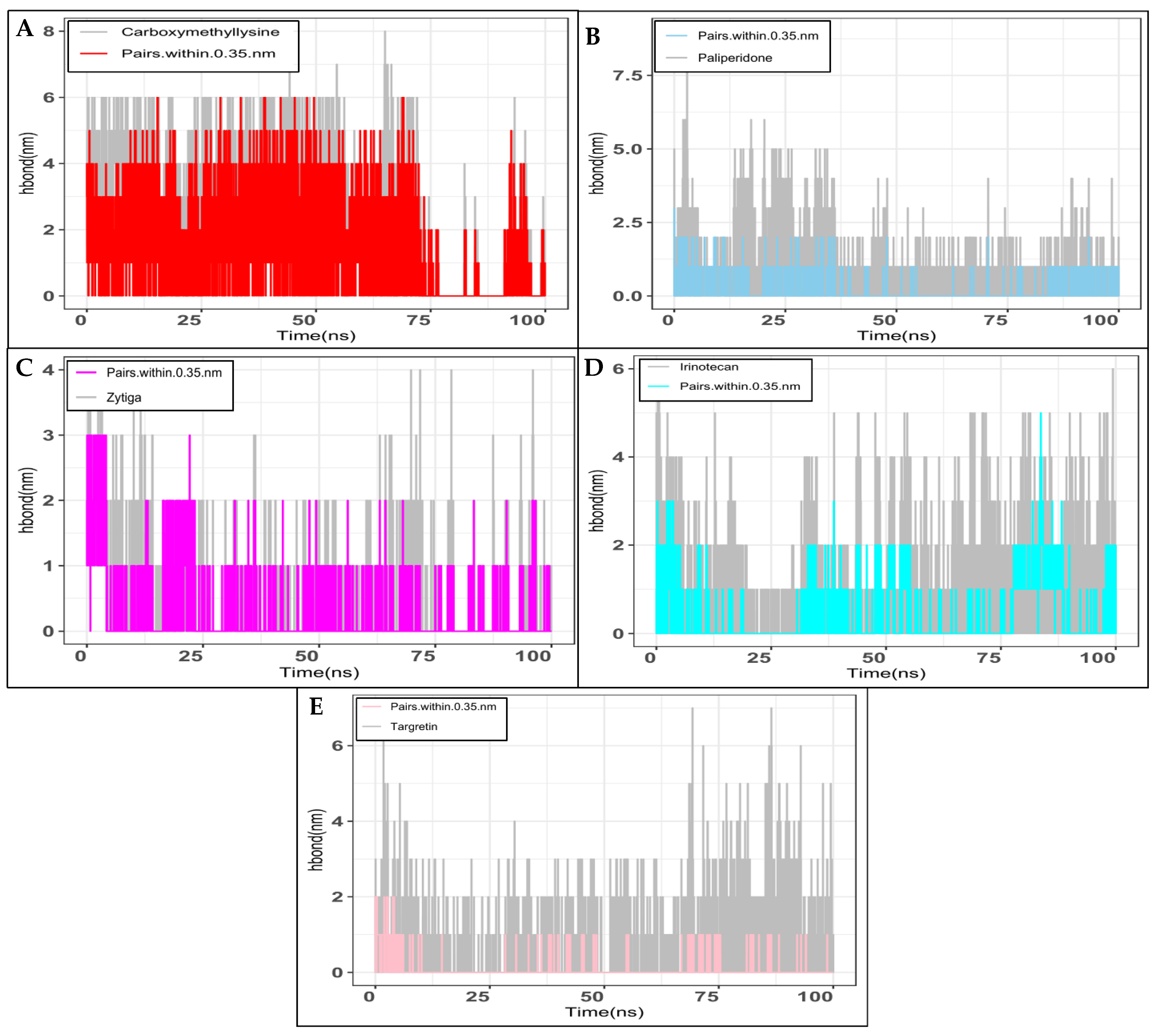
2.2. Molecular Dynamics Simulation Studies
2.3. MMPBSA Analysis
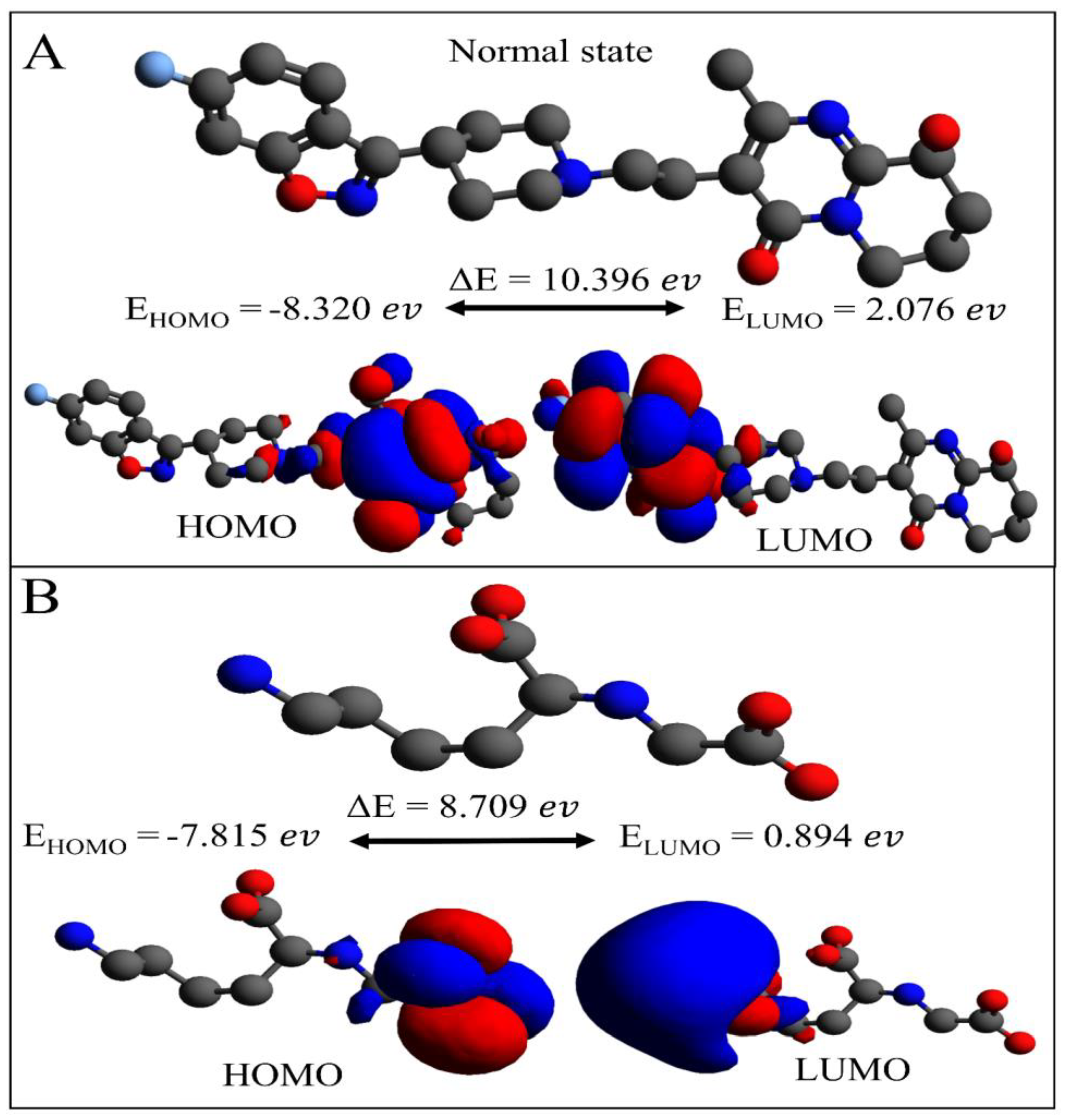
2.4. DFT Computations: Paliperidone and Control Carboxymethyllysine Comparison
2.5. ADME Properties of Selected Paliperidone Molecules
2.6. Effect of Paliperidone on RAGE Expression with/Without HMGB1 Stimulation in MCF7 Cells
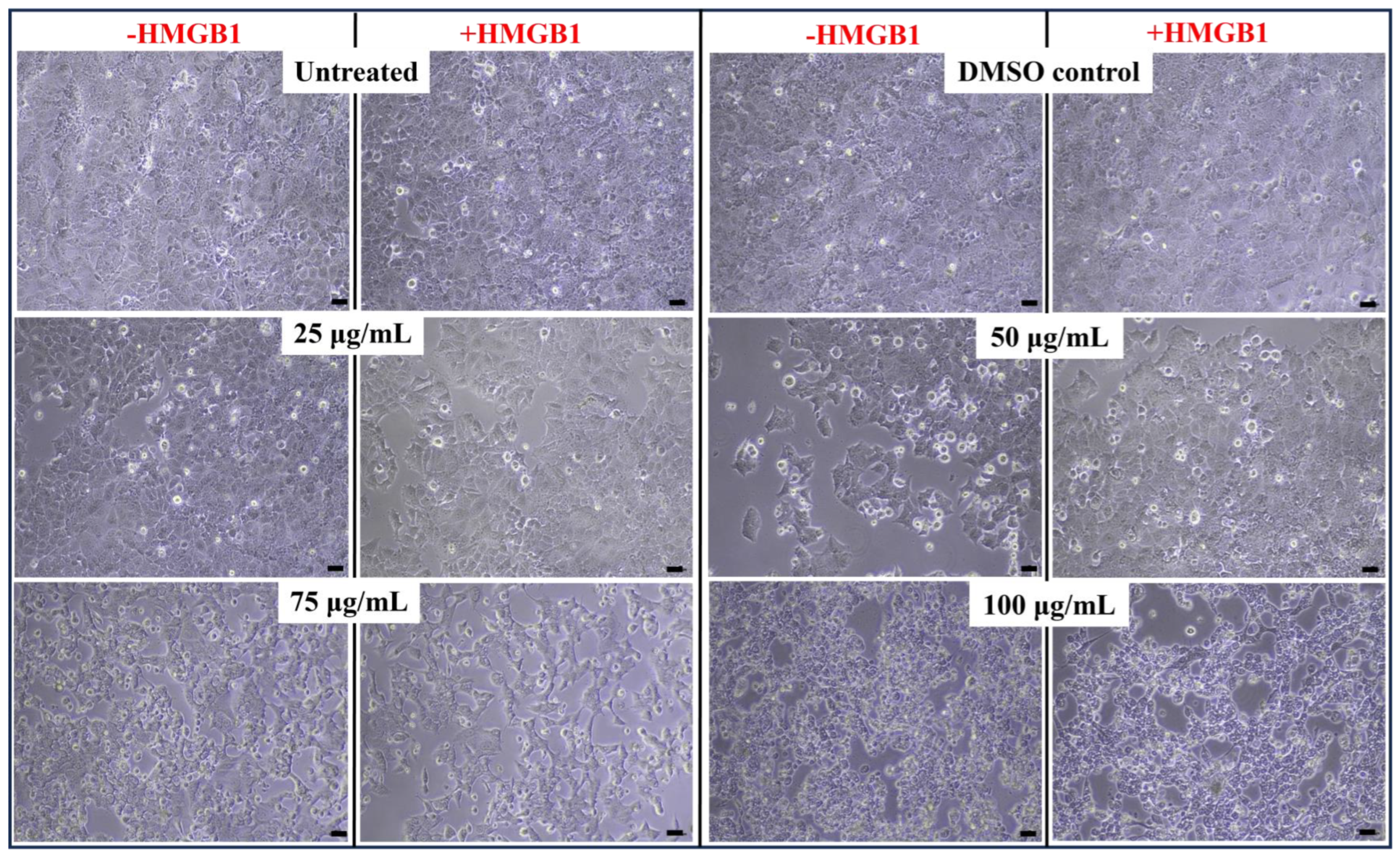
2.7. Cell Viability Effect on HMGB1-Stimulated MCF7 with Paliperidone Treatment
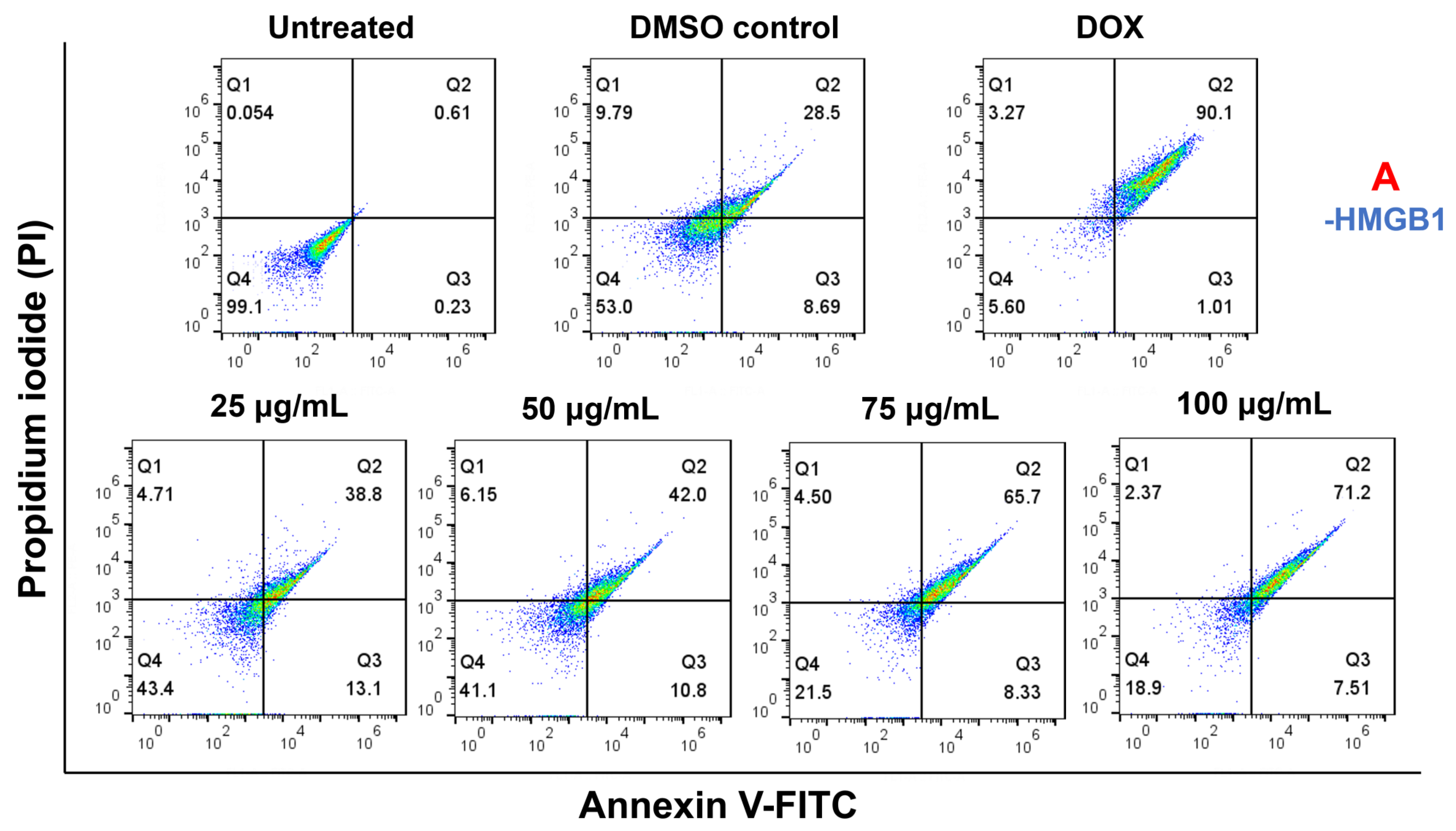
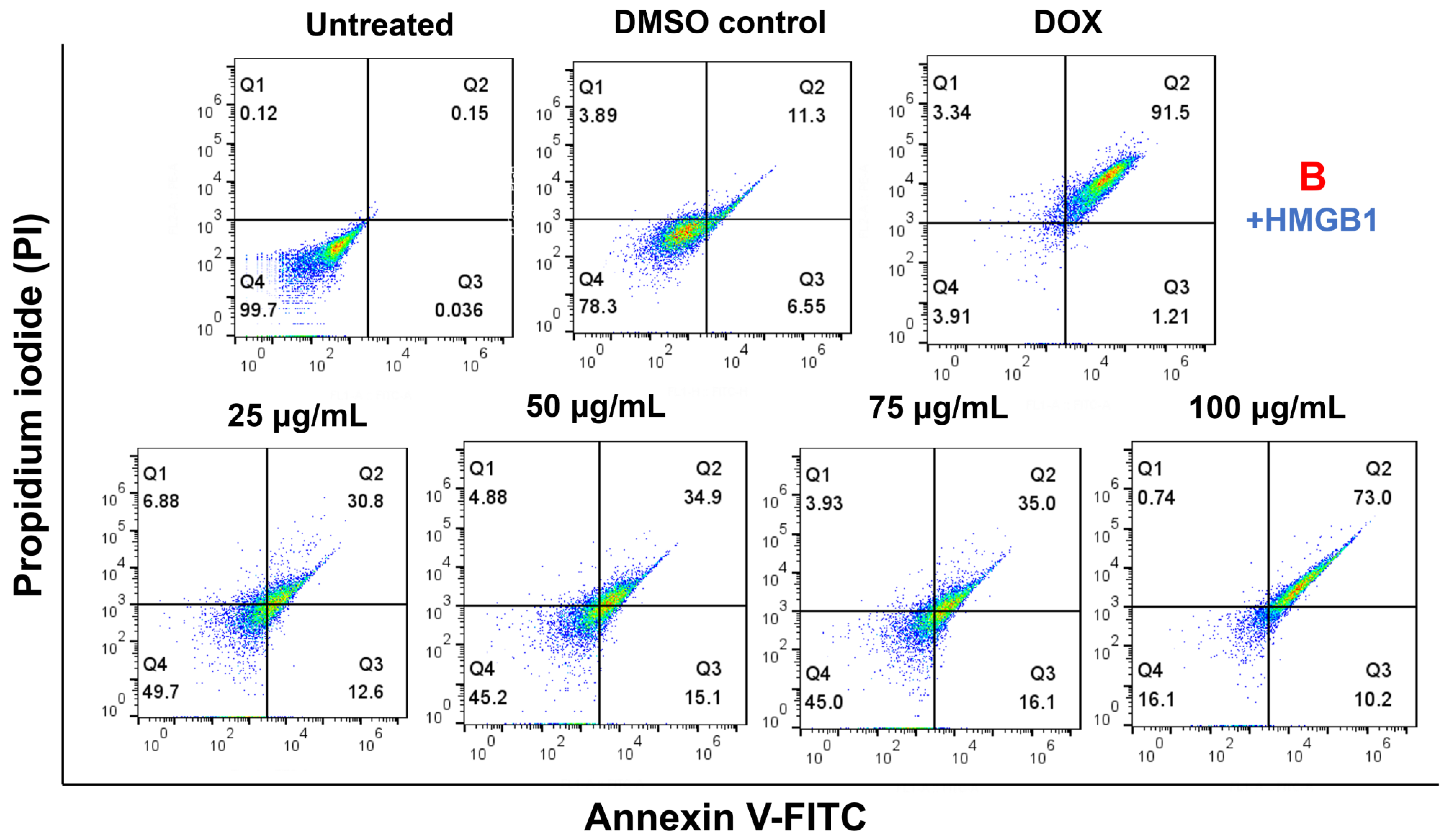
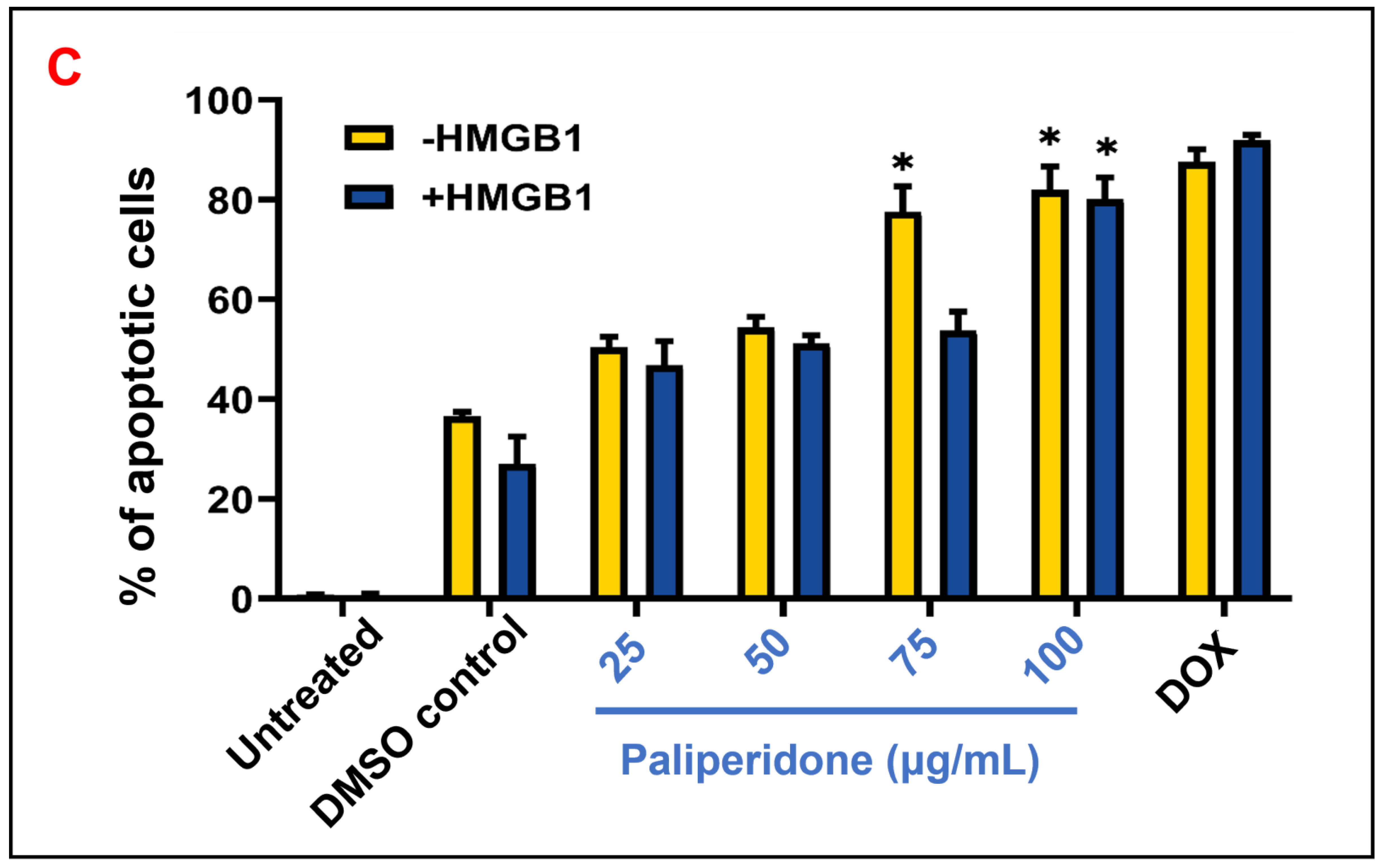
2.8. Cell Apoptosis Effect on HMGB1-Stimulated MCF7 with Paliperidone Treatment
3. Materials and Methods
3.1. Retrieval of Protein and Active Site Prediction
3.2. Ligand Preparation, Selection, and ADME Analysis
3.3. Molecular Docking RAGEs with FDA Compounds
3.4. Molecular Dynamics Simulation of RAGE and FDA-Approved Compounds
3.5. MMPBSA Calculations
3.6. Density Functional Theory Analysis
3.7. Chemicals and Reagents
3.8. Cell Culture and Drug Treatment
3.9. Cell Proliferation Assay (MTT)
3.10. Apoptosis Assay (Annexin-V/PI Staining)
3.11. Immunoblotting
3.12. Statistical Analysis
4. Discussion
5. Conclusions
6. Limitations of This Study and Avenues for Future Research
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhattacharya, S.; Heidler, P.; Varshney, S. Incorporating neglected non-communicable diseases into the national health program-A review. Front. Public Health 2022, 10, 1093170. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan JC, N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Ugai, T.; Sasamoto, N.; Lee, H.Y.; Ando, M.; Song, M.; Tamimi, R.M.; Kawachi, I.; Campbell, P.T.; Giovannucci, E.L.; Weiderpass, E.; et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat. Rev. Clin. Oncol. 2022, 19, 656–673. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de González, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Lega, I.C.; Lipscombe, L.L. Review: Diabetes, Obesity, and Cancer-Pathophysiology and Clinical Implications. Endocr. Rev. 2020, 41, 33–52. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, K. An extensive review on lung cancer therapeutics using machine learning techniques: State-of-the-art and perspectives. J. Drug Target 2024, 32, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Singh, A.P.; Bano, N.; Raza, K.; Singh, J.; Medigeshi, G.R.; Pandey, R.; Gautam, H.K. Integrative analysis discovers Imidurea as dual multitargeted inhibitor of CD69, CD40, SHP2, lysozyme, GATA3, cCBL, and S-cysteinase from SARS-CoV-2 and M. tuberculosis. Int. J. Biol. Macromol. 2024, 270, 132332. [Google Scholar] [CrossRef]
- Famuyiwa, S.O.; Ahmad, S.; Olufolabo, K.O.; Olanudun, E.A.; Bano, N.; Oguntimehin, S.A.; Adesida, S.A.; Oyelekan, E.I.; Raza, K.; Faloye, K.O. Investigating the multitargeted anti-diabetic potential of cucurbitane-type triterpenoid from Momordica charantia: An LC-MS, docking-based MM\GBSA and MD simulation study. J. Biomol. Struct. Dyn. 2023, 1–12. [Google Scholar] [CrossRef]
- Ruiz, H.H.; Ramasamy, R.; Schmidt, A.M. Advanced Glycation End Products: Building on the Concept of the “Common Soil” in Metabolic Disease. Endocrinology 2020, 161, bqz006. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed]
- Sasahira, T.; Kirita, T.; Bhawal, U.K.; Yamamoto, K.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Receptor for advanced glycation end products (RAGE) is important in the prediction of recurrence in human oral squamous cell carcinoma. Histopathology 2007, 51, 166–172. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hasu, M.; Popov, D.; Zhang, J.H.; Chen, J.; Yan, S.D.; Brett, J.; Cao, R.; Kuwabara, K.; Costache, G.; et al. Receptor for advanced glycation end products (AGEs) has a central role in vessel wall interactions and gene activation in response to circulating AGE proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 8807–8811. [Google Scholar] [CrossRef]
- Lue, L.F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; D’Agati, V.; Schmidt, A.M.; Ramasamy, R. Receptor for Advanced Glycation Endproducts (RAGE): A formidable force in the pathogenesis of the cardiovascular complications of diabetes & aging. Curr. Mol. Med. 2007, 7, 699–710. [Google Scholar]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [PubMed]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Dattilo, B.M.; Fritz, G.; Leclerc, E.; Kooi, C.W.; Heizmann, C.W.; Chazin, W.J. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry 2007, 46, 6957–6970. [Google Scholar] [CrossRef] [PubMed]
- HaJeung, P.; Jeffrey, C.B. The 1.5 Å Crystal Structure of Human Receptor for Advanced Glycation Endproducts (RAGE) Ectodomains Reveals Unique Features Determining Ligand Binding. J. Biol. Chem. 2010, 285, 40762–40770. [Google Scholar]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Razak, N.A.; Abu, N.; Ho, W.Y.; Zamberi, N.R.; Tan, S.W.; Alitheen, N.B.; Long, K.; Yeap, S.K. Cytotoxicity of eupatorin in MCF-7 and MDA-MB-231 human breast cancer cells via cell cycle arrest, anti-angiogenesis and induction of apoptosis. Sci. Rep. 2019, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Singh, A.P.; Ahmad, S.; Raza, K.; Gautam, H.K. Computational screening and MM/GBSA-based MD simulation studies reveal the high binding potential of FDA-approved drugs against Cutibacterium acnes sialidase. J. Biomol. Struct. Dyn. 2023, 42, 6245–6255. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Ahmad, S.; Tyagi, R.; Dahiya, V.; Yadav, M.K. Fundamentals of molecular modeling in drug design. In Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Elsevier: Amsterdam, The Netherlands, 2022; pp. 125–155. [Google Scholar]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Chadha, D.; Mehrotra, R. Molecular modeling and spectroscopic studies of semustine binding with DNA and its comparison with lomustine–DNA adduct formation. J. Biomol. Struct. Dyn. 2014, 33, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Baeshen, N.A.; Albeshri, A.O.; Baeshen, N.N.; Attar, R.; Karkashan, A.; Abbas, B.; Bouback, T.A.; Aljaddawi, A.A.; Refai, M.Y.; Abdelkader, H.S.; et al. In silico screening of some compounds derived from the desert medicinal plant Rhazya stricta for the potential treatment of COVID-19. Sci. Rep. 2022, 12, 11120. [Google Scholar] [CrossRef] [PubMed]
- Sharp, M.E.; Vázquez, F.X.; Wagner, J.W.; Dannenhoffer-Lafage, T.; Voth, G.A. Multiconfigurational Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput. 2019, 15, 3306–3315. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterisation and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, K. Identification of 5-nitroindazole as a multitargeted inhibitor for CDK and transferase kinase in lung cancer: A multisampling algorithm-based structural study. Mol. Divers. 2023, 28, 1189–1202. [Google Scholar] [CrossRef]
- Faloye, K.O.; Ahmad, S.; Oyasowo, O.T.; Shalom, E.O.; Bano, N.; Olanudun, E.A.; Kelani, T.O.; Aliyu, H.E.; Raza, K.; Makinde, B.I. Deciphering the influenza neuraminidase inhibitory potential of naturally occurring biflavonoids: An in silico approach. Open Chem. 2024, 22, 20240053. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Buddensiek, D.; Mloston, G.; Matczak, P.; Voss, J. A DFT study on the mechanism of the formation of 1,4,2,3-dithiadiazinanes by head-to-head 3 + 3 cyclodimerization of thiocarbonyl S -imides. J. Phys. Org. Chem. 2020, 34. [Google Scholar] [CrossRef]
- Vantangoli, M.M.; Madnick, S.J.; Huse, S.M.; Weston, P.; Boekelheide, K. MCF-7 Human Breast Cancer Cells Form Differentiated Microtissues in Scaffold-Free Hydrogels. PLoS ONE 2015, 10, e0135426. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Hsing, H.W.; Lai, T.C.; Chen, Y.W.; Lee, T.R.; Chan, H.T.; Lyu, P.C.; Wu, C.L.; Lu, Y.C.; Lin, S.T.; et al. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J. Biomed. Sci. 2010, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Characterisation of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localisation, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell. Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef]
- Karakaş, D.; Ari, F.; Ulukaya, E. The MTT viability assay yields strikingly false-positive viabilities although the cells are killed by some plant extracts. Turk. J. Biol. 2017, 41, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Tangpong, J.; Miriyala, S.; Noel, T.; Sinthupibulyakit, C.; Jungsuwadee, P.; Clair, D.S. Doxorubicin-induced central nervous system toxicity and protection by xanthone derivative of Garcinia mangostana. Neuroscience 2011, 175, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Xie, X.; Cao, P. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed. Pharmacother. 2020, 121, 109139. [Google Scholar]
- He, Q.; You, H.; Li, X.-M.; Liu, T.-H.; Wang, P.; Wang, B.-E. HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by activation of p38 MAPK and NF-κB through receptors for advanced glycation end-products in macrophages. Asian Pac. J. Cancer Prev. 2012, 13, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Srisawat, T.; Sukpondma, Y.; Graidist, P.; Chimplee, S.; Kanokwiroon, K. The dose dependent in vitro responses of MCF-7 and MDA-MB-231 cell lines to extracts of Vatica diospyroides symington type SS fruit include effects on mode of cell death. Pharmacogn. Mag. 2015, 11 (Suppl. S1), S148–S155. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Jain, N.; Kumar, R.; Jain, A.; Singh, N.K.; Rai, V. A comparative method for protein extraction and 2-D gel electrophoresis from different tissues of Cajanus cajan. Front. Plant Sci. 2015, 6, 606. [Google Scholar] [CrossRef]
- Ramlal, A.; Ahmad, S.; Kumar, L.; Khan, F.N.; Chongtham, R. From molecules to patients: The clinical applications of biological databases and electronic health records. In Translational Bioinformatics in Healthcare and Medicine; Academic Press: Cambridge, MA, USA, 2021; pp. 107–125. [Google Scholar]
- Sahu, A.; Ahmad, S.; Imtiyaz, K.; Kizhakkeppurath Kumaran, A.; Islam, M.; Raza, K.; Easwaran, M.; Kurukkan Kunnath, A.; Rizvi, M.A.; Verma, S. In-silico and in-vitro study reveals Ziprasidone as a potential aromatase inhibitor against breast carcinoma. Sci. Rep. 2023, 13, 16545. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.K.; Ahmad, S.; Raza, K.; Kumar, S.; Eswaran, M.; Pasha, K.M.M. Predictive modeling and therapeutic repurposing of natural compounds against the receptor-binding domain of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 41, 1527–1539. [Google Scholar] [CrossRef]
- Chhipa, A.S.; Borse, S.P.; Baksi, R.; Lalotra, S.; Nivsarkar, M. Targeting receptors of advanced glycation end products (RAGE): Preventing diabetes induced cancer and diabetic complications. Pathol. Res. Pract. 2019, 215, 152643. [Google Scholar] [CrossRef]
- Chen, J.; Jing, J.; Yu, S.; Song, M.; Tan, H.; Cui, B.; Huang, L. Advanced glycation endproducts induce apoptosis of endothelial progenitor cells by activating receptor RAGE and NADPH oxidase/JNK signaling axis. Am. J. Transl. Res. 2016, 8, 2169–2178. [Google Scholar]
- López-Díez, R.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Cellular mechanisms and consequences of glycation in atherosclerosis and obesity. Biochim. Biophys. Acta 2016, 1862, 2244–2252. [Google Scholar] [CrossRef]
- Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Glycation & the RAGE axis: Targeting signal transduction through DIAPH1. Expert Rev. Proteom. 2017, 14, 147–156. [Google Scholar]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Du Yan, S.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.A.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.L.; Knight, D.A.; et al. The Role of Pathological Aging in Cardiac and Pulmonary Fibrosis. Aging Dis. 2019, 10, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Singh, V.; Gautam, H.K.; Raza, K. Multisampling-based docking reveals Imidazolidinyl urea as a multitargeted inhibitor for lung cancer: An optimisation followed multi-simulation and in-vitro study. J. Biomol. Struct. Dyn. 2024, 42, 2494–2511. [Google Scholar] [CrossRef] [PubMed]
- Bharti, J.; Sahil, S.M.; Ahmad, S.; Singh, B.; Padhy, A.K.; Srivastava, N.; Pandey, V. Mitogen-Activated Protein Kinase, Plants, and Heat Stress. In Harsh Environment and Plant Resilience: Molecular and Functional Aspects; Springer: Berlin/Heidelberg, Germany, 2021; Volume 1, pp. 323–354. [Google Scholar]
- Khuntia, B.K.; Sharma, V.; Wadhawan, M.; Chhabra, V.; Kidambi, B.; Rathore, S.; Agarwal, A.; Ram, A.; Qazi, S.; Ahmad, S. Antiviral potential of Indian medicinal plants against influenza and SARS-CoV: A systematic review. Nat. Prod. Commun. 2022, 17, 1934578X221086988. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Docking Score | MMPBSA | Analysis | |||||
|---|---|---|---|---|---|---|---|---|
| VDWAALS | EEL | EPB | ENPOLAR | GGAS | GSOLV | TOTAL | ||
| Zytiga | −11.2 | −21.05 | −1.9 | 7.64 | −2.81 | −22.95 | 9.84 | −12.12 |
| Paliperidone | −11.2 | −39.84 | 4.27 | 26.93 | −4.84 | −35.58 | 22.09 | −13.49 |
| Targretin | −11.2 | −25.18 | −2.1 | 17.99 | −3.52 | −27.27 | 14.47 | −12.8 |
| Irinotecan | −11.1 | −41.25 | −11.34 | 46.02 | −4.96 | −52.59 | 41.07 | −11.52 |
| Carboxymethyllysine | −5.0 | −0.81 | −39.19 | 37.13 | −0.87 | −40.0 | 36.27 | −3.73 |
| Categories | Descriptors | Values | Categories | Descriptors | Values |
|---|---|---|---|---|---|
| Physicochemical Properties | Formula | C23H27FN4O3 | Lipophilicity | Log S (ESOL) | −3.95 |
| Molecular weight | 426.48 g/mol | Log Po/w (iLOGP) | 3.86 | ||
| Num. heavy atoms | 31 | Log Po/w (XLOGP3) | 2.17 | ||
| Num. arom. heavy atoms | 15 | Log Po/w (WLOGP) | 2.8 | ||
| Fraction Csp3 | 0.52 | Log Po/w (MLOGP) | 2.39 | ||
| Num. rotatable bonds | 4 | Log Po/w (SILICOS-IT) | 3.58 | ||
| Num. H-bond acceptors | 7 | Consensus Log Po/w | 2.96 | ||
| Num. H-bond donors | 1 | Pharmacokinetics | BBB permeant | No | |
| Molar refractivity | 118.87 | P-gp substrate | Yes | ||
| TPSA | 84.39 Å2 | CYP1A2 inhibitor | No | ||
| Water Solubility | GI absorption | High | CYP2C19 inhibitor | No | |
| Solubility | 4.84 × 10−2 mg/mL; 1.13 × 10−4 mol/L | CYP2C9 inhibitor | No | ||
| Class | Soluble | CYP2D6 inhibitor | Yes | ||
| Log S (Ali) | −3.58 | CYP3A4 inhibitor | Yes | ||
| Solubility | 1.13 × 10−1 mg/mL; 2.66 × 10−4 mol/L | Drug Likeness | Lipinski | Yes; 0 violation | |
| Class | Soluble | Ghose | Yes | ||
| Log S (SILICOS-IT) | −5.94 | Veber | Yes | ||
| Solubility | 4.90 × 10−4 mg/mL; 1.15 × 10−6 mol/L | Egan | Yes | ||
| Class | Moderately soluble | Muegge | Yes | ||
| Log Kp (skin permeation) | −7.36 cm/s | Bioavailability score | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.P.; Ahmad, S.; Roy, A.; Raza, K.; Gautam, H.K. Investigating the Inhibitory Effects of Paliperidone on RAGEs: Docking, DFT, MD Simulations, MMPBSA, MTT, Apoptosis, and Immunoblotting Studies. Int. J. Mol. Sci. 2025, 26, 1060. https://doi.org/10.3390/ijms26031060
Singh AP, Ahmad S, Roy A, Raza K, Gautam HK. Investigating the Inhibitory Effects of Paliperidone on RAGEs: Docking, DFT, MD Simulations, MMPBSA, MTT, Apoptosis, and Immunoblotting Studies. International Journal of Molecular Sciences. 2025; 26(3):1060. https://doi.org/10.3390/ijms26031060
Chicago/Turabian StyleSingh, Akash Pratap, Shaban Ahmad, Ahona Roy, Khalid Raza, and Hemant K. Gautam. 2025. "Investigating the Inhibitory Effects of Paliperidone on RAGEs: Docking, DFT, MD Simulations, MMPBSA, MTT, Apoptosis, and Immunoblotting Studies" International Journal of Molecular Sciences 26, no. 3: 1060. https://doi.org/10.3390/ijms26031060
APA StyleSingh, A. P., Ahmad, S., Roy, A., Raza, K., & Gautam, H. K. (2025). Investigating the Inhibitory Effects of Paliperidone on RAGEs: Docking, DFT, MD Simulations, MMPBSA, MTT, Apoptosis, and Immunoblotting Studies. International Journal of Molecular Sciences, 26(3), 1060. https://doi.org/10.3390/ijms26031060