
Integrated Transcriptomics and Metabolomics Analysis Reveals Convergent and Divergent Key Molecular Networks of Dominant Genic Male Sterility and Cytoplasmic Male Sterility in Cabbage


 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. TEM Differences Between Ogura CMS20-2-5 and DGMS20-2-5
2.2. Transcriptome Sequencing and Differential Gene Screening
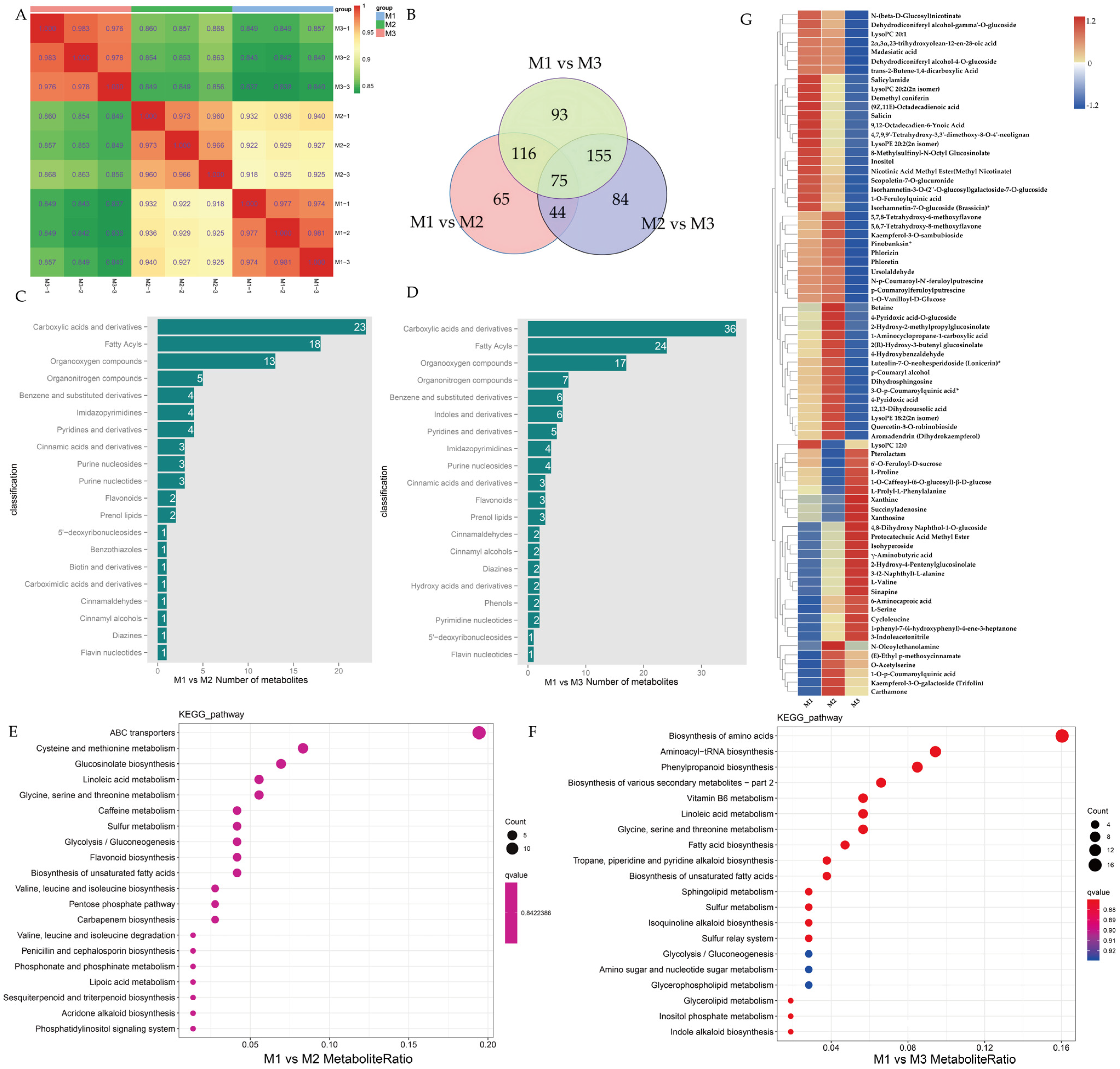
2.3. Metabolomic Measurements and Analysis
2.4. Transcriptional Regulatory Network Analysis of Pollen Wall Development in Cabbage
2.5. Carbohydrate Metabolic Pathway Transcriptome Metabolome Integration Analysis
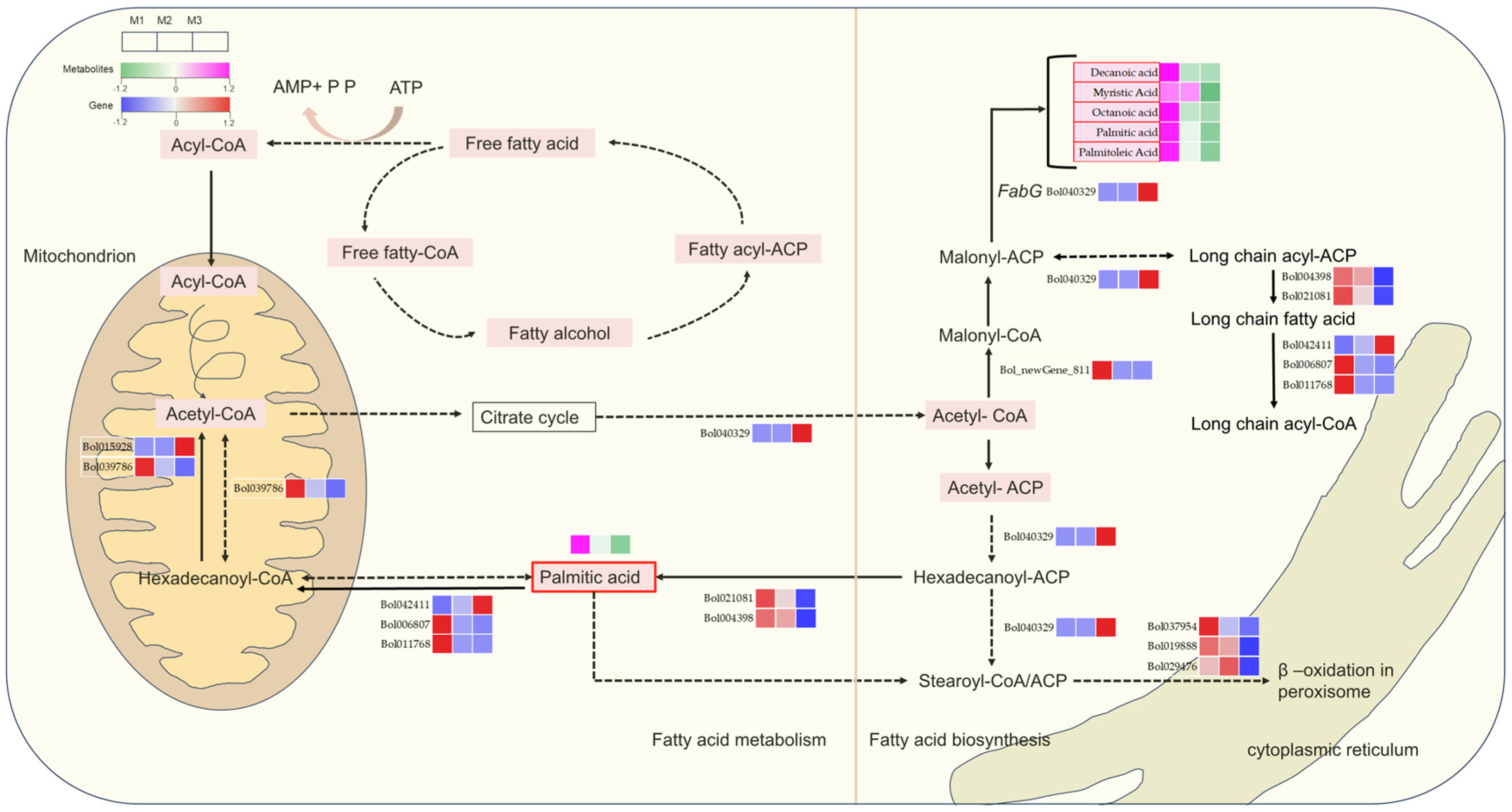
2.6. The Critical Role of Lipid Biosynthesis and Metabolism in Male Gametophyte Development
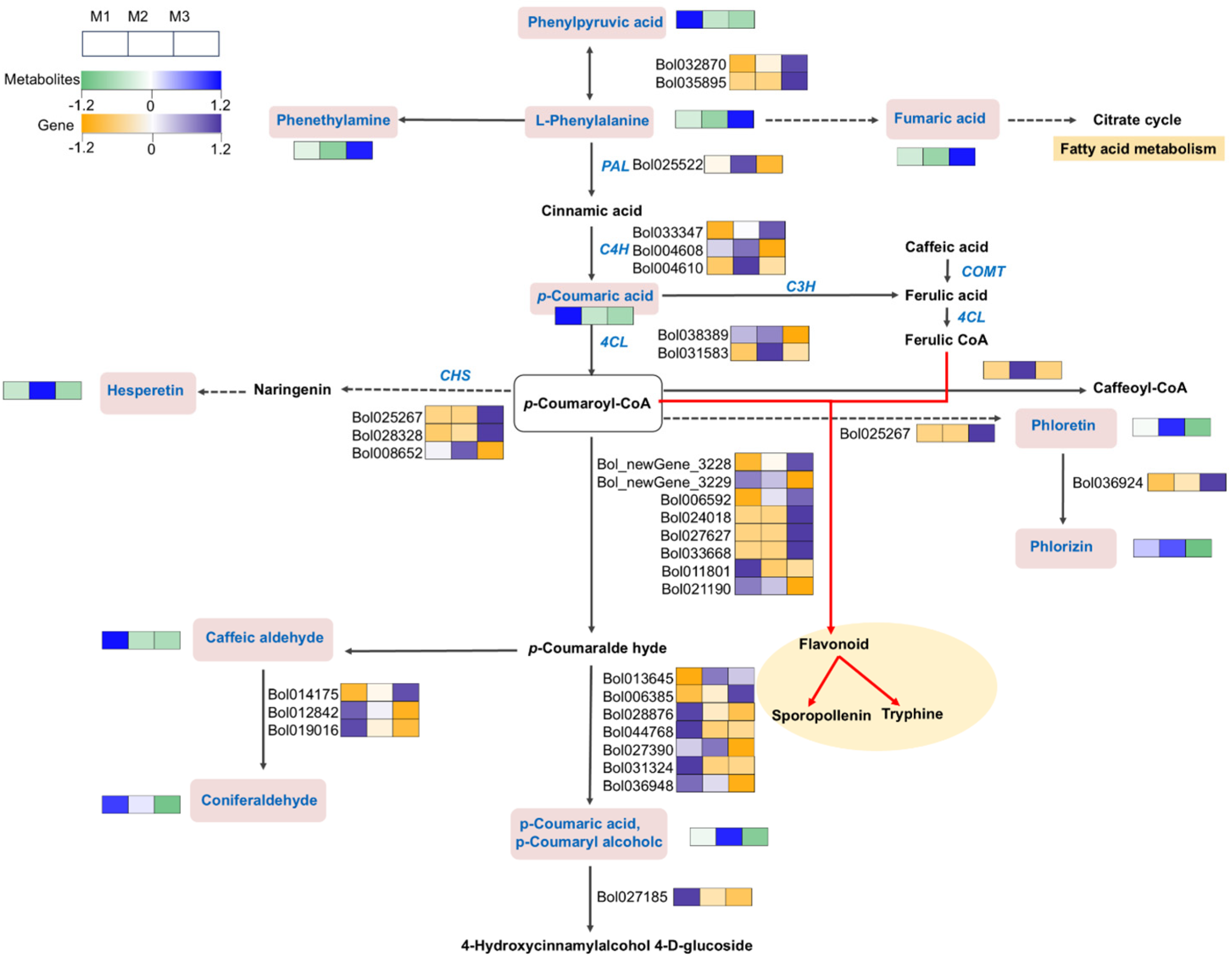
2.7. Disruption of Phenylpropanoid Metabolism and Synthesis (Flavonoids) in CMS Lineage Development
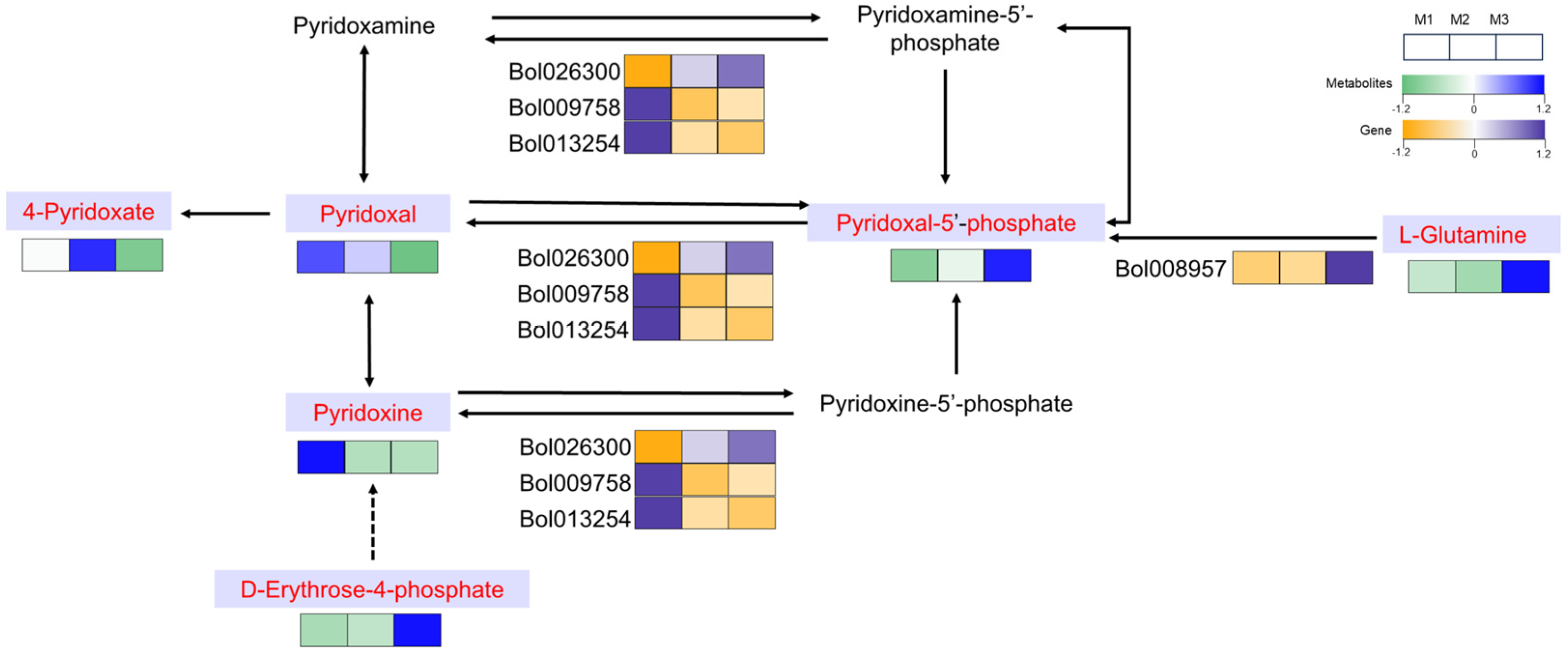
2.8. The Unique Role of Vitamin B6 Metabolism in the CMS Lineage
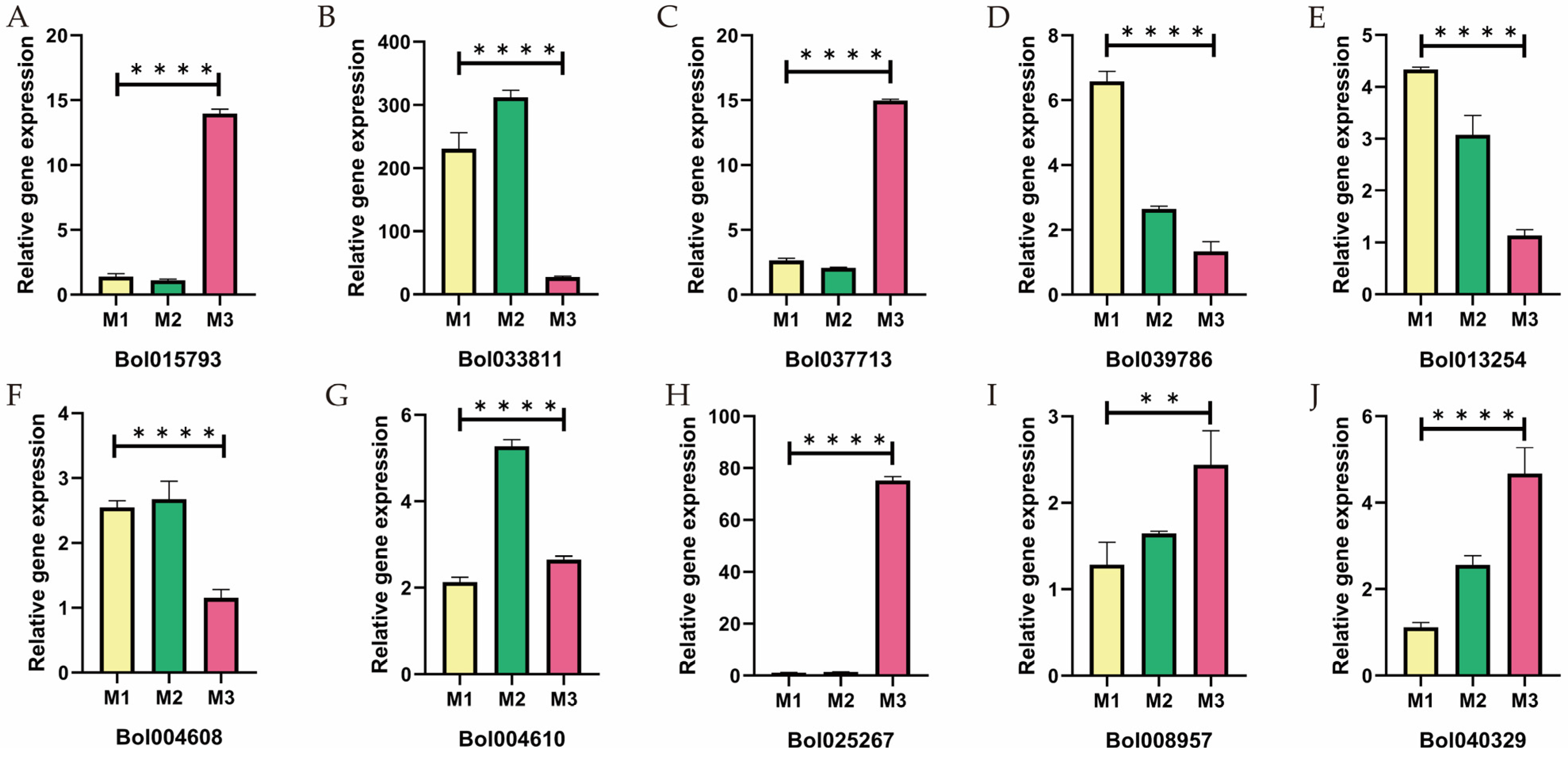
2.9. qRT-PCR Analysis
3. Discussion
3.1. Differences in the Regulatory Network of Pollen Wall Development Between the DGMS and CMS Lines Contribute to the Differences in Their Respective Sterility Phenotypes
3.2. Carbohydrate Metabolism and Fertility
3.3. Impaired Lipid Metabolic Pathways Are Closely Related to Pollen Wall Mechanisms
3.4. Importance of Phenylpropanoid Compounds in Maintaining Fertility
3.5. Future Research Directions and Applications
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Cytological Microscopy
4.3. RNA Extraction and Transcriptome Sequencing
4.4. Metabolite Extraction and Analysis Methods
4.5. Bioinformatic and Statistical Analysis
4.6. Real-Time Quantitative PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Ling, J.; Xiao, Z.; Xie, B.; Fang, Z.; Yang, L.; Zhang, Y.; Lv, H.; Yang, Y. Characterization of emerging populations of Fusarium oxysporum f. sp. conglutinans causing cabbage wilt in China. J. Phytopathol. 2017, 165, 813–821. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, L.; Li, Z.; Cheng, M.; Chen, X.; Wang, A.; Wang, C.; Zhang, X. DIA-based proteome profiling with PRM verification reveals the involvement of ER-associated protein processing in pollen abortion in Ogura CMS cabbage. Hortic. Plant J. 2024, 10, 755–770. [Google Scholar] [CrossRef]
- Sinskaja, E. The Oleiferous Plants and Root Crops of the Family Cruciferae; Instituta Irikladnoi Botaniki i Novich Kultur: Leningrad, Russia, 1928. [Google Scholar]
- Schiemann, E. Entstehung der Kulturpflanzen; Borntraeger: Berlin, Germany, 1932. [Google Scholar]
- Tanaka, N.; Niikura, S. Genetic analysis of the developmental characteristics related to the earliness of head formation in cabbage (Brassica oleracea L.). Breed. Sci. 2006, 56, 147–153. [Google Scholar] [CrossRef]
- Cheng, F.; Sun, R.; Hou, X.; Zheng, H.; Zhang, F.; Zhang, Y.; Liu, B.; Liang, J.; Zhuang, M.; Liu, Y. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat. Genet. 2016, 48, 1218–1224. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Zheng, R.; Song, X. Research progress on biological functions of lncRNAs in major vegetable crops. Veg. Res. 2022, 2, 1–14. [Google Scholar] [CrossRef]
- Tang, M.; Li, Z.; Luo, D.; Wei, F.; Kashif, M.H.; Lu, H.; Hu, Y.; Yue, J.; Huang, Z.; Tan, W. A comprehensive integrated transcriptome and metabolome analyses to reveal key genes and essential metabolic pathways involved in CMS in kenaf. Plant Cell Rep. 2021, 40, 223–236. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, T.; Li, X.; Duan, M.; Wang, J.; Qiu, Y.; Wang, H.; Song, J.; Shen, D. Interspecific hybridization, polyploidization and backcross of Brassica oleracea var. alboglabra with B. rapa var. purpurea morphologically recapitulate the evolution of Brassica vegetables. Sci. Rep. 2016, 6, 18618. [Google Scholar] [CrossRef]
- Dhall, R. Status of male sterility in vegetables for hybrid development: A review. Adv. Hortic. Sci. [Riv. Dell’ortofloroftutticoltura Ital.] 2010, 24, 1000–1017. [Google Scholar]
- Lv, J.; Deng, M.; Li, Z.; Zhu, H.; Wang, Z.; Yue, Y.; Yang, Z.; Xu, J.; Jiang, S.; Zhao, W. Integrative analysis of the transcriptome and metabolome reveals the response mechanism to tomato spotted wilt virus. Hortic. Plant J. 2023, 9, 958–970. [Google Scholar] [CrossRef]
- Yamagishi, H.; Bhat, S.R. Cytoplasmic male sterility in Brassicaceae crops. Breed. Sci. 2014, 64, 38–47. [Google Scholar] [CrossRef]
- Ogura, H. Studies on a New Male-Sterility in Japanese Radish, with Special Reference to the Utilization of This Sterility Towards the Practical Resing of Hybrid Seeds. Ph.D. Thesis, Kyushu University, Fukuoka, Japan, 1967. [Google Scholar]
- Bannerot, H.; Boulidard, L.; Cauderon, Y.; Tempe, J. Transfer of cytoplasmic male sterility from Raphanus sativus to Brassica oleracea. Proc. Eucarpia Meet. Crucif. 1974, 25, 52–54. [Google Scholar]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Tester, M.; Langridge, P. Breeding technologies to increase crop production in a changing world. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kumari, V. A review on male sterility-concepts and utilization in vegetable crops. Int. J. Curr. Microbiol. Appl. Sci. 2018, 7, 3016–3034. [Google Scholar] [CrossRef]
- Walters, T.W.; Mutschler, M.A.; Earle, E.D. Protoplast fusion-derived Ogura male sterile cauliflower with cold tolerance. Plant Cell Rep. 1992, 10, 624–628. [Google Scholar] [CrossRef]
- Yu, H.-l.; Li, Z.-y.; Ren, W.-j.; Han, F.-q.; Yang, L.-m.; Zhuang, M.; Lv, H.-h.; Liu, Y.-m.; Fang, Z.-y.; Zhang, Y.-y. Creation of fertility-restored materials for Ogura CMS in Brassica oleracea by introducing Rfo gene from Brassica napus via an allotriploid strategy. Theor. Appl. Genet. 2020, 133, 2825–2837. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, S.; Singh, B. Heterosis for mineral elements in single cross-hybrids of cabbage (Brassica oleracea var. capitata L.). Sci. Hortic. 2009, 122, 32–36. [Google Scholar] [CrossRef]
- Fang, Z.; Sun, P.; Liu, Y.; Yang, L.; Wang, X.; Hou, A.; Bian, C. A male sterile line with dominant gene (Ms) in cabbage (Brassica oleracea var. capitata) and its utilization for hybrid seed production. Euphytica 1997, 97, 265–268. [Google Scholar] [CrossRef]
- Dey, S.S.; Bhatia, R.; Parkash, C.; Sharma, S.; Dabral, M.; Mishra, V.; Bhardwaj, I.; Sharma, K.; Sharma, V.K.; Kumar, R. Alteration in important quality traits and antioxidant activities in Brassica oleracea with Ogura cybrid cytoplasm. Plant Breed. 2017, 136, 400–409. [Google Scholar] [CrossRef]
- Singh, S.; Bhatia, R.; Kumar, R.; Behera, T.K.; Kumari, K.; Pramanik, A.; Ghemeray, H.; Sharma, K.; Bhattacharya, R.; Dey, S.S. Elucidating mitochondrial DNA markers of Ogura-based CMS lines in Indian cauliflowers (Brassica oleracea var. botrytis L.) and their floral abnormalities due to diversity in cytonuclear interactions. Front. Plant Sci. 2021, 12, 631489. [Google Scholar] [CrossRef]
- Tang, B.; Yang, H.; Zhang, X.; Du, J.; Xie, L.; Dai, X.; Zou, X.; Liu, F. A global view of transcriptome dynamics during flower development in Capsicum annuum L. Hortic. Plant J. 2023, 9, 999–1012. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Srivastava, D. High frequency organogenesis in hypocotyl, cotyledon, leaf and petiole explants of broccoli (Brassica oleracea L. var. italica), an important vegetable crop. Physiol. Mol. Biol. Plants 2015, 21, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Shi, J.; Cui, M.; Yang, L.; Kim, Y.-J.; Zhang, D. Genetic and biochemical mechanisms of pollen wall development. Trends Plant Sci. 2015, 20, 741–753. [Google Scholar] [CrossRef]
- Xu, J.; Yang, C.; Yuan, Z.; Zhang, D.; Gondwe, M.Y.; Ding, Z.; Liang, W.; Zhang, D.; Wilson, Z.A. The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell 2010, 22, 91–107. [Google Scholar] [CrossRef]
- Wang, H.; Zong, C.; Bai, Y.; Yuan, S.; Li, Y.; Han, T.; Xu, H.; Hu, C.; Hou, X.; Li, Y. Comparative transcriptome-based identification and expression analysis of ATP-binding cassette (ABC) transporters reveals a likely role in the transport of β-caryophyllene and response to abiotic stress in Brassica campestris. Veg. Res. 2023, 3, 13. [Google Scholar] [CrossRef]
- Clément, C.; Pacini, E. Anther plastids in angiosperms. Bot. Rev. 2001, 67, 54–73. [Google Scholar] [CrossRef]
- Li, H.; Pinot, F.; Sauveplane, V.; Werck-Reichhart, D.; Diehl, P.; Schreiber, L.; Franke, R.; Zhang, P.; Chen, L.; Gao, Y. Cytochrome P450 family member CYP704B2 catalyzes the ω-hydroxylation of fatty acids and is required for anther cutin biosynthesis and pollen exine formation in rice. Plant Cell 2010, 22, 173–190. [Google Scholar] [CrossRef]
- Ariizumi, T.; Toriyama, K. Genetic regulation of sporopollenin synthesis and pollen exine development. Annu. Rev. Plant Biol. 2011, 62, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, D. Biosynthesis of anther cuticle and pollen exine in rice. Plant Signal. Behav. 2010, 5, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lou, Y.; Xu, X.; Yang, Z.N. A genetic pathway for tapetum development and function in Arabidopsis. J. Integr. Plant Biol. 2011, 53, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Yuan, K.; Sun, W.; Zhang, X.; Liu, X.; Zhao, X.; Yang, L.; Wang, Y.; Ji, J.; Liu, Y. A natural mutation in the promoter of Ms-cd1 causes dominant male sterility in Brassica oleracea. Nat. Commun. 2023, 14, 6212. [Google Scholar] [CrossRef]
- Post-Beittenmiller, D.; Roughan, G.; Ohlrogge, J.B. Regulation of plant fatty acid biosynthesis: Analysis of acyl-coenzyme a and acyl-acyl carrier protein substrate pools in spinach and pea chloroplasts. Plant Physiol. 1992, 100, 923–930. [Google Scholar] [CrossRef]
- Shi, J.; Tan, H.; Yu, X.-H.; Liu, Y.; Liang, W.; Ranathunge, K.; Franke, R.B.; Schreiber, L.; Wang, Y.; Kai, G. Defective pollen wall is required for anther and microspore development in rice and encodes a fatty acyl carrier protein reductase. Plant Cell 2011, 23, 2225–2246. [Google Scholar] [CrossRef]
- Huang, H.; Yang, X.; Zheng, M.; Chen, Z.; Yang, Z.; Wu, P.; Jenks, M.A.; Wang, G.; Feng, T.; Liu, L. An ancestral role for 3-KETOACYL-COA SYNTHASE3 as a negative regulator of plant cuticular wax synthesis. Plant Cell 2023, 35, 2251–2270. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Zheng, X.; Yang, J.; Imamichi, T.; Stephens, R.; Lempicki, R.A. Extracting biological meaning from large gene lists with DAVID. Curr. Protoc. Bioinform. 2009, 27, 13.11.1–13.11.13. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Clean Reads | Clean Bases | GC (%) | ≥Q30 (%) |
|---|---|---|---|---|
| M1-1 | 22073512 | 6588876216 | 47.27 | 93.62 |
| M1-2 | 25223728 | 7547719730 | 46.88 | 91.61 |
| M1-3 | 19246583 | 5759013734 | 46.93 | 92.26 |
| M2-1 | 25705343 | 7665178646 | 47.55 | 94.13 |
| M2-2 | 31271530 | 9327533842 | 47.39 | 94.21 |
| M2-3 | 22920781 | 6856184676 | 47.25 | 93.19 |
| M3-1 | 19767572 | 5914870538 | 47.31 | 92.97 |
| M3-2 | 21603206 | 6462452108 | 47.32 | 92.81 |
| M3-3 | 22816605 | 6826911388 | 47.42 | 93.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Kuang, L.; Yang, L.; Wang, Y.; Han, F.; Zhang, Y.; Wang, S.; Lv, H.; Ji, J. Integrated Transcriptomics and Metabolomics Analysis Reveals Convergent and Divergent Key Molecular Networks of Dominant Genic Male Sterility and Cytoplasmic Male Sterility in Cabbage. Int. J. Mol. Sci. 2025, 26, 1259. https://doi.org/10.3390/ijms26031259
Zhang N, Kuang L, Yang L, Wang Y, Han F, Zhang Y, Wang S, Lv H, Ji J. Integrated Transcriptomics and Metabolomics Analysis Reveals Convergent and Divergent Key Molecular Networks of Dominant Genic Male Sterility and Cytoplasmic Male Sterility in Cabbage. International Journal of Molecular Sciences. 2025; 26(3):1259. https://doi.org/10.3390/ijms26031259
Chicago/Turabian StyleZhang, Nan, Linqian Kuang, Limei Yang, Yong Wang, Fengqing Han, Yangyong Zhang, Shaohui Wang, Honghao Lv, and Jialei Ji. 2025. "Integrated Transcriptomics and Metabolomics Analysis Reveals Convergent and Divergent Key Molecular Networks of Dominant Genic Male Sterility and Cytoplasmic Male Sterility in Cabbage" International Journal of Molecular Sciences 26, no. 3: 1259. https://doi.org/10.3390/ijms26031259
APA StyleZhang, N., Kuang, L., Yang, L., Wang, Y., Han, F., Zhang, Y., Wang, S., Lv, H., & Ji, J. (2025). Integrated Transcriptomics and Metabolomics Analysis Reveals Convergent and Divergent Key Molecular Networks of Dominant Genic Male Sterility and Cytoplasmic Male Sterility in Cabbage. International Journal of Molecular Sciences, 26(3), 1259. https://doi.org/10.3390/ijms26031259