
A Novel Human Anti-FV mAb as a Potential Tool for Diagnostic and Coagulation Inhibitory Approaches
 , ,
, ,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
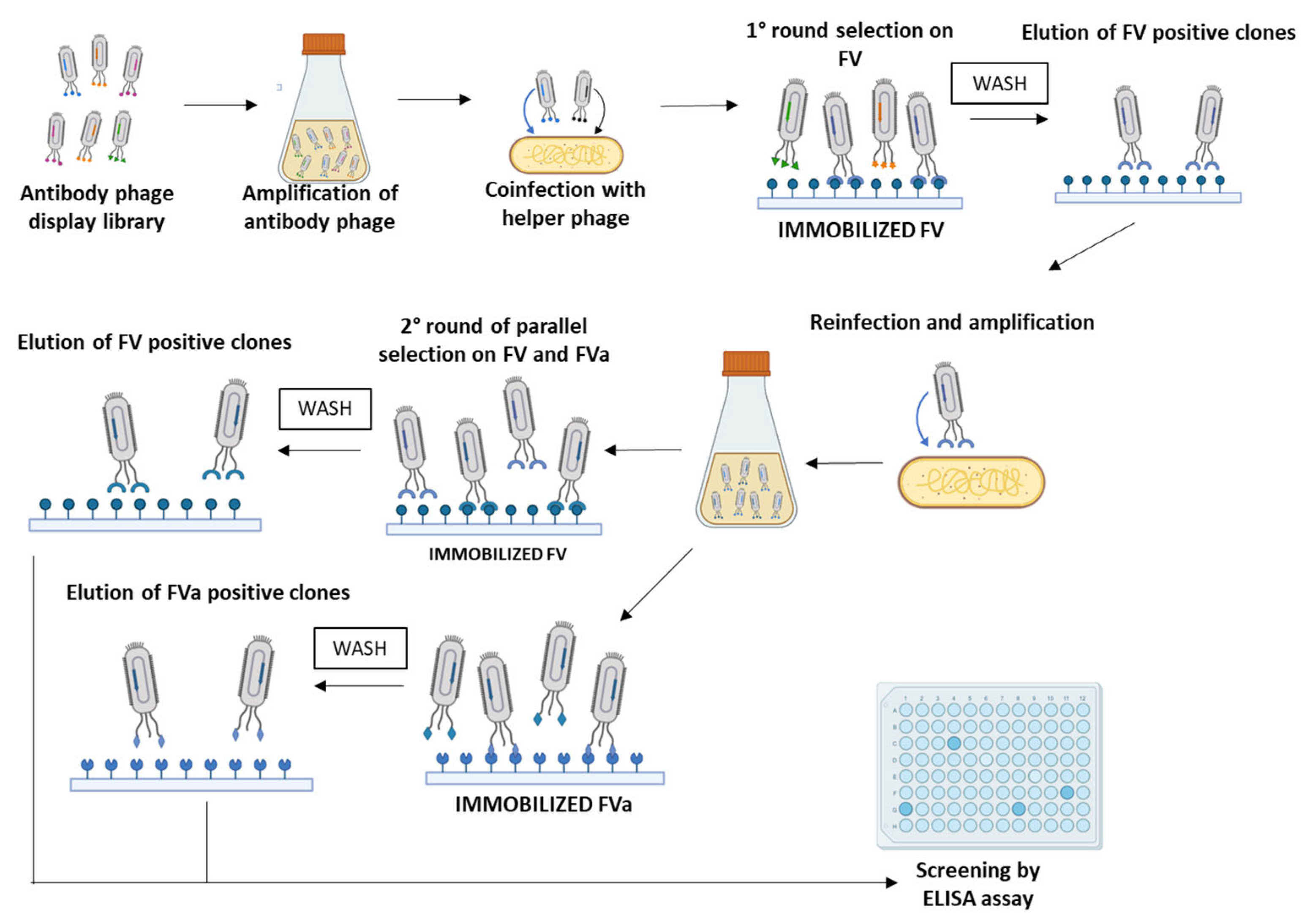
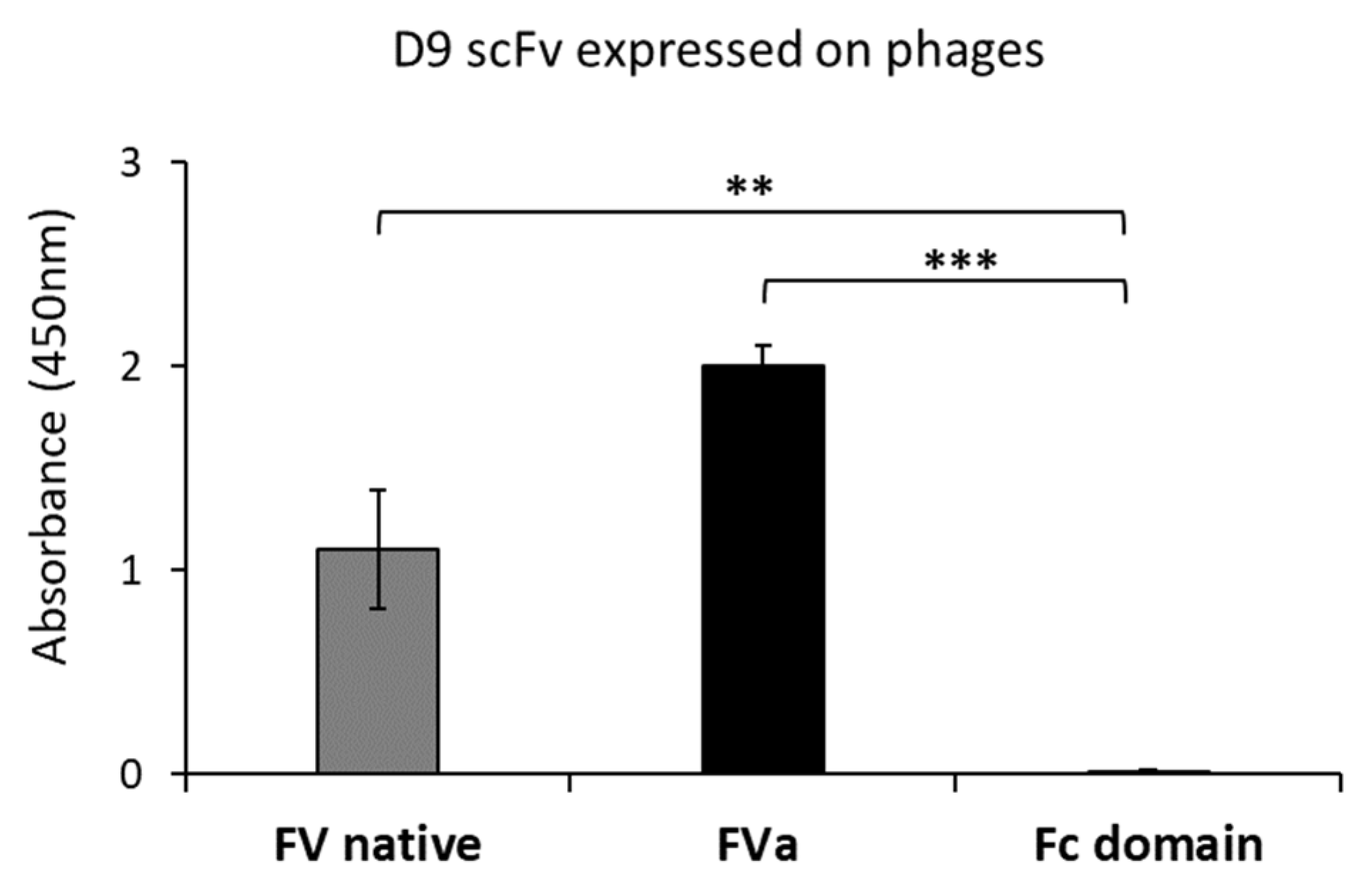
2.1. Isolation and Screening of Novel Human scFvs Specific to Human Factor V
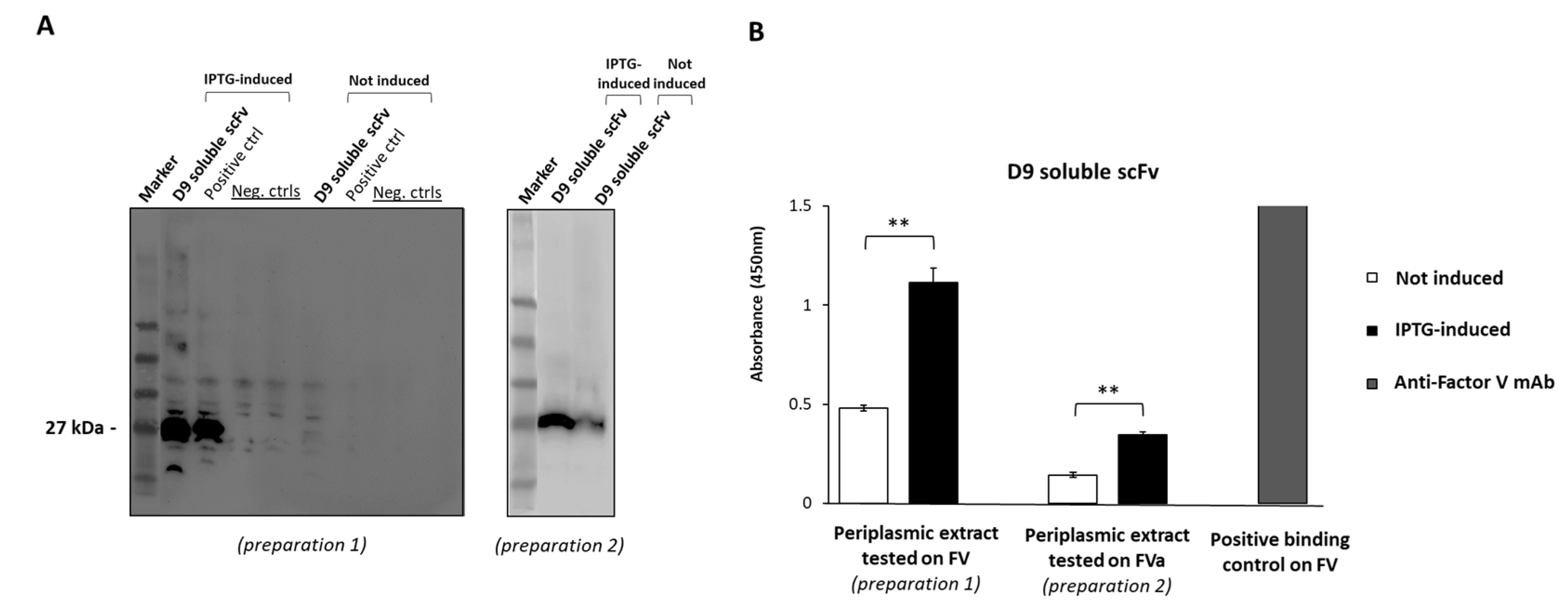
2.2. Expression of D9 Clone as Soluble scFv
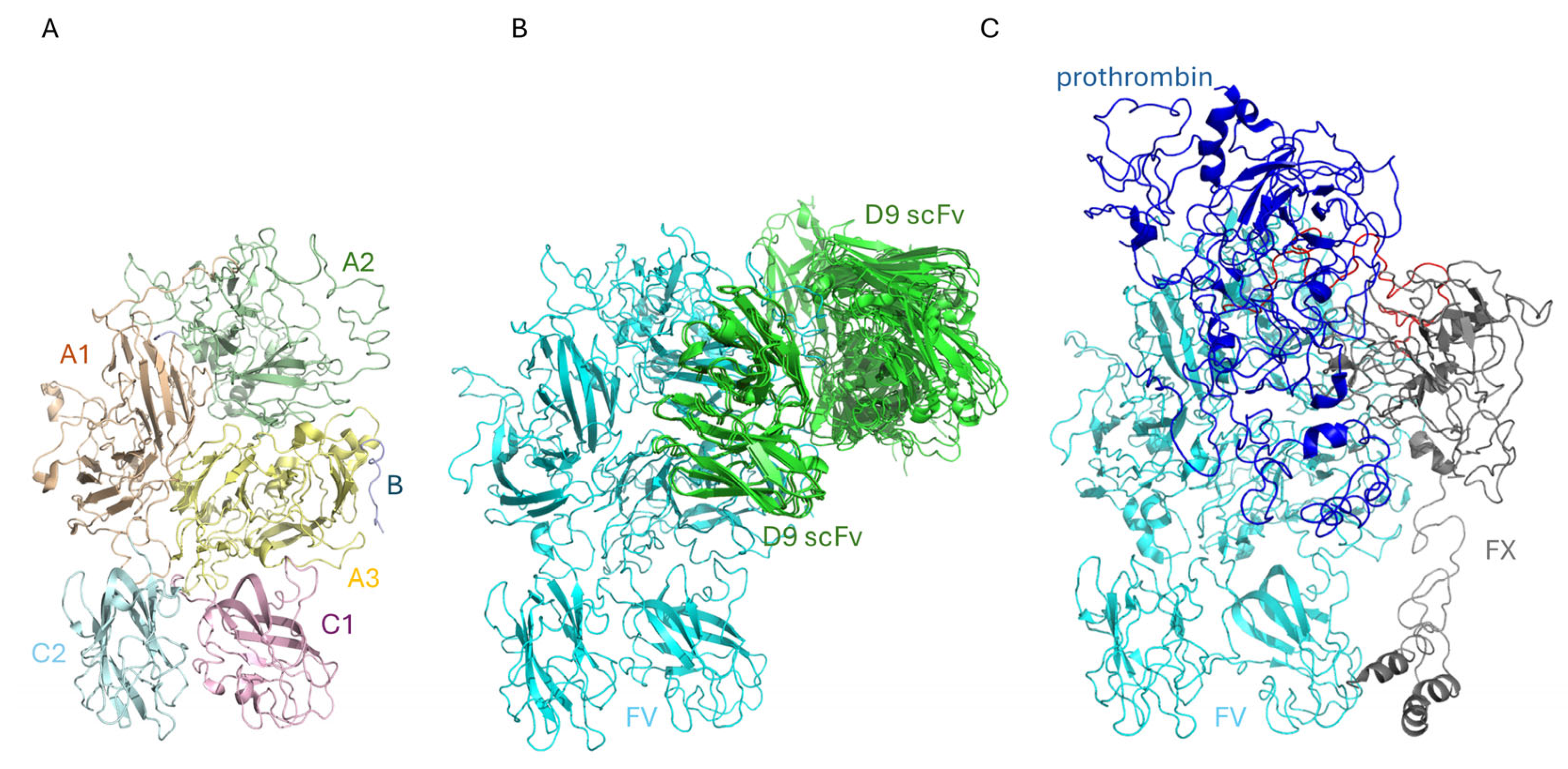
2.3. The Binding of D9 to FV: An In Silico Approach
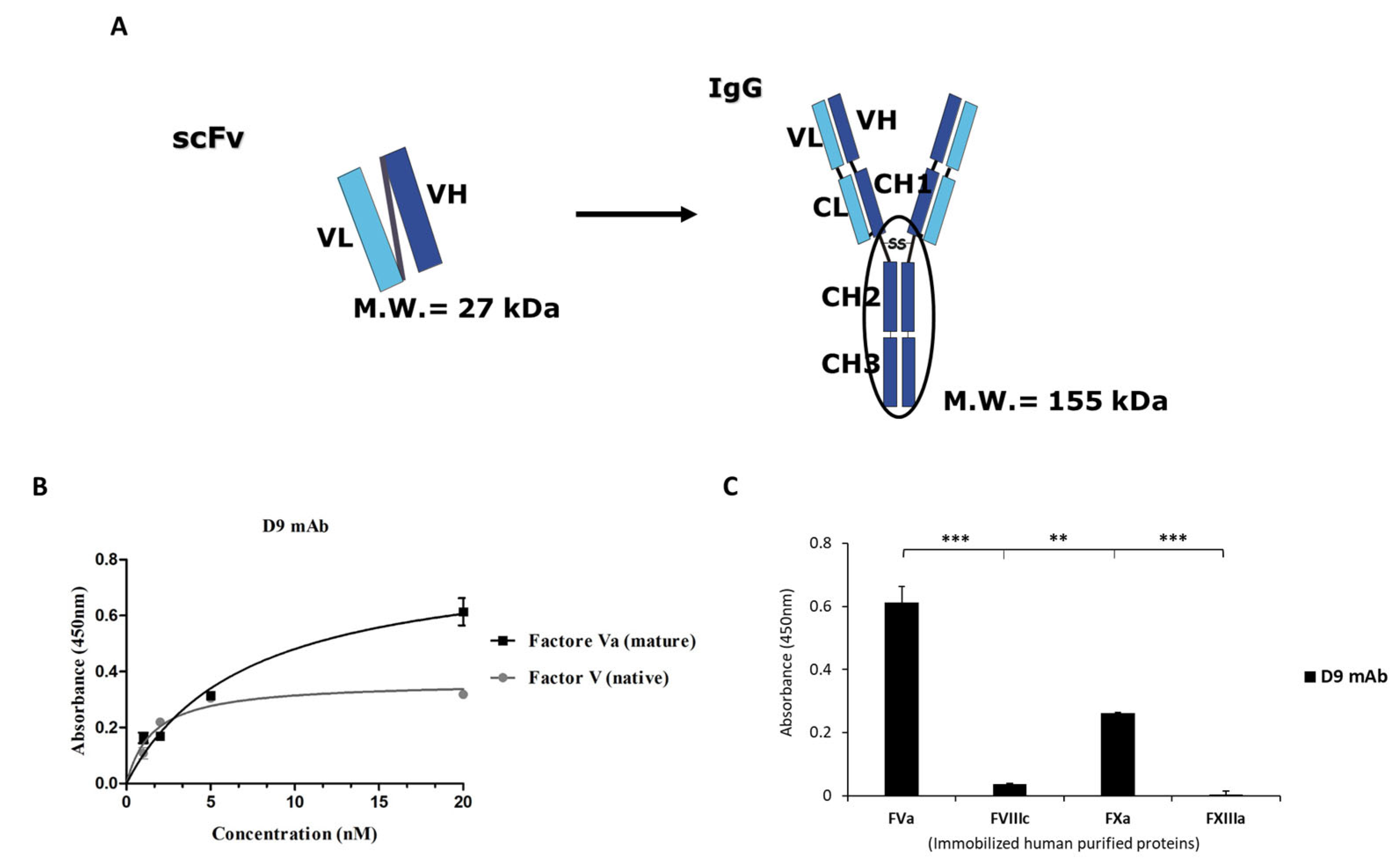
2.4. Generation and Characterization of the Novel Anti-FVa D9 Full-Size mAb
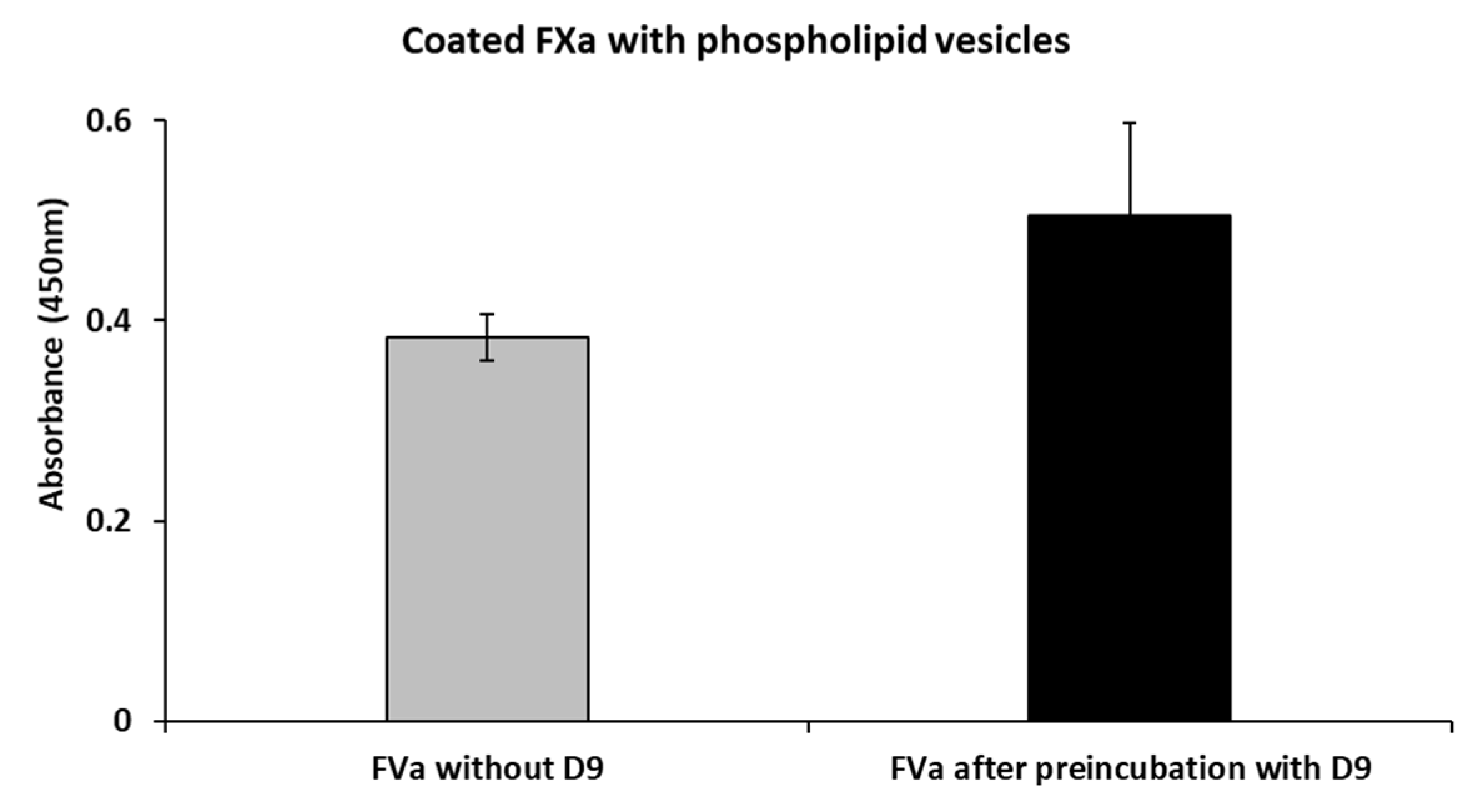
2.5. Effects of D9 on the Complex Formation Between FVa and FXa
2.6. Effects of the D9 mAb on the Coagulation Cascade
3. Discussion
4. Materials and Methods
4.1. Antibodies, Human Recombinant Proteins, and Phospholipids
4.2. Bacterial Strains, Culture Media, and Antibiotics
4.3. Selection of scFv-Phage Clones
4.4. Preparation of scFv-Phages for ELISA and Analysis of Positive Clones
4.5. Soluble scFv Expression and Partial Purification
4.6. ELISA Assays of D9 Soluble scFv
4.7. Conversion of scFv into Full-Size mAb
4.8. Binding Specificity of D9 mAb by ELISA Assays
4.9. Phospholipid Vesicles Preparation
4.10. Competitive ELISA Assays
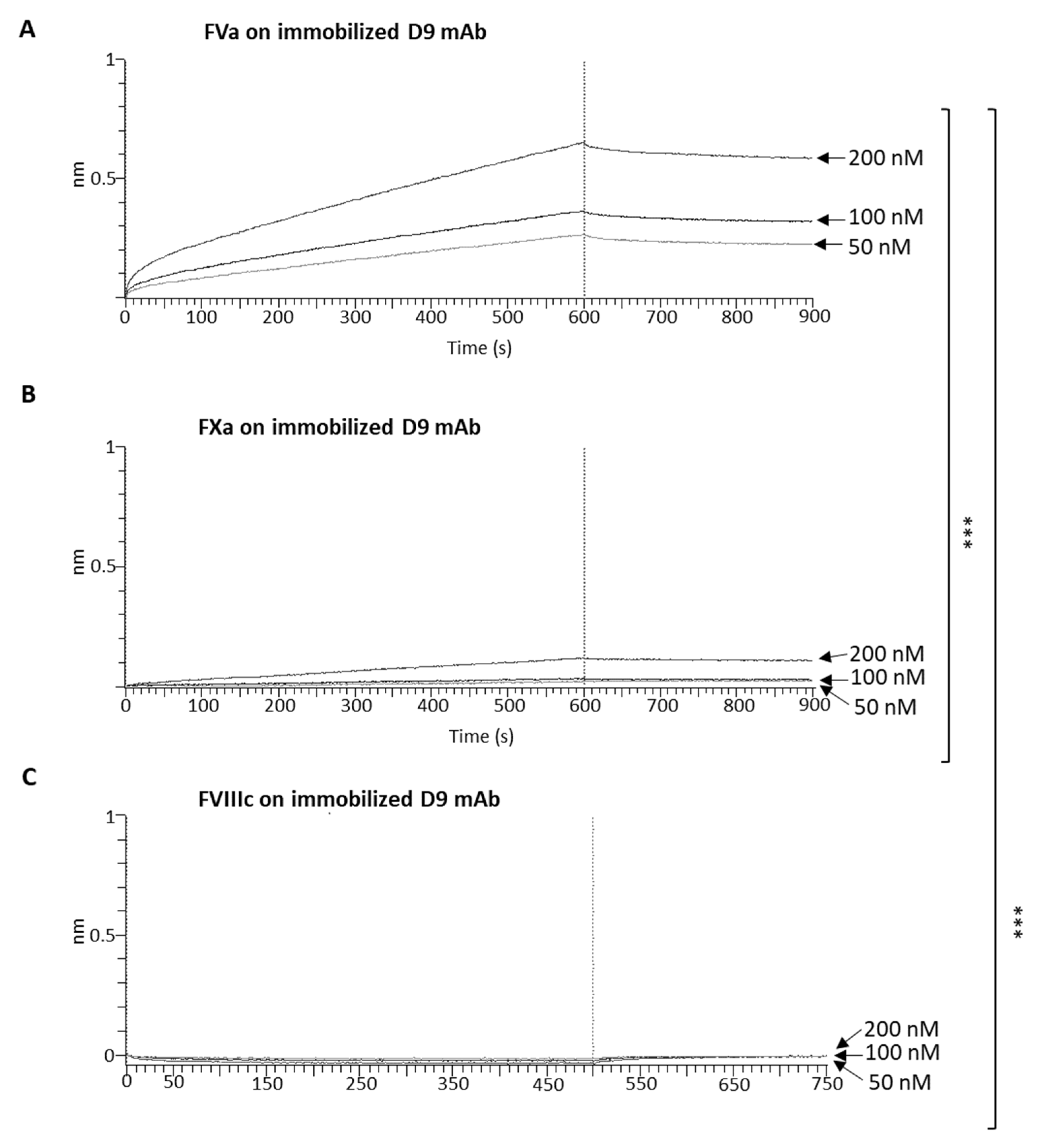
4.11. Biolayer Interferometry (BLI) Analyses
4.12. Effects of D9 on Blood Coagulation
4.13. Effects of D9 on FV Activation
4.13.1. Thrombin Mediated Digestion of FV in the Absence or Presence of D9
4.13.2. Western Blotting Analysis (WB)
4.14. Statistics
4.15. Docking Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aPTT | Activated partial thromboplastin time |
| BLI | Biolayer Interferometry |
| DOACs | Direct oral anticoagulants |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPS | 1,2-dioctadecenoyl-sn-glycero-3-phosphoserine |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| FII | Factor II |
| FIX | Factor IX |
| FV | Factor V |
| FVa | Activated Factor V |
| FVIIIc | Factor VIIIc |
| FXa | Activated Factor X |
| FXI | Factor XI |
| FXII | Factor XII |
| FXIIIa | Activated Factor XIII |
| HRP | Horseradish peroxidase |
| IPTG | Isopropyl β-d-1-thiogalactopyranoside |
| KB | Kinetic Buffer |
| LMWH | Low-molecular-weight heparins |
| mAbs | Monoclonal antibodies |
| ProA | Protein A |
| PT | Prothrombin time |
| scFv | Single-chain variable fragment |
| VKA | Vitamin K antagonists |
| WB | Western Blotting |
References
- Ashorobi, D.; Ameer, M.A.; Fernandez, R. Thrombosis; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Senst, B.; Tadi, P.; Basit, H.; Jan, A. Hypercoagulability; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Pastori, D.; Cormaci, V.M.; Marucci, S.; Franchino, G.; Del Sole, F.; Capozza, A.; Fallarino, A.; Corso, C.; Valeriani, E.; Menichelli, D.; et al. A Comprehensive Review of Risk Factors for Venous Thromboembolism: From Epidemiology to Pathophysiology. Int. J. Mol. Sci. 2023, 24, 3169. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Franchini, M.; Montagnana, M.; Favaloro, E.J. Inherited disorders of blood coagulation. Ann. Med. 2011, 44, 405–418. [Google Scholar] [CrossRef]
- del Zoppo, G.J. Virchow’s triad: The vascular basis of cerebral injury. Rev. Neurol. Dis. 2008, 5, S12–S21. [Google Scholar]
- Morange, P.E.; Suchon, P.; Trégouët, D.A. Genetics of Venous Thrombosis: Update in 2015. Thromb. Haemost. 2015, 114, 910–919. [Google Scholar]
- Martinelli, I.; Bucciarelli, P.; Mannucci, P.M. Thrombotic risk factors: Basic pathophysiology. Crit. Care Med. 2010, 38, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Troisi, R.; Balasco, N.; Autiero, I.; Sica, F.; Vitagliano, L. New insight into the traditional model of the coagulation cascade and its regulation: Illustrated review of a three-dimensional view. Res. Pract. Thromb. Haemost. 2023, 7, 102160. [Google Scholar] [CrossRef]
- Franchini, M.; Liumbruno, G.M.; Bonfanti, C.; Lippi, G. The evolution of anticoagulant therapy. Blood Transfus. 2016, 14, 175–184. [Google Scholar] [PubMed]
- Schulman, S.; Bijsterveld, N.R. Anticoagulants and their reversal. Transfus. Med. Rev. 2007, 21, 37–48. [Google Scholar] [CrossRef]
- Fredenburgh, J.C.; Weitz, J.I. New anticoagulants: Moving beyond the direct oral anticoagulants. J. Thromb. Haemost. 2021, 19, 20–29. [Google Scholar] [CrossRef]
- Bentounes, N.K.; Melicine, S.; Martin, A.C.; Smadja, D.M.; Gendron, N. Development of new anticoagulant in 2023: Prime time for anti-factor XI and XIa inhibitors. J. Med. Vasc. 2023, 48, 69–80. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; García-Fuentes, M.; Suárez-Gea, M.L. Discovery methods of coagulation-inhibiting drugs. Expert. Opin. Drug Discov. 2017, 12, 1195–1205. [Google Scholar] [CrossRef]
- Chai-Adisaksopha, C.; Hillis, C.; Isayama, T.; Lim, W.; Iorio, A.; Crowther, M. Mortality outcomes in patients receiving direct oral anticoagulants: A systematic review and meta-analysis of randomized controlled trials. J. Thromb. Haemost. 2015, 13, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Burness, C.B. Idarucizumab: First Global Approval. Drugs 2015, 75, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Andexanet Alfa: First Global Approval. Drugs 2018, 78, 1285. [Google Scholar] [CrossRef]
- Liu, M.; Zaman, K.; Fortenberry, Y.M. Overview of the Therapeutic Potential of Aptamers Targeting Coagulation Factors. Int. J. Mol. Sci. 2021, 22, 3897. [Google Scholar] [CrossRef]
- Derszniak, K.; Przyborowski, K.; Matyjaszczyk, K.; Moorlag, M.; de Laat, B.; Nowakowska, M.; Chlopicki, S. Comparison of Effects of Anti-thrombin Aptamers HD1 and HD22 on Aggregation of Human Platelets, Thrombin Generation, Fibrin Formation, and Thrombus Formation Under Flow Conditions. Front. Pharmacol. 2019, 10, 68. [Google Scholar] [CrossRef]
- Mao, Y.; Gu, J.; Chang, D.; Wang, L.; Yao, L.; Ma, Q.; Luo, Z.; Qu, H.; Li, Y.; Zheng, L. Evolution of a highly functional circular DNA aptamer in serum. Nucleic Acids Res. 2020, 48, 10680–10690. [Google Scholar] [CrossRef]
- Dahlbäck, B. Novel insights into the regulation of coagulation by factor V isoforms, tissue factor pathway inhibitorα, and protein S. J. Thromb. Haemost. 2017, 15, 1241–1250. [Google Scholar] [CrossRef]
- Schreuder, M.; Reitsma, P.H.; Bos, M.H.A. Blood coagulation factor Va’s key interactive residues and regions for prothrombinase assembly and prothrombin binding. J. Thromb. Haemost. 2019, 17, 1229–1239. [Google Scholar] [CrossRef]
- De Lorenzo, C.; Palmer, D.B.; Piccoli, R.; Ritter, M.A.; D’Alessio, G. A new human antitumor immunoreagent specific for ErbB2. Clin. Cancer Res. 2002, 8, 1710–1719. [Google Scholar]
- Sasso, E.; D’Avino, C.; Passariello, M.; D’Alise, A.M.; Siciliano, D.; Esposito, M.L.; Froechlich, G.; Cortese, R.; Scarselli, E.; Zambrano, N.; et al. Massive parallel screening of phage libraries for the generation of repertoires of human immunomodulatory monoclonal antibodies. mAbs 2018, 10, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Akbar, R.; Robert, P.A.; Weber, C.R.; Widrich, M.; Frank, R.; Pavlović, M.; Scheffer, L.; Chernigovskaya, M.; Snapkov, I.; Slabodkin, A.; et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. mAbs 2022, 14, 2031482. [Google Scholar] [CrossRef] [PubMed]
- Hummer, A.M.; Abanades, B.; Deane, C.M. Advances in computational structure-based antibody design. Curr. Opin. Struct. Biol. 2022, 74, 102379. [Google Scholar] [CrossRef]
- Ruben, E.A.; Rau, M.J.; Fitzpatrick, J.A.J.; Di Cera, E. Cryo-EM structures of human coagulation factors V and Va. Blood 2021, 137, 3137–3144. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, T.; Ayombil, F.; Van’t Veer, C.; Camire, R.M. Regulation of factor V and factor V-short by TFPIα: Relationship between B-domain proteolysis and binding. J. Biol. Chem. 2021, 296, 100234. [Google Scholar] [CrossRef]
- Ruben, E.A.; Summers, B.; Rau, M.J.; Fitzpatrick, J.A.J.; Di Cera, E. Cryo-EM structure of the prothrombin-prothrombinase complex. Blood 2022, 139, 3463–3473. [Google Scholar] [CrossRef]
- Beck, D.O.; Bukys, M.A.; Singh, L.S.; Szabo, K.A.; Kalafatis, M. The contribution of amino acid region ASP695-TYR698 of factor V to procofactor activation and factor Va function. J. Biol. Chem. 2004, 279, 3084–3095. [Google Scholar] [CrossRef]
- Shim, J.Y.; Lee, C.J.; Wu, S.; Pedersen, L.G. A model for the unique role of factor Va A2 domain extension in the human ternary thrombin-generating complex. Biophys. Chem. 2015, 199, 46–50. [Google Scholar] [CrossRef]
- Qiao, S.P.; Liu, Z.N.; Li, H.C.; He, X.; Pan, H.; Gao, Y.Z. Construction of a CRISPR-Biolayer Interferometry Platform for Real-Time, Sensitive, and Specific DNA Detection. Chembiochem 2021, 22, 1974–1984. [Google Scholar] [CrossRef]
- Lindhout, T.; Govers-Riemslag, J.W.; van de Waart, P.; Hemker, H.C.; Rosing, J. Factor Va-factor Xa interaction. Effects of phospholipid vesicles of varying composition. Biochemistry 1982, 21, 5494–5502. [Google Scholar] [CrossRef]
- Rosing, J.; Tans, G.; Govers-Riemslag, J.W.; Zwaal, R.F.; Hemker, H.C. The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 1980, 255, 274–283. [Google Scholar] [CrossRef]
- Sidonio, R.F., Jr.; Hoffman, M.; Kenet, G.; Dargaud, Y. Thrombin generation and implications for hemophilia therapies: A narrative review. Res. Pract. Thromb. Haemost. 2022, 7, 100018. [Google Scholar] [CrossRef]
- Lakshmanan, H.H.S.; Estonilo, A.; Reitsma, S.E.; Melrose, A.R.; Subramanian, J.; Zheng, T.J.; Maddala, J.; Tucker, E.I.; Gailani, D.; McCarty, O.J.T.; et al. Revised model of the tissue factor pathway of thrombin generation: Role of the feedback activation of FXI. J. Thromb. Haemost. 2022, 20, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Passariello, M.; Gentile, C.; Ferrucci, V.; Sasso, E.; Vetrei, C.; Fusco, G.; Viscardi, M.; Brandi, S.; Cerino, P.; Zambrano, N.; et al. Novel human neutralizing mAbs specific for Spike-RBD of SARS-CoV-2. Sci. Rep. 2021, 11, 11046. [Google Scholar] [CrossRef]
- Passariello, M.; Vetrei, C.; Sasso, E.; Froechlich, G.; Gentile, C.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Nicosia, A.; De Lorenzo, C. Isolation of Two Novel Human Anti-CTLA-4 mAbs with Intriguing Biological Properties on Tumor and NK Cells. Cancers 2020, 12, 2204. [Google Scholar] [CrossRef] [PubMed]
- de Kruijff, B.; Cullis, P.R.; Radda, G.K. Differential scanning calorimetry and 31P NMR studies on sonicated and unsonicated phosphatidylcholine liposomes. Biochim. Biophys. Acta 1975, 406, 6–20. [Google Scholar] [CrossRef]
- Passariello, M.; Esposito, S.; Manna, L.; Rapuano Lembo, R.; Zollo, I.; Sasso, E.; Amato, F.; De Lorenzo, C. Comparative Analysis of a Human Neutralizing mAb Specific for SARS-CoV-2 Spike-RBD with Cilgavimab and Tixagevimab for the Efficacy on the Omicron Variant in Neutralizing and Detection Assays. Int. J. Mol. Sci. 2023, 24, 10053. [Google Scholar] [CrossRef]
- Manna, L.; Rapuano Lembo, R.; Yoshioka, A.; Nakamura, K.; Passariello, M.; De Lorenzo, C. A Comparison of the Antitumor Efficacy of Novel Multi-Specific Tribodies with Combinations of Approved Immunomodulatory Antibodies. Cancers 2023, 15, 5345. [Google Scholar] [CrossRef]
- Orthwein, T.; Huergo, L.F.; Forchhammer, K.; Selim, K.A. Kinetic Analysis of a Protein-protein Complex to Determine its Dissociation Constant (KD) and the Effective Concentration (EC50) of an Interplaying Effector Molecule Using Bio-layer Interferometry. Bio Protocol. 2021, 11, e4152. [Google Scholar] [CrossRef]
- Gelardi, T.; Damiano, V.; Rosa, R.; Bianco, R.; Cozzolino, R.; Tortora, G.; Laccetti, P.; D’Alessio, G.; De Lorenzo, C. Two novel human anti-ErbB2 immunoagents are active on trastuzumab-resistant tumours. Br.J. Cancer 2010, 102, 513–519. [Google Scholar] [CrossRef]
- Motulsky, H.J. Prism 5 Statistics Guide; GraphPad Software Inc.: San Diego, CA, USA, 2007; Available online: www.graphpad.com (accessed on 17 February 2025).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garcia, B.; Pons, C.; Fernandez-Recio, J. pyDockWEB: A web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) D9 mAb effects on the coagulation time | ||||||||||||||
| Sample | PT (v.r. 0.8–1.20 Ratio) | PT seconds | PT INR (v.r. 0.8–1.20 Ratio) | aPTT (v.r. 0.8–1.20 Ratio) | aPTT seconds | |||||||||
| NORMAL POOL PLASMA (untreated) | 1.01 | 11.5 | 1.01 | 1.02 | 29.3 | |||||||||
| NORMAL POOL PLASMA (+100 nM D9 mAb) | 0.98 | 11.2 | 0.97 | 1.07 | 30.6 | |||||||||
| NORMAL POOL PLASMA (+1 μM D9 mAb) | 0.95 | 10.9 | 0.95 | 1.47 | 42.4 | |||||||||
| NORMAL POOL PLASMA (+2 μM D9 mAb) | 0.96 | 11 | 0.96 | 1.9 | 54 | |||||||||
| (B) D9 mAb effects on the coagulation factors | ||||||||||||||
| (% activity) | (seconds) | |||||||||||||
| NORMAL POOL PLASMA | FII (v.r. 60–120%) | FV (v.r. 60–120%) | FVIII (v.r. 50–130%) | FIX (v.r. 50–120%) | FX (v.r. 60–120%) | FXI (v.r. 50–120%) | FXII (v.r. 50–120%) | FII | FV | FVIII | FIX | FX | FXI | FXII |
| (untreated) | 95 | 102 | 84 | 109 | 96 | 98 | 83 | 14.2 | 16.9 | 53.5 | 51.3 | 21 | 55 | 50.2 |
| (+100 nM D9 mAb) | 105.6 | 97 | 73 | 97 | 96 | 94 | 77 | 13.9 | 17.1 | 54.6 | 52.2 | 21 | 55 | 51 |
| (+1 μM D9 mAb) | 101 | 82 | 53 | 67 | 96 | 57 | 61 | 14.1 | 17.8 | 58 | 55.2 | 21.3 | 59.6 | 53 |
| (+2 μM D9 mAb) | 98 | 73 | 40 | 47 | 93 | 40 | 49 | 14.2 | 18.3 | 60.3 | 58.3 | 21.6 | 64.3 | 54.5 |
| p-value | ns | ** | ** | ** | ns | ** | ** | ns | ** | ** | ** | ns | ** | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passariello, M.; Rapuano Lembo, R.; Manna, L.; Miele, C.; Merlino, A.; Mazzaccara, C.; Leonardi, A.; De Lorenzo, C. A Novel Human Anti-FV mAb as a Potential Tool for Diagnostic and Coagulation Inhibitory Approaches. Int. J. Mol. Sci. 2025, 26, 2721. https://doi.org/10.3390/ijms26062721
Passariello M, Rapuano Lembo R, Manna L, Miele C, Merlino A, Mazzaccara C, Leonardi A, De Lorenzo C. A Novel Human Anti-FV mAb as a Potential Tool for Diagnostic and Coagulation Inhibitory Approaches. International Journal of Molecular Sciences. 2025; 26(6):2721. https://doi.org/10.3390/ijms26062721
Chicago/Turabian StylePassariello, Margherita, Rosa Rapuano Lembo, Lorenzo Manna, Ciro Miele, Antonello Merlino, Cristina Mazzaccara, Antonio Leonardi, and Claudia De Lorenzo. 2025. "A Novel Human Anti-FV mAb as a Potential Tool for Diagnostic and Coagulation Inhibitory Approaches" International Journal of Molecular Sciences 26, no. 6: 2721. https://doi.org/10.3390/ijms26062721
APA StylePassariello, M., Rapuano Lembo, R., Manna, L., Miele, C., Merlino, A., Mazzaccara, C., Leonardi, A., & De Lorenzo, C. (2025). A Novel Human Anti-FV mAb as a Potential Tool for Diagnostic and Coagulation Inhibitory Approaches. International Journal of Molecular Sciences, 26(6), 2721. https://doi.org/10.3390/ijms26062721