
Exploiting the Molecular Properties of Fibrinogen to Control Bleeding Following Vascular Injury

Abstract
1. Introduction
2. Fibrinogen Structure and Function
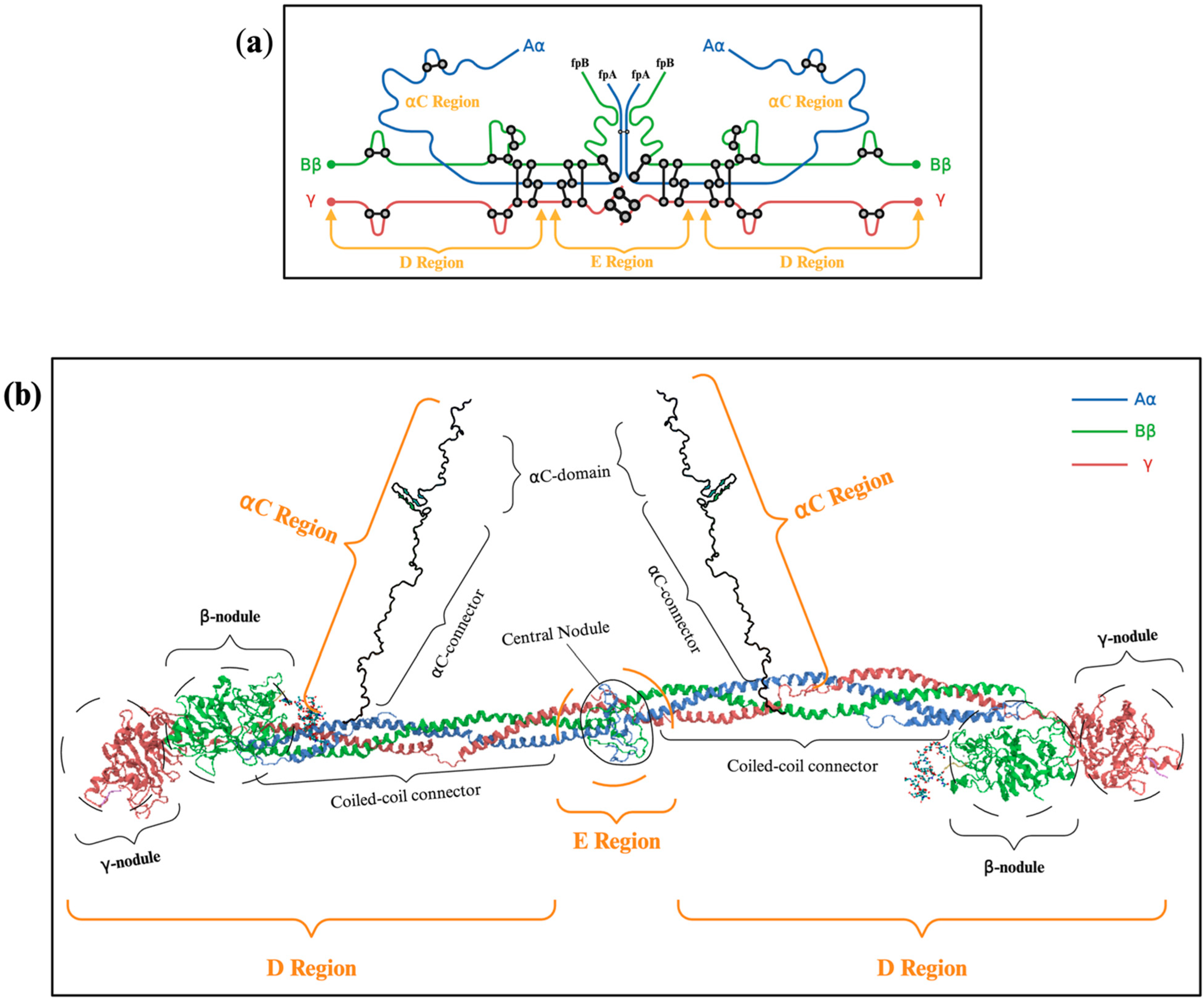
2.1. Subsection Description of Fibrinogen’s Molecular Structure: Aα, Bβ γ Chains
2.2. Mechanism of Conversion from Fibrinogen to Fibrin by Thrombin
2.3. Structural Role of Fibrin in Blood Clot Formation and Wound Healing
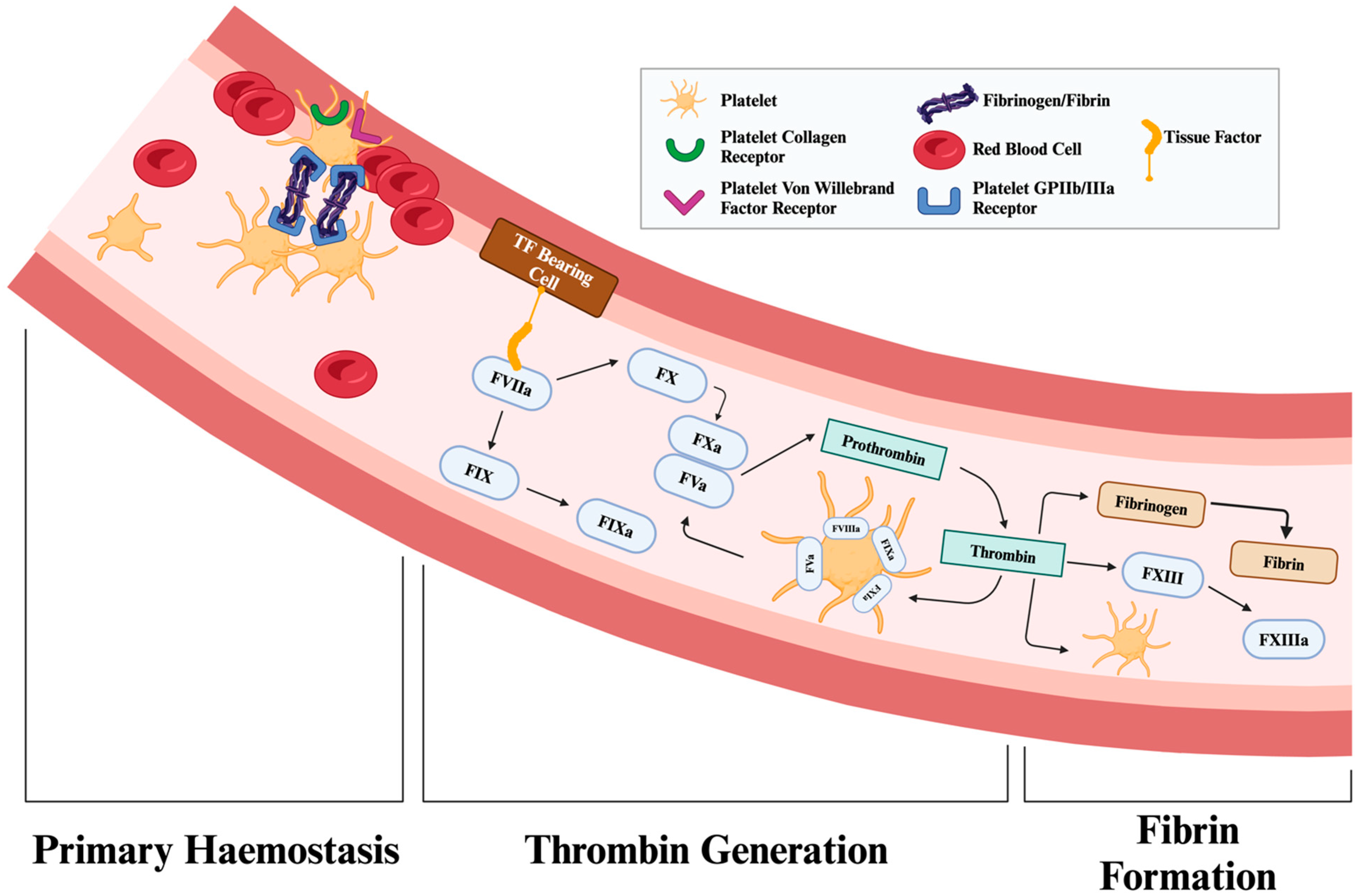
3. Molecular Mechanisms of Fibrinogen in Bleeding Reduction
3.1. Cross-Linking Fibrin
3.2. Platelet Aggregation and Clot Retraction: Molecular Interplay with Fibrin
3.2.1. Fibrin Mechanisms of Platelet Aggregation
3.2.2. Clot Retraction
3.3. Role of Fibrinolysis
3.3.1. Activation of Plasmin and Fibrin Degradation
3.3.2. Anti-Fibrinolytic Mechanisms
3.4. Clot Permeability and Contribution to Wound Healing
4. Clinical Applications of Fibrinogen
4.1. Trauma and Acute Bleeding
4.2. Obstetric Haemorrhage
4.3. Cardiac and General Surgery
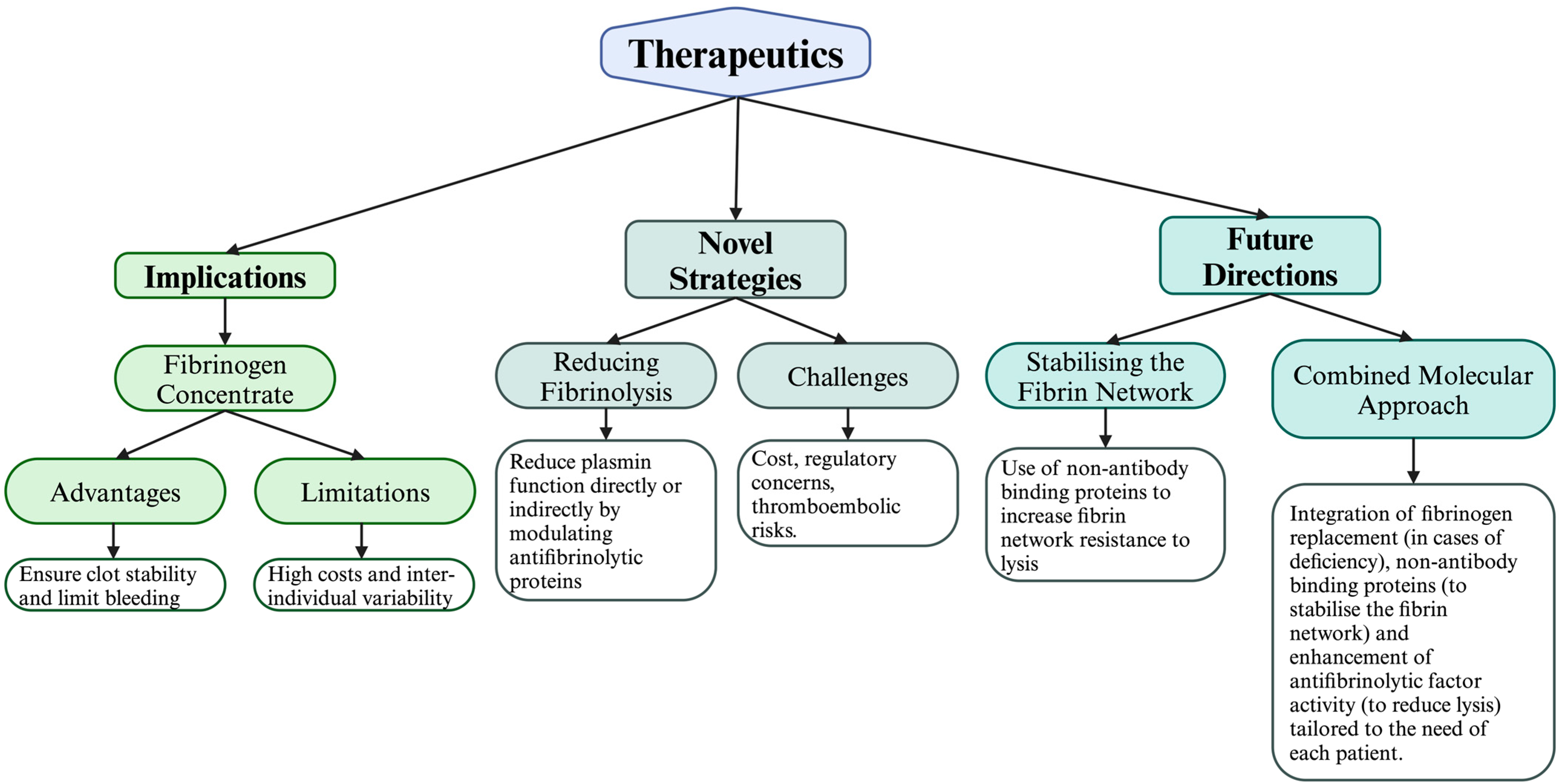
5. Therapeutic Implications and Future Research
5.1. The Role of TEG/ROTEM
5.2. Fibrinogen Replacement
5.2.1. Advantages of Fibrinogen Concentrates
5.2.2. Observational Evidence in Postpartum Haemorrhage and Cardiac Surgery
5.2.3. Empirical and Observational Evidence from the FIBRES Trial and Post Hoc Analyses
5.3. Tranexamic Acid Administration
5.3.1. Trauma
5.3.2. Postpartum Haemorrhage
5.4. Novel Strategies for Enhancing Fibrin Network Stability and Reducing Fibrinolysis
5.5. Personalised Medicine and Genetic Variations in Fibrinogen Genes
6. Future Directions
6.1. Stabilising the Fibrin Network with Affimers
6.2. Increasing Resistance to Lysis via α2-Antiplasmin
6.3. A Combined Molecular Approach
7. Summary of Key Findings
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaur, J.; Jain, A. Fibrinogen. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537184/#:~:text=%5B9%5D%20The%20minimum%20amount%20of (accessed on 1 January 2022).
- Pechlivani, N.; Kearney, K.J.; Ajjan, R.A. Fibrinogen and Antifibrinolytic Proteins: Interactions and Future Therapeutics. Int. J. Mol. Sci. 2021, 22, 12537. [Google Scholar] [CrossRef]
- Rossaint, R.; Bouillon, B.; Cerny, V.; Coats, T.J.; Duranteau, J.; Fernández-Mondéjar, E.; Hunt, B.J.; Komadina, R.; Nardi, G.; Neugebauer, E.; et al. Management of bleeding following major trauma: An updated European guideline. Crit. Care 2010, 14, R52. [Google Scholar] [CrossRef]
- Hakkenbrak, N.A.G.; Mikdad, S.Y.; Zuidema, W.P.; Halm, J.A.; Schoonmade, L.J.; Reijnders, U.J.L.; Bloemers, F.W.; Giannakopoulos, G.F. Preventable death in trauma: A systematic review on definition and classification. Injury 2021, 52, 2768–2777. [Google Scholar] [CrossRef] [PubMed]
- Gaule, T.G.; Ajjan, R.A. Fibrin(ogen) as a Therapeutic Target: Opportunities and Challenges. Int. J. Mol. Sci. 2021, 22, 6916. [Google Scholar] [CrossRef]
- Fries, D.; Martini, W.Z. Role of fibrinogen in trauma-induced coagulopathy. BJA Br. J. Anaesth. 2010, 105, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Goodnough, L.T. How I use fibrinogen replacement therapy in acquired bleeding. Blood 2015, 125, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Henschen, A.; Lottspeich, F.; Kehl, M.; Southan, C. Covalent Structure of Fibrinogen. Ann. N. Y. Acad. Sci. 1983, 408, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Cao, Z.Y.; Davie, E.W. The Role of Amino-Terminal Disulfide Bonds in the Structure and Assembly of Human Fibrinogen. Biochem. Biophys. Res. Commun. 1993, 190, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Pieters, M.; Wolberg, A.S. Fibrinogen and fibrin: An illustrated review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172. [Google Scholar] [CrossRef]
- Wolberg, A.S. Fibrinogen and fibrin: Synthesis, structure, and function in health and disease. J. Thromb. Haemost. 2023, 21, 3005–3015. [Google Scholar] [CrossRef]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between platelets and coagulation. Blood Rev. 2020, 46, 100733. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef]
- Spraggon, G.; Everse, S.J.; Doolittle, R.F. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature 1997, 389, 455–462. [Google Scholar] [CrossRef]
- Brass, L.F.; Diamond, S.L. Transport physics and biorheology in the setting of hemostasis and thrombosis. J. Thromb. Haemost. 2016, 14, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Varjú, I.; Sótonyi, P.; Machovich, R.; Szabó, L.; Tenekedjiev, K.; Silva, M.M.C.G.; Longstaff, C.; Kolev, K. Hindered dissolution of fibrin formed under mechanical stress. J. Thromb. Haemost. 2011, 9, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Eyisoylu, H.; Hazekamp, E.D.; Cruts, J.; Koenderink, G.H.; de Maat, M.P.M. Flow affects the structural and mechanical properties of the fibrin network in plasma clots. J. Mater. Sci. Mater. Med. 2024, 35, 8. [Google Scholar] [CrossRef]
- Miszta, A.; Huskens, D.; Donkervoort, D.; Roberts, M.J.M.; Wolberg, A.S.; de Laat, B. Assessing Plasmin Generation in Health and Disease. Int. J. Mol. Sci. 2021, 22, 2758. [Google Scholar] [CrossRef]
- Zhmurov, A.; Protopopova, A.D.; Litvinov, R.I.; Zhukov, P.; Mukhitov, A.R.; Weisel, J.W.; Barsegov, V. Structural Basis of Interfacial Flexibility in Fibrin Oligomers. Structure 2016, 24, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Kearney, K.J.; Ariëns, R.A.S.; Macrae, F.L. The Role of Fibrin(ogen) in Wound Healing and Infection Control. Semin. Thromb. Hemost. 2021, 48, 174–187. [Google Scholar] [CrossRef]
- Cialdai, F.; Risaliti, C.; Monici, M. Role of fibroblasts in wound healing and tissue remodeling on Earth and in space. Front. Bioeng. Biotechnol. 2022, 10, 958381. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komáromi, I.; Katona, É. Factor XIII: A Coagulation Factor With Multiple Plasmatic and Cellular Functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef]
- Soria, J.; Mirshahi, S.; Mirshahi, S.Q.; Varin, R.; Pritchard, L.L.; Soria, C.; Mirshahi, M. Fibrinogen αC domain: Its importance in physiopathology. Res. Pract. Thromb. Haemost. 2019, 3, 173–183. [Google Scholar] [CrossRef] [PubMed]
- McPherson, H.R.; Duval, C.; Baker, S.R.; Hindle, M.S.; Cheah, L.T.; Asquith, N.L.; Domingues, M.M.; Ridger, V.C.; Connell, S.D.; Naseem, K.M.; et al. Fibrinogen αC-subregions critically contribute blood clot fibre growth, mechanical stability, and resistance to fibrinolysis. eLife 2021, 10, e68761. [Google Scholar] [CrossRef] [PubMed]
- Domingues, M.M.; Carvalho, F.A.; Santos, N.C. Nanomechanics of Blood Clot and Thrombus Formation. Annu. Rev. Biophys. 2022, 51, 201–221. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Sang, Y. Fibrinogen and Factor XIII in Venous Thrombosis and Thrombus Stability. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 931–941. [Google Scholar] [CrossRef]
- Bennett, J.S.; Berger, B.W.; Billings, P.C. The structure and function of platelet integrins. J. Thromb. Haemost. 2009, 7, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Savage, B.; Cattaneo, M.; Ruggeri, Z.M. Mechanisms of platelet aggregation. Curr. Opin. Hematol. 2001, 8, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Guo, Y.; Shen, L.; Li, Y. Fibrinogen binding to activated platelets and its biomimetic thrombus-targeted thrombolytic strategies. Int. J. Biol. Macromol. 2024, 274, 133286. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Sun, C.W.; Woodley, A.B.; Dong, J. Clot Retraction and Its Correlation with the Function of Platelet Integrin αIIbβ3. Biomedicines 2023, 11, 2345. [Google Scholar] [CrossRef]
- Risman, R.A.; Paynter, B.; Percoco, V.; Shroff, M.; Bannish, B.E.; Tutwiler, V. Internal fibrinolysis of fibrin clots is driven by pore expansion. Sci. Rep. 2024, 14, 2623. [Google Scholar] [CrossRef]
- Kietsiriroje, N.; Ariëns, R.A.S.; Ajjan, R.A. Fibrinolysis in Acute and Chronic Cardiovascular Disease. Semin. Thromb. Hemost. 2021, 47, 490–505. [Google Scholar] [CrossRef]
- Risman, R.A.; Kirby, N.C.; Bannish, B.E.; Hudson, N.E.; Tutwiler, V. Fibrinolysis: An illustrated review. Res. Pract. Thromb. Haemost. 2023, 7, 100081. [Google Scholar] [CrossRef] [PubMed]
- Jilani, T.N.; Siddiqui, A.H. Tissue Plasminogen Activator. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507917/ (accessed on 17 September 2024).
- Katz, J.M.; Tadi, P. Physiology, Plasminogen Activation. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539745/ (accessed on 17 September 2024).
- Longstaff, C.; Thelwell, C.; Williams, S.C.; Silva, M.M.C.G.; Szabó, L.; Kolev, K. The interplay between tissue plasminogen activator domains and fibrin structures in the regulation of fibrinolysis: Kinetic and microscopic studies. Blood 2011, 117, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Hudson, N.E. Biophysical Mechanisms Mediating Fibrin Fiber Lysis. BioMed Res. Int. 2017, 2017, 2748340. [Google Scholar] [CrossRef] [PubMed]
- Sillen, M.; Declerck, P.J. A Narrative Review on Plasminogen Activator Inhibitor-1 and Its (Patho)Physiological Role: To Target or Not to Target? Int. J. Mol. Sci. 2021, 22, 2721. [Google Scholar] [CrossRef]
- Singh, S.; Saleem, S.; Reed, G.L. Alpha2-Antiplasmin: The Devil You Don’t Know in Cerebrovascular and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 608899. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Suzuki, Y.; Iwaki, T.; Sano, H.; Honkura, N.; Castellino, F.J. Recognition of Plasminogen Activator Inhibitor Type 1 as the Primary Regulator of Fibrinolysis. Curr. Drug Targets 2019, 20, 1695–1701. [Google Scholar] [CrossRef]
- Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170. [Google Scholar] [CrossRef]
- Mihalko, E.; Brown, A.C. Clot Structure and Implications for Bleeding and Thrombosis. Semin. Thromb. Hemost. 2019, 46, 096–104. [Google Scholar] [CrossRef]
- Lagrange, J.; Lecompte, T.; Knopp, T.; Lacolley, P.; Regnault, V. Alpha-2-macroglobulin in hemostasis and thrombosis: An underestimated old double-edged sword. J. Thromb. Haemost. 2022, 20, 806–815. [Google Scholar] [CrossRef]
- Napolitano, F.; Giudice, V.; Selleri, C.; Montuori, N. Plasminogen System in the Pathophysiology of Sepsis: Upcoming Biomarkers. Int. J. Mol. Sci. 2023, 24, 12376. [Google Scholar] [CrossRef]
- Jain, S.; Acharya, S.S. Inherited disorders of the fibrinolytic pathway. Transfus. Apher. Sci. 2019, 58, 572–577. [Google Scholar] [CrossRef]
- Sillen, M.; Declerck, P.J. Thrombin Activatable Fibrinolysis Inhibitor (TAFI): An Updated Narrative Review. Int. J. Mol. Sci. 2021, 22, 3670. [Google Scholar] [CrossRef]
- Nagashima, M.; Yin, Z.-F.; Jr, G.J.B.; Morser, J. Thrombin-activatable fibrinolysis inhibitor TAFI deficient mice. Front. Biosci. 2002, 7, d556-68. [Google Scholar] [CrossRef]
- Wyseure, T.; Yang, T.; Zhou, J.Y.; Cooke, E.J.; Wanko, B.; Olmer, M.; Agashe, R.; Morodomi, Y.; Behrendt, N.; Lotz, M.; et al. TAFI deficiency causes maladaptive vascular remodeling after hemophilic joint bleeding. JCI Insight 2019, 4, e128379. [Google Scholar] [CrossRef]
- Vandooren, J.; Itoh, Y. Alpha-2-Macroglobulin in Inflammation, Immunity and Infections. Front. Immunol. 2021, 12, 803244. [Google Scholar] [CrossRef]
- Yaron, J.R.; Zhang, L.; Guo, Q.; Haydel, S.E.; Lucas, A.R. Fibrinolytic Serine Proteases, Therapeutic Serpins and Inflammation: Fire Dancers and Firestorms. Front. Cardiovasc. Med. 2021, 8, 648947. [Google Scholar] [CrossRef]
- Pereira, R.V.S.; EzEldeen, M.; Ugarte-Berzal, E.; Martens, E.; Malengier-Devlies, B.; Vandooren, J.; Vranckx, J.J.; Matthys, P.; Opdenakker, G. Physiological fibrin hydrogel modulates immune cells and molecules and accelerates mouse skin wound healing. Front. Immunol. 2023, 14, 1170153. [Google Scholar] [CrossRef]
- Bayer, I.S. Advances in Fibrin-Based Materials in Wound Repair: A Review. Molecules 2022, 27, 4504. [Google Scholar] [CrossRef]
- Tottoli, E.M.; Dorati, R.; Genta, I.; Chiesa, E.; Pisani, S.; Conti, B. Skin Wound Healing Process and New Emerging Technologies for Skin Wound Care and Regeneration. Pharmaceutics 2020, 12, 735. [Google Scholar] [CrossRef]
- Solarte David, V.A.; Güiza-Argüello, V.R.; Arango-Rodríguez, M.L.; Sossa, C.L.; Becerra-Bayona, S.M. Decellularized Tissues for Wound Healing: Towards Closing the Gap Between Scaffold Design and Effective Extracellular Matrix Remodeling. Front. Bioeng. Biotechnol. 2022, 10, 821852. [Google Scholar] [CrossRef]
- Raza, I.; Davenport, R.; Rourke, C.; Platton, S.; Manson, J.; Spoors, C.; Khan, S.; De’Ath, H.D.; Allard, S.; Hart, D.P.; et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J. Thromb. Haemost. 2013, 11, 307–314. [Google Scholar] [CrossRef]
- Curry, N.; Rourke, C.; Davenport, R.; Stanworth, S.; Brohi, K. Fibrinogen replacement in trauma haemorrhage. Scand. J. Trauma Resusc. Emerg. Med. 2014, 22, A5. [Google Scholar] [CrossRef]
- Nencini, F.; Bettiol, A.; Argento, F.R.; Borghi, S.; Giurranna, E.; Emmi, G.; Prisco, D.; Taddei, N.; Fiorillo, C.; Becatti, M. Post-translational modifications of fibrinogen: Implications for clotting, fibrin structure and degradation. Mol. Biomed. 2024, 5, 45. [Google Scholar] [CrossRef]
- Collaborators, C.T.; Shakur, H.; Roberts, I.; Bautista, R.; Caballero, J.; Coats, T.; Dewan, Y.; El-Sayed, H.; Gogichaishvili, T.; Gupta, S.; et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): A randomised, placebo-controlled trial. Lancet 2010, 376, 23–32. [Google Scholar] [CrossRef]
- Innerhofer, N.; Treichl, B.; Rugg, C.; Fries, D.; Mittermayr, M.; Hell, T.; Oswald, E.; Innerhofer, P.; on behalf of the RETIC Study Group. First-Line Administration of Fibrinogen Concentrate in the Bleeding Trauma Patient: Searching for Effective Dosages and Optimal Post-Treatment Levels Limiting Massive Transfusion—Further Results of the RETIC Study. J. Clin. Med. 2021, 10, 3930. [Google Scholar] [CrossRef]
- Obaid, O.; Anand, T.; Nelson, A.; Reina, R.; Ditillo, M.; Stewart, C.; Douglas, M.; Friese, R.; Gries, L.; Joseph, B. Fibrinogen supplementation for the trauma patient: Should you choose fibrinogen concentrate over cryoprecipitate? J. Trauma. Acute Care Surg. 2022, 93, 453–460. [Google Scholar] [CrossRef]
- Wormer, K.C.; Jamil, R.T.; Bryant, S.B. Acute Postpartum Hemorrhage. Available online: https://pubmed.ncbi.nlm.nih.gov/29763164/ (accessed on 22 October 2024).
- Sahin, A.S.; Ozkan, S. Treatment of Obstetric Hemorrhage with Fibrinogen Concentrate. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 1814–1821. [Google Scholar] [CrossRef]
- Bienstock, J.L.; Eke, A.C.; Hueppchen, N.A. Postpartum Hemorrhage. N. Engl. J. Med. 2021, 384, 1635–1645. [Google Scholar] [CrossRef]
- McNamara, H.; Kenyon, C.; Smith, R.; Mallaiah, S.; Barclay, P. Four years’ experience of a ROTEM ®-guided algorithm for treatment of coagulopathy in obstetric haemorrhage. Anaesthesia 2019, 74, 984–991. [Google Scholar] [CrossRef]
- Wikkelsø, A.J.; Edwards, H.M.; Afshari, A.; Stensballe, J.; Langhoff-Roos, J.; Albrechtsen, C.; Ekelund, K.; Hanke, G.; Secher, E.L.; Sharif, H.F.; et al. Pre-emptive treatment with fibrinogen concentrate for postpartum haemorrhage: Randomized controlled trial. Br. J. Anaesth. 2015, 114, 623–633. [Google Scholar] [CrossRef]
- Vermeulen, T.; Van De Velde, M. The role of fibrinogen in postpartum hemorrhage. Best. Pract. Res. Clin. Anaesthesiol. 2022, 36, 399–410. [Google Scholar] [CrossRef]
- Okerberg, C.K.; Williams, L.A.; Kilgore, M.L.; Kim, C.; Marques, M.B.; Schwartz, J.-C.; Pham, H.P. Cryoprecipitate AHF vs. fibrinogen concentrates for fibrinogen replacement in acquired bleeding patients—An economic evaluation. Vox Sang. 2016, 111, 292–298. [Google Scholar] [CrossRef]
- Lam, T.; Medcalf, R.L.; Cloud, G.; Myles, P.S.; Keragala, C.B. Tranexamic acid for haemostasis and beyond: Does dose matter? Thromb. J. 2023, 21, 94. [Google Scholar] [CrossRef]
- Sentilhes, L.; Sénat, M.V.; Le Lous, M.; Winer, N.; Rozenberg, P.; Kayem, G.; Verspyck, E.; Fuchs, F.; Azria, E.; Gallot, D.; et al. Tranexamic Acid for the Prevention of Blood Loss after Cesarean Delivery. N. Engl. J. Med. 2021, 384, 1623–1634. [Google Scholar] [CrossRef]
- Shakur-Still, H.; Roberts, I.; Fawole, B.; Kuti, M.; Olayemi, O.O.; Bello, A.; Huque, S.; Ogunbode, O.; Kotila, T.; Aimakhu, C.; et al. Effect of tranexamic acid on coagulation and fibrinolysis in women with postpartum haemorrhage (WOMAN-ETAC): A single-centre, randomised, double-blind, placebo-controlled trial. Wellcome Open Res. 2018, 3, 100. [Google Scholar] [CrossRef]
- Callum, J.; Farkouh, M.E.; Scales, D.C.; Heddle, N.M.; Crowther, M.; Rao, V.; Hucke, H.-P.; Carroll, J.; Grewal, D.; Brar, S.; et al. Effect of Fibrinogen Concentrate vs Cryoprecipitate on Blood Component Transfusion After Cardiac Surgery. JAMA 2019, 322, 1966–1976. [Google Scholar] [CrossRef]
- Jeppsson, A.; Waldén, K.; Roman-Emanuel, C.; Thimour-Bergström, L.; Karlsson, M. Preoperative supplementation with fibrinogen concentrate in cardiac surgery: A randomized controlled study. Br. J. Anaesth. 2016, 116, 208–214. [Google Scholar] [CrossRef]
- Rahe-Meyer, N.; Levy, J.H.; Mazer, C.D.; Schramko, A.; Klein, A.A.; Brat, R.; Okita, Y.; Ueda, Y.; Schmidt, D.S.; Ranganath, R.; et al. Randomized evaluation of fibrinogen vs placebo in complex cardiovascular surgery (REPLACE): A double-blind phase III study of haemostatic therapy. Br. J. Anaesth. 2016, 117, 41–51. [Google Scholar] [CrossRef]
- Hamada, S.R.; Pirracchio, R.; Beauchesne, J.; Benlaldj, M.N.; Meaudre, E.; Leone, M.; Pottecher, J.; Abback, P.S.; Gauss, T.; Boutonnet, M.; et al. Effect of fibrinogen concentrate administration on early mortality in traumatic hemorrhagic shock: A propensity score analysis. J. Trauma. Acute Care Surg. 2020, 88, 661–670. [Google Scholar] [CrossRef]
- Scala, E.; Marcucci, C. Massive Hemorrhage: The Role of Whole Blood Viscoelastic Assays. Hämostaseologie 2020, 40, 515–523. [Google Scholar] [CrossRef]
- Rossaint, R.; Afshari, A.; Bouillon, B.; Cerny, V.; Cimpoesu, D.; Curry, N.; Duranteau, J.; Filipescu, D.; Grottke, O.; Grønlykke, L.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Sixth edition. Crit. Care 2023, 27, 80. [Google Scholar] [CrossRef]
- Zheng, Z.; Mukhametova, L.; Boffa, M.B.; Moore, E.E.; Wolberg, A.S.; Urano, T.; Kim, P.Y. Assays to quantify fibrinolysis: Strengths and limitations. Communication from the International Society on Thrombosis and Haemostasis Scientific and Standardization Committee on fibrinolysis. J. Thromb. Haemost. 2023, 21, 1043–1054. [Google Scholar] [CrossRef]
- Drotarova, M.; Zolkova, J.; Belakova, K.M.; Brunclikova, M.; Skornova, I.; Stasko, J.; Simurda, T. Basic Principles of Rotational Thromboelastometry (ROTEM®) and the Role of ROTEM—Guided Fibrinogen Replacement Therapy in the Management of Coagulopathies. Diagnostics 2023, 13, 3219. [Google Scholar] [CrossRef]
- Dias, J.D.; Butwick, A.J.; Hartmann, J.; Waters, J.H. Viscoelastic haemostatic point-of-care assays in the management of postpartum haemorrhage: A narrative review. Anaesthesia 2022, 77, 700–711. [Google Scholar] [CrossRef]
- Bareille, M.; Lecompte, T.; Mullier, F.; Roullet, S. Are Viscoelastometric Assays of Old Generation Ready for Disposal? Comment on Volod et al. Viscoelastic Hemostatic Assays: A Primer on Legacy and New Generation Devices. J. Clin. Med. 2022, 11, 860. J. Clin. Med. 2023, 12, 477. [Google Scholar] [CrossRef]
- Reardon, B.; Pasalic, L.; Favaloro, E.J. The Role of Viscoelastic Testing in Assessing Hemostasis: A Challenge to Standard Laboratory Assays? J. Clin. Med. 2024, 13, 3612. [Google Scholar] [CrossRef]
- Curry, N.S.; Davenport, R.; Pavord, S.; Mallett, S.V.; Kitchen, D.; Klein, A.A.; Maybury, H.; Collins, P.W.; Laffan, M. The use of viscoelastic haemostatic assays in the management of major bleeding. Br. J. Haematol. 2018, 182, 789–806. [Google Scholar] [CrossRef]
- Rigouzzo, A.; Louvet, N.; Favier, R.; Ore, M.-V.; Piana, F.; Girault, L.; Farrugia, M.; Sabourdin, N.; Constant, I. Assessment of Coagulation by Thromboelastography During Ongoing Postpartum Hemorrhage: A Retrospective Cohort Analysis. Anesth. Analg. 2020, 130, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Kodali, B.S.; Karuppiah, A.; Bharadwaj, S.; Chow, J.; Tanaka, K. Efficacy of sonorheometry point of the care device in determining low fibrinogen levels in pregnant blood: An invitro dilution and reconstitution study. J. Clin. Monit. Comput. 2021, 36, 1423–1431. [Google Scholar] [CrossRef]
- Baulig, W. Comparison of the Resonance Sonorheometry Based Quantra® System with Rotational Thromboelastometry ROTEM® Sigma in Cardiac Surgery—A Prospective Observational Study. Available online: https://www.academia.edu/67965645/Comparison_of_the_resonance_sonorheometry_based_Quantra_system_with_rotational_thromboelastometry_ROTEM_sigma_in_cardiac_surgery_a_prospective_observational_study? (accessed on 1 January 2025).
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Duranteau, J.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Maegele, M.; Nardi, G.; Riddez, L.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef]
- Winearls, J.; Wullschleger, M.; Wake, E.; McQuilten, Z.; Reade, M.; Hurn, C.; Ryan, G.; Trout, M.; Walsham, J.; Holley, A.; et al. Fibrinogen Early In Severe Trauma studY (FEISTY): Results from an Australian multicentre randomised controlled pilot trial. Crit. Care Resusc. 2021, 23, 32–46. [Google Scholar] [CrossRef]
- Keltner, N.M.; Cushing, M.M.; Haas, T.; Spinella, P.C. Analyzing and modeling massive transfusion strategies and the role of fibrinogen—How much is the patient actually receiving? Transfusion 2024, 64, S136–S145. [Google Scholar] [CrossRef]
- Kleber, C.; Sablotzki, A.; Casu, S.; Olivieri, M.; Thoms, K.-M.; Horter, J.; Schmitt, F.C.F.; Birschmann, I.; Fries, D.; Maegele, M.; et al. The impact of acquired coagulation factor XIII deficiency in traumatic bleeding and wound healing. Crit. Care 2022, 26, 69. [Google Scholar] [CrossRef]
- Abrahamyan, L.; Tomlinson, G.; Callum, J.; Carcone, S.; Grewal, D.; Bartoszko, J.; Krahn, M.; Karkouti, K. Cost-effectiveness of Fibrinogen Concentrate vs Cryoprecipitate for Treating Acquired Hypofibrinogenemia in Bleeding Adult Cardiac Surgical Patients. JAMA Surg. 2023, 158, 245–253. [Google Scholar] [CrossRef]
- National Clinical Guideline Centre (UK). Blood Transfusion. London: National Institute for Health and Care Excellence (NICE); 2015 Nov. (NICE Guideline, No. 24.) Appendix N, Unit Costs. Available online: https://www.ncbi.nlm.nih.gov/books/NBK338809/ (accessed on 30 January 2025).
- Jacobs, J.W.; Diaz, M.; Arevalo Salazar, D.E.; Tang, A.; Stephens, L.D.; Booth, G.S.; Lehmann, C.U.; Adkins, B.D. United States blood pricing: A cross-sectional analysis of charges and reimbursement at 200 US hospitals. Am. J. Hematol. 2023, 98, E179–E182. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, C.; Cairns, J.; Perel, P.; Shakur, H.; Roberts, I. Cost-Effectiveness Analysis of Administering Tranexamic Acid to Bleeding Trauma Patients Using Evidence from the CRASH-2 Trial. PLoS ONE 2011, 6, e18987. [Google Scholar] [CrossRef] [PubMed]
- Volod, O.; Bunch, C.M.; Zackariya, N.; Moore, E.E.; Moore, H.B.; Kwaan, H.C.; Neal, M.D.; Al-Fadhl, M.D.; Patel, S.S.; Wiarda, G.; et al. Viscoelastic Hemostatic Assays: A Primer on Legacy and New Generation Devices. J. Clin. Med. 2022, 11, 860. [Google Scholar] [CrossRef]
- Cushing, M.M.; Haas, T. Fibrinogen concentrate for perioperative bleeding: What can we learn from the clinical trials? Transfusion 2019, 59, 3295–3297. [Google Scholar] [CrossRef]
- Lier, H.; Krep, H.; Schroeder, S.; Stuber, F. Preconditions of Hemostasis in Trauma: A Review. The Influence of Acidosis, Hypocalcemia, Anemia, and Hypothermia on Functional Hemostasis in Trauma. J. Trauma Inj. Infect. Crit. Care 2008, 65, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Narick, C.; Triulzi, D.J.; Yazer, M.H. Transfusion-associated circulatory overload after plasma transfusion. Transfusion 2011, 52, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Innerhofer, P.; Fries, D.; Mittermayr, M.; Innerhofer, N.; von Langen, D.; Hell, T.; Gruber, G.; Schmid, S.; Friesenecker, B.; Lorenz, I.H.; et al. Reversal of trauma-induced coagulopathy using first-line coagulation factor concentrates or fresh frozen plasma (RETIC): A single-centre, parallel-group, open-label, randomised trial. Lancet Haematol. 2017, 4, e258–e271. [Google Scholar] [CrossRef]
- Akbari, E.; Safari, S.; Hatamabadi, H. The effect of fibrinogen concentrate and fresh frozen plasma on the outcome of patients with acute traumatic coagulopathy: A quasi-experimental study. Am. J. Emerg. Med. 2018, 36, 1947–1950. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.; Roy, A.; Cecil, T.; Hegener, O.; Schulz, P.; Turaj, A.; Lim, S.; Arbuthnot, E. Differences in coagulation-relevant parameters: Comparing cryoprecipitate and a human fibrinogen concentrate. PLoS ONE 2023, 18, e0290571. [Google Scholar] [CrossRef] [PubMed]
- Green, L.; Daru, J.; Dodds, J.; Carreras, F.J.G.; Lanz, D.; Zamora, J.; Llorente, M.d.C.P.; Pérez, T.P.; Sweeney, L.; Thangaratinam, S.; et al. Effect of early cryoprecipitate transfusion versus standard care in women who develop severe postpartum haemorrhage (ACROBAT) in the UK: A protocol for a pilot cluster randomised trial. BMJ Open 2020, 10, e036416. [Google Scholar] [CrossRef] [PubMed]
- Green, L.; Daru, J.; Carreras, F.J.G.; Lanz, D.; Pardo, M.C.; Pérez, T.; Philip, S.; Tanqueray, T.; Khan, K.S. Early cryoprecipitate transfusion versus standard care in severe postpartum haemorrhage: A pilot cluster-randomised trial. Anaesthesia 2021, 77, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kamidani, R.; Miyake, T.; Okada, H.; Yoshimura, G.; Kusuzawa, K.; Miura, T.; Shimaoka, R.; Oiwa, H.; Yamaji, F.; Mizuno, Y.; et al. Effect of cryoprecipitate transfusion therapy in patients with postpartum hemorrhage: A retrospective cohort study. Sci. Rep. 2021, 11, 18458. [Google Scholar] [CrossRef] [PubMed]
- Bartoszko, J.; Martinez-Perez, S.; Callum, J.; Karkouti, K.; Farouh, M.E.; Scales, D.C.; Heddle, N.M.; Crowther, M.; Rao, V.; Hucke, H.-P.; et al. Impact of cardiopulmonary bypass duration on efficacy of fibrinogen replacement with cryoprecipitate compared with fibrinogen concentrate: A post hoc analysis of the Fibrinogen Replenishment in Surgery (FIBRES) randomised controlled trial. Br. J. Anaesth. 2022, 129, 294–307. [Google Scholar] [CrossRef]
- Karkouti, K.; Callum, J.; Rao, V.; Heddle, N.; Farkouh, M.E.; Crowther, M.A.; Scales, D.C. Protocol for a phase III, non-inferiority, randomised comparison of a new fibrinogen concentrate versus cryoprecipitate for treating acquired hypofibrinogenaemia in bleeding cardiac surgical patients: The FIBRES trial. BMJ Open 2018, 8, e020741. [Google Scholar] [CrossRef] [PubMed]
- Seebold, J.A.; Campbell, D.; Wake, E.; Walters, K.; Ho, D.; Chan, E.; Bulmer, A.C.; Wullschleger, M.; Winearls, J. Targeted fibrinogen concentrate use in severe traumatic haemorrhage. Crit. Care Resusc. 2019, 21, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.; Shakur, H.; Coats, T.; Hunt, B.; Balogun, E.; Barnetson, L.; Cook, L.; Kawahara, T.; Perel, P.; Prieto-Merino, D.; et al. The CRASH-2 trial: A randomised controlled trial and economic evaluation of the effects of tranexamic acid on death, vascular occlusive events and transfusion requirement in bleeding trauma patients. Health Technol. Assess. 2013, 17, 1–79. [Google Scholar] [CrossRef]
- Roberts, I.; Shakur-Still, H.; Aeron-Thomas, A.; Belli, A.; Brenner, A.; Chaudary, M.A.; Chaudhri, R.; Jamaluddin, S.F.B.; Frimley, L.; Javaid, K.; et al. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): A randomised, placebo-controlled trial. Lancet 2019, 394, 1713–1723. [Google Scholar] [CrossRef]
- Fouche, P.F.; Stein, C.; Nichols, M.; Meadley, B.; Bendall, J.C.; Smith, K.; Anderson, D.; Doi, S.A. Tranexamic Acid for Traumatic Injury in the Emergency Setting: A Systematic Review and Bias-Adjusted Meta-Analysis of Randomized Controlled Trials. Ann. Emerg. Med. 2023, 83, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Ageron, F.-X.D. Tranexamic Acid in Trauma Care: Who Should Be Treated, When and Where? Ph.D. Thesis, London School of Hygiene & Tropical Medicine, London, England, 2021. [Google Scholar] [CrossRef]
- Monge, K.M.; Domene, S.S.; Mendoza, D.L.D.; Vidal-Gallardo, A.; Llique, A.M.A.; Rodriguez, M.; Premchandra, P.; Pandya, S.A.; Arruarana, V.S.; Paredes, K.A.; et al. Effectiveness of Tranexamic Acid in Trauma Patients: A Systematic Review. Cureus 2024, 16, e52111. [Google Scholar] [CrossRef]
- Rohwer, C.; Rohwer, A.C.; Cluver, C.; Ker, K.; Hofmeyr, G.J. Tranexamic acid for preventing postpartum haemorrhage after vaginal birth. Cochrane Database Syst. Rev. 2025, 1, CD007872. [Google Scholar] [CrossRef]
- Rohwer, C.; Rohwer, A.; Cluver, C.; Ker, K.; Hofmeyr, G.J. Tranexamic acid for preventing postpartum haemorrhage after caesarean section. Cochrane Libr. 2024, 2024, CD016278. [Google Scholar] [CrossRef]
- Akbulut, A.C.; Arisz, R.A.; Baaten, C.C.F.M.J.; Baidildinova, G.; Barakzie, A.; Bauersachs, R.; Berg, J.T.; Broek, W.W.A.v.D.; de Boer, H.C.; Bonifay, A.; et al. Blood coagulation and beyond: Position paper from the Fourth Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2023, 123, 808–839. [Google Scholar] [CrossRef]
- Hensley, N.B.; Mazzeffi, M.A. Pro-Con Debate: Fibrinogen Concentrate or Cryoprecipitate for Treatment of Acquired Hypofibrinogenemia in Cardiac Surgical Patients. Anesth. Analg. 2021, 133, 19–28. [Google Scholar] [CrossRef]
- Cardenas, J.C.; Matijevic, N.; Baer, L.A.; Holcomb, J.B.; Cotton, B.A.; Wade, C.E. Elevated Tissue Plasminogen Activator and Reduced Plasminogen Activator Inhibitor Promote Hyperfibrinolysis in Trauma Patients. Shock 2014, 41, 514–521. [Google Scholar] [CrossRef]
- Cotton, B.A.; Harvin, J.A.; Kostousouv, V.; Minei, K.M.; Radwan, Z.A.; Schöchl, H.; Wade, C.E.; Holcomb, J.B.; Matijevic, N. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J. Trauma Acute Care Surg. 2012, 73, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Bodary, P.F.; Wickenheiser, K.J.; Eitzman, D.T. Recent advances in understanding endogenous fibrinolysis: Implications for molecular-based treatment of vascular disorders. Expert Rev. Mol. Med. 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Atasever, A.G.; Eerens, M.; Van den Eynde, R.; Faraoni, D.; Rex, S. Efficacy and safety of aprotinin in paediatric cardiac surgery. Eur. J. Anaesthesiol. 2022, 39, 352–367, publish ahead of print. [Google Scholar] [CrossRef] [PubMed]
- De Hert, S.; Ouattara, A.; Royston, D.; Zacharowski, K. Use and safety of aprotinin in routine clinical practice. Eur. J. Anaesthesiol. 2022, 39, 685–694. [Google Scholar] [CrossRef]
- Erdoes, G.; Koster, A.; Levy, J.H. Retrospective aprotinin cardiac surgical studies and their limitations: Time for a prospective randomized clinical trial. Eur. J. Cardio-Thorac. Surg. 2024, 65, ezae038. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.-L.; Kwok, J.Y.-Y.; Chung, K.-Y.; Cheung, K.-W.; Chiu, K.-H.; Chau, W.-W.; Ho, K.K.-W. Prospective randomized trial comparing efficacy and safety of intravenous and intra-articular tranexamic acid in total knee arthroplasty. Knee Surg. Relat. Res. 2020, 32, 62. [Google Scholar] [CrossRef] [PubMed]
- Annals Admin Association Between Genetic Polymorphisms in Fibrinogen Genes and Bleeding Risk in Patients Treated with Direct Oral Anticoagulants—Annals Singapore. Available online: https://annals.edu.sg/association-between-genetic-polymorphisms-in-fibrinogen-genes-and-bleeding-risk-in-patients-treated-with-direct-oral-anticoagulants/ (accessed on 1 January 2025).
- Jacquemin, B.; Antoniades, C.; Nyberg, F.; Plana, E.; Müller, M.; Greven, S.; Salomaa, V.; Sunyer, J.; Bellander, T.; Chalamandaris, A.-G.; et al. Common Genetic Polymorphisms and Haplotypes of Fibrinogen Alpha, Beta, and Gamma Chains Affect Fibrinogen Levels and the Response to Proinflammatory Stimulation in Myocardial Infarction Survivors: The AIRGENE Study. J. Am. Coll. Cardiol. 2008, 52, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Coats, T.J.; Morsy, M. Biological mechanisms and individual variation in fibrinolysis after major trauma. Emerg. Med. J. 2020, 37, 135–140. [Google Scholar] [CrossRef]
- Huffman, J.E.; Nicholas, J.; Hahn, J.; Heath, A.S.; Raffield, L.M.; Yanek, L.R.; Brody, J.A.; Thibord, F.; Almasy, L.; Bartz, T.M.; et al. Whole-genome analysis of plasma fibrinogen reveals population-differentiated genetic regulators with putative liver roles. Blood 2024, 144, 2248–2265. [Google Scholar] [CrossRef]
- Levy, J.H.; Koster, A.; Quinones, Q.J.; Milling, T.J.; Key, N.S. Antifibrinolytic Therapy and Perioperative Considerations. Anesthesiol. J. Am. Soc. Anesthesiol. 2018, 128, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Congenital Fibrinogen Deficiency via the FGG Gene Test—PreventionGenetics. Available online: https://www.preventiongenetics.com/testInfo?val=Congenital-Fibrinogen-Deficiency-via-the-FGG-Gene (accessed on 1 January 2025).
- Kearney, K.J.; Pechlivani, N.; King, R.; Tiede, C.; Phoenix, F.; Cheah, R.; Macrae, F.L.; Simmons, K.J.; Manfield, I.W.; Smith, K.A.; et al. Affimer proteins as a tool to modulate fibrinolysis, stabilize the blood clot, and reduce bleeding complications. Blood 2019, 133, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Pechlivani, N.; Alsayejh, B.; Almutairi, M.K.; Simmons, K.; Gaule, T.; Phoenix, F.; Kietsiriroje, N.; Ponnambalam, S.; Duval, C.; Ariëns, R.A.S.; et al. Use of Affimer Technology for Inhibition of α2-antiplasmin and Enhancement of Fibrinolysis. Blood Adv. 2024, 9, 89–100. [Google Scholar] [CrossRef]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Pechlivani, N.; Kearney, K.J.; Tiede, C.; Cheah, R.; Phoenix, F.; Ponnambalam, S.; Ault, J.R.; McPherson, M.J.; Tomlinson, D.C.; Ajjan, R.A. Affinity purification of fibrinogen using an Affimer column. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2022, 1866, 130115. [Google Scholar] [CrossRef] [PubMed]
- Good Manufacturing Practice|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compliance-research-development/good-manufacturing-practice?utm_source=chatgpt.com (accessed on 1 January 2025).
- Morrow, G.B.; Flannery, S.; Charles, P.D.; Heilig, R.; Feller, T.; McQuilten, Z.; Wake, E.; Ariens, R.A.S.; Winearls, J.; Mutch, N.J.; et al. A novel method to quantify fibrin–fibrin and fibrin–α2-antiplasmin cross-links in thrombi formed from human trauma patient plasma. J. Thromb. Haemost. 2024, 22, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Alsayejh, B.; Kietsiriroje, N.; Almutairi, M.; Simmons, K.; Pechlivani, N.; Ponnambalam, S.; Ajjan, R.A. Plasmin Inhibitor in Health and Diabetes: Role of the Protein as a Therapeutic Target. TH Open 2022, 6, e396–e407. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Otto, E.; Eder, C.; Youssef, Y.; Kaufner, L.; Märdian, S. Coagulopathy management of multiple injured patients—A comprehensive literature review of the European guideline 2019. EFORT Open Rev. 2022, 7, 710–726. [Google Scholar] [CrossRef]
- Thaventhiran, A.; Thiemermann, C.; Brohi, K.; Tremoleda, J.; Davenport, R. O1: Discovering Novel Therapeutics to Treat Acute Traumatic Coagulopathy (ATC). Br. J. Surg. 2021, 108, 7. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Name | Class | Mechanism |
|---|---|---|
| Factor XIII | Protein transglutaminase | Activated by thrombin; catalyses bonds between g chains of fibrin molecules; crosslinks molecules to anti fibrinolytic molecules, e.g., α2-antiplasmin |
| GPIIb/IIIa | Integrin | One fibrinogen molecule links two receptors via g chain sequence, forms platelet aggregates |
| Calcium (Ca2+) | Ion (Ca2+) | Binds with varying levels of affinity to different fibrinogen chains, highest affinity on the γ1 chain, increase the rate of lateral aggregation between fibres of fibrinogen |
| α2AP | Serine protease inhibitor | Main physiological inhibitor of plasmin; Factor XIII cross-links it to fibrinogen |
| TAFI | Zinc-dependent metallocarboxypeptidase | Found in platelet α-granules, accumulates when platelets gather; cleaves off C-terminal lysine residues in fibrinogen, thus reducing binding of plasminogen |
| PAI-1 | Serine protease inhibitor | The main inhibitor of both tPA and uPA, thereby modulating plasmin generation and controlling fibrin degradation to avert excessive fibrinolysis. |
| PAI-2 | Serine protease inhibitor | Endogenous inhibitor of urokinase-type plasminogen activator; cross-linking to Lysine residues through glutamine residues on the PAI-2 chain |
| α2-Macroglobulin (α2M) | Broad-spectrum protease inhibitor | Binds and inactivates a range of proteases, including plasmin, through a conformational trap mechanism. By sequestering proteolytic enzymes, α2M adds an additional layer of protection against excessive fibrinolysis. |
| Treatment Modality | Mechanism of Action | Applications | Advantages | Drawbacks | Cost |
|---|---|---|---|---|---|
| Fibrinogen concentrate | Directly replenishes fibrinogen levels for clot formation and stability | Trauma, obstetric haemorrhage, cardiac surgery | Rapid, precise dosing; predictable effects; low infection risk | High cost; limited availability in resource-limited settings | USD 400–USD 800 per gram [78] |
| Cryoprecipitate | Supplies fibrinogen along with Factor VIII, XIII and vWF | Trauma, obstetric haemorrhage, cardiac surgery | Widely available; cost-effective in many settings | Variable composition; freezing requirements; infection risk | USD 400–USD 700 [78,79] |
| Fresh Frozen Plasma (FFP) | Provides a broad range of clotting factors to address coagulopathy | Massive transfusion in trauma or surgery | Provides broad coagulation support; widely available | Requires large volumes; risk of transfusion reactions | USD 132–USD 477 [80] |
| Tranexamic Acid (TXA) | Inhibits plasminogen activation to reduce fibrinolysis | Trauma, obstetric haemorrhage, surgical bleeding | Simple administration; effective in reducing fibrinolysis | inconsistent action; efficacy depends on fibrinogen levels | USD 2–USD 10 (USD) per dose [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, T.; Hasan, M.; Gaule, T.G.; Ajjan, R.A. Exploiting the Molecular Properties of Fibrinogen to Control Bleeding Following Vascular Injury. Int. J. Mol. Sci. 2025, 26, 1336. https://doi.org/10.3390/ijms26031336
Singh T, Hasan M, Gaule TG, Ajjan RA. Exploiting the Molecular Properties of Fibrinogen to Control Bleeding Following Vascular Injury. International Journal of Molecular Sciences. 2025; 26(3):1336. https://doi.org/10.3390/ijms26031336
Chicago/Turabian StyleSingh, Tanjot, Muhammad Hasan, Thembaninkosi G. Gaule, and Ramzi A. Ajjan. 2025. "Exploiting the Molecular Properties of Fibrinogen to Control Bleeding Following Vascular Injury" International Journal of Molecular Sciences 26, no. 3: 1336. https://doi.org/10.3390/ijms26031336
APA StyleSingh, T., Hasan, M., Gaule, T. G., & Ajjan, R. A. (2025). Exploiting the Molecular Properties of Fibrinogen to Control Bleeding Following Vascular Injury. International Journal of Molecular Sciences, 26(3), 1336. https://doi.org/10.3390/ijms26031336