
Enhancing CAR T-Cell Function with Domains of Innate Immunity Sensors


Abstract
:1. Introduction
2. CAR T-Cell Therapy and Its Challenges
3. Innate Immunity Receptors and Their Domains
4. CAR T-Cells with Innate Immunity Domains
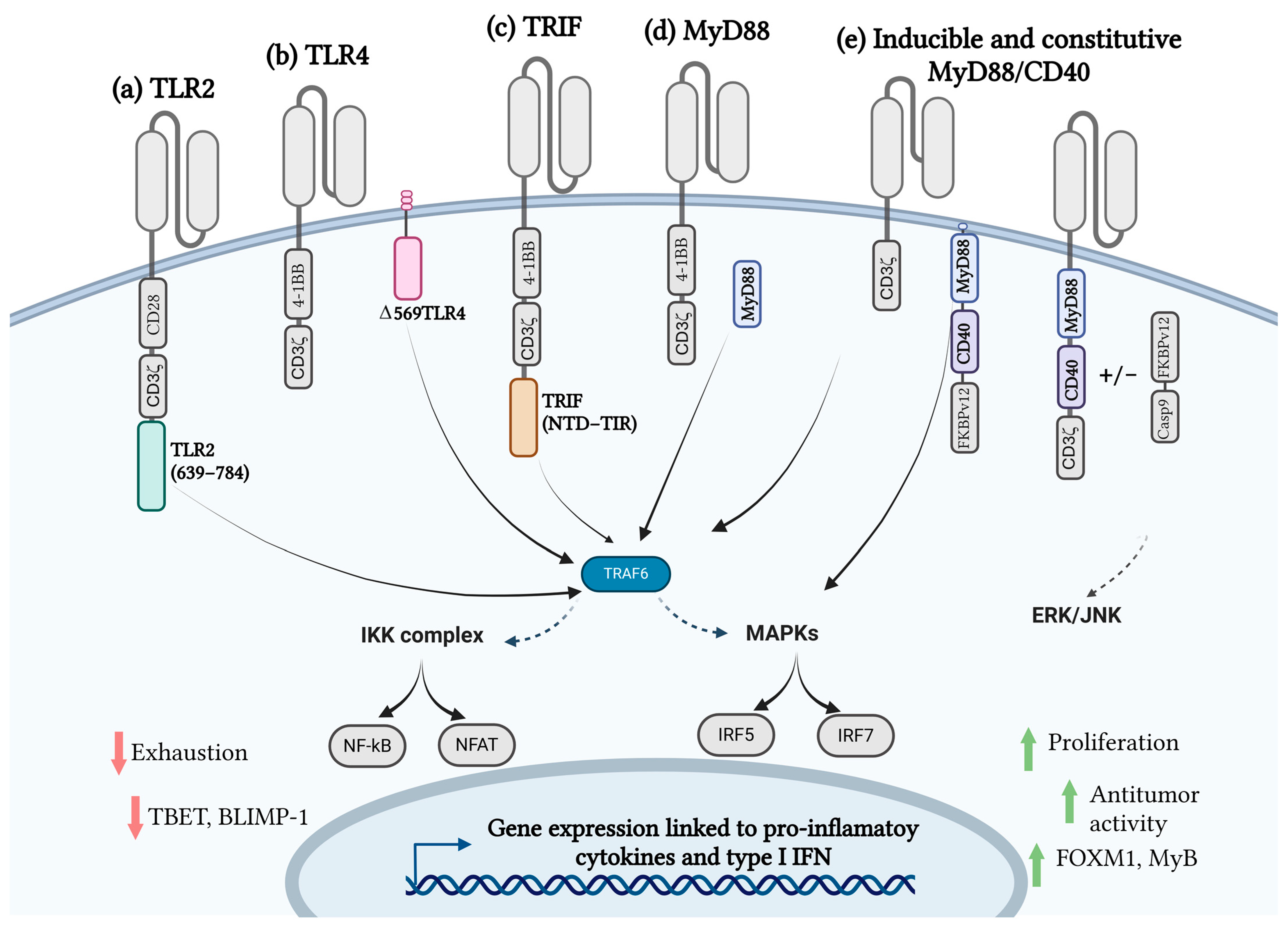
4.1. Toll-like Receptors and CAR T-Cell Therapy
4.2. MyD88/CD40 CAR T-Cell Therapy
4.3. TRIF CAR T-Cell Therapy
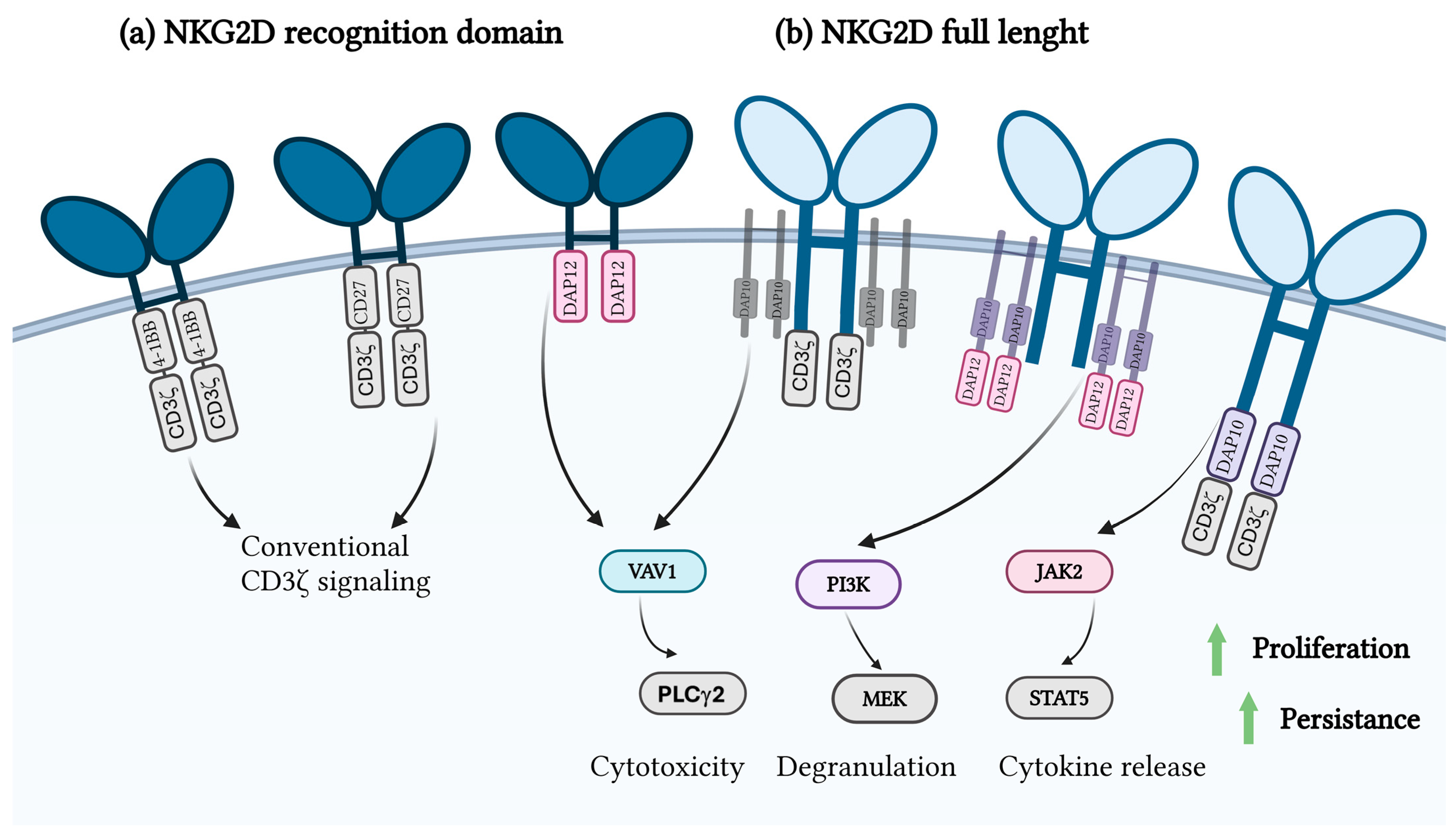
4.4. NKG2D and CAR T-Cell Therapy
5. Advantages and Limitations of Incorporating Innate Immune Domains in CAR T-Cells
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, L.A.; June, C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017, 27, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.R.; Devan, A.R.; Nair, B.; Vinod, B.S.; Nath, L.R. Harnessing the immune system against cancer: Current immunotherapy approaches and therapeutic targets. Mol. Biol. Rep. 2021, 48, 8075–8095. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Afamitresgene Autoleucel: First Approval. Mol. Diagn. Ther. 2024, 28, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Almåsbak, H.; Aarvak, T.; Vemuri, M.C. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J. Immunol. Res. 2016, 2016, 5474602. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, W.; Li, D.; Niu, T.; Wang, W. BCMA-targeting chimeric antigen receptor T-cell therapy for multiple myeloma. Cancer Lett. 2023, 553, 215949. [Google Scholar] [CrossRef] [PubMed]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The Long Road to the First FDA Approved Gene Therapy: Chimeric Antigen Receptor T Cells Targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Kagoya, Y. Cytokine signaling in chimeric antigen receptor T-cell therapy. Int. Immunol. 2023, 36, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Pradeu, T.; Thomma, B.P.H.J.; Girardin, S.E.; Lemaitre, B. The conceptual foundations of innate immunity: Taking stock 30 years later. Immunity 2024, 57, 613–631. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, N.; Li, C.; Cao, Y.; Xiao, Y.; Xiao, M.; Zhang, Y.; Zhang, T.; Zhou, J. Sequential Infusion of Anti-CD22 and Anti-CD19 Chimeric Antigen Receptor T Cells for Adult Patients with Refractory/Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2017, 130, 846. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]
- Globerson Levin, A.; Rawet Slobodkin, M.; Waks, T.; Horn, G.; Ninio-Many, L.; Deshet Unger, N.; Ohayon, Y.; Suliman, S.; Cohen, Y.; Tartakovsky, B.; et al. Treatment of Multiple Myeloma Using Chimeric Antigen Receptor T Cells with Dual Specificity. Cancer Immunol. Res. 2020, 8, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Mueller, K.T.; Waldron, E.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Hamilton, J.; Awasthi, R.; et al. Clinical Pharmacology of Tisagenlecleucel in B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 6175–6184. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, C.; Hudson, K.L.; Anderson, K.V. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 1988, 52, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; O’Neill, L.A.J. From periphery to center stage: 50 years of advancements in innate immunity. Cell 2024, 187, 2030–2051. [Google Scholar] [CrossRef]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.-M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette spätzle/Toll/cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Nouri, Y.; Weinkove, R.; Perret, R. T-cell intrinsic Toll-like receptor signaling: Implications for cancer immunotherapy and CAR T-cells. J. Immunother. Cancer 2021, 9, e003065. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L. Important aspects of Toll-like receptors, ligands and their signaling pathways. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. Al 2010, 59, 791–808. [Google Scholar] [CrossRef]
- Ranoa, D.R.E.; Kelley, S.L.; Tapping, R.I. Human Lipopolysaccharide-binding Protein (LBP) and CD14 Independently Deliver Triacylated Lipoproteins to Toll-like Receptor 1 (TLR1) and TLR2 and Enhance Formation of the Ternary Signaling Complex. J. Biol. Chem. 2013, 288, 9729–9741. [Google Scholar] [CrossRef]
- Pohar, J.; Kužnik Krajnik, A.; Jerala, R.; Benčina, M. Minimal sequence requirements for oligodeoxyribonucleotides activating human TLR9. J. Immunol. 2015, 194, 3901–3908. [Google Scholar] [CrossRef]
- Pohar, J.; Lainšček, D.; Fukui, R.; Yamamoto, C.; Miyake, K.; Jerala, R.; Benčina, M. Species-Specific Minimal Sequence Motif for Oligodeoxyribonucleotides Activating Mouse TLR9. J. Immunol. 2015, 195, 4396–4405. [Google Scholar] [CrossRef]
- Enokizono, Y.; Kumeta, H.; Funami, K.; Horiuchi, M.; Sarmiento, J.; Yamashita, K.; Standley, D.M.; Matsumoto, M.; Seya, T.; Inagaki, F. Structures and interface mapping of the TIR domain-containing adaptor molecules involved in interferon signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 19908–19913. [Google Scholar] [CrossRef] [PubMed]
- Nimma, S.; Gu, W.; Maruta, N.; Li, Y.; Pan, M.; Saikot, F.K.; Lim, B.Y.J.; McGuinness, H.Y.; Zaoti, Z.F.; Li, S.; et al. Structural Evolution of TIR-Domain Signalosomes. Front. Immunol. 2021, 12, 784484. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, L.; Chen, P.; Liang, G. Myeloid Differentiation Primary Response Protein 88 (MyD88): The Central Hub of TLR/IL-1R Signaling. J. Med. Chem. 2020, 63, 13316–13329. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Fu, Y. The critical role of Toll-like receptor-mediated signaling in cancer immunotherapy. Med. Drug Discov. 2022, 14, 100122. [Google Scholar] [CrossRef]
- Lai, Y.; Weng, J.; Wei, X.; Qin, L.; Lai, P.; Zhao, R.; Jiang, Z.; Li, B.; Lin, S.; Wang, S.; et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia 2018, 32, 801–808. [Google Scholar] [CrossRef]
- Pereira, M.; Gazzinelli, R.T. Regulation of innate immune signaling by IRAK proteins. Front. Immunol. 2023, 14, 1133354. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Yamamoto, T.; Yamazaki, K.; Gohda, J.; Akiyama, T.; Semba, K.; Goto, H.; Kato, A.; Yujiri, T.; Imai, T.; et al. TRAF6 establishes innate immune responses by activating NF-kappaB and IRF7 upon sensing cytosolic viral RNA and DNA. PLoS ONE 2009, 4, e5674. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Jelenčić, V.; Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Mikolič, V.; Pantović-Žalig, J.; Malenšek, Š.; Sever, M.; Lainšček, D.; Jerala, R. Toll-like receptor 4 signaling activation domains promote CAR T cell function against solid tumors. Mol. Ther. Oncol. 2024, 32, 200815. [Google Scholar] [CrossRef]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Collinson-Pautz, M.R.; Chang, W.C.; Lu, A.; Khalil, M.; Crisostomo, J.W.; Lin, P.Y.; Mahendravada, A.; Shinners, N.P.; Brandt, M.E.; Zhang, M.; et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia 2019, 33, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Komai-Koma, M.; Jones, L.; Ogg, G.S.; Xu, D.; Liew, F.Y. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Crellin, N.K.; Garcia, R.V.; Hadisfar, O.; Allan, S.E.; Steiner, T.S.; Levings, M.K. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J. Immunol. 2005, 175, 8051–8059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Ma, Z.; Lu, M. Contribution of T- and B-cell intrinsic toll-like receptors to the adaptive immune response in viral infectious diseases. Cell. Mol. Life Sci. 2022, 79, 547. [Google Scholar] [CrossRef]
- Reynolds, J.M.; Pappu, B.P.; Peng, J.; Martinez, G.J.; Zhang, Y.; Chung, Y.; Ma, L.; Yang, X.O.; Nurieva, R.I.; Tian, Q.; et al. Toll-like receptor 2 signaling in CD4+ T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010, 32, 692–702. [Google Scholar] [CrossRef]
- Zhang, E.; Ma, Z.; Li, Q.; Yan, H.; Liu, J.; Wu, W.; Guo, J.; Zhang, X.; Kirschning, C.J.; Xu, H.; et al. TLR2 Stimulation Increases Cellular Metabolism in CD8+ T Cells and Thereby Enhances CD8+ T Cell Activation, Function, and Antiviral Activity. J. Immunol. 2019, 203, 2872–2886. [Google Scholar] [CrossRef]
- Zahm, C.D.; Colluru, V.T.; McIlwain, S.J.; Ong, I.M.; McNeel, D.G. TLR Stimulation during T-cell Activation Lowers PD-1 Expression on CD8+ T Cells. Cancer Immunol. Res. 2018, 6, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Jorge, O.; Kempis-Calanis, L.A.; Abou-Jaoudé, W.; Gutiérrez-Reyna, D.Y.; Hernandez, C.; Ramirez-Pliego, O.; Thomas-Chollier, M.; Spicuglia, S.; Santana, M.A.; Thieffry, D. Cooperation between T cell receptor and Toll-like receptor 5 signaling for CD4+ T cell activation. Sci. Signal. 2019, 12, eaar3641. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Lai, P.; Qin, L.; Lai, Y.; Jiang, Z.; Luo, C.; Huang, X.; Wu, S.; Shao, D.; Deng, C.; et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J. Hematol. Oncol. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- George, P.; Dasyam, N.; Giunti, G.; Mester, B.; Bauer, E.; Andrews, B.; Perera, T.; Ostapowicz, T.; Frampton, C.; Li, P.; et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: A phase I clinical trial protocol (ENABLE). BMJ Open 2020, 10, e034629. [Google Scholar] [CrossRef] [PubMed]
- Lei, A.; Yu, H.; Lu, S.; Lu, H.; Ding, X.; Tan, T.; Zhang, H.; Zhu, M.; Tian, L.; Wang, X.; et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy. Nat. Immunol. 2024, 25, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Li, Z.; Wang, Z.; Chen, C.; Luo, Y. Chimeric antigen receptor macrophages activated through TLR4 or IFN-γ receptors suppress breast cancer growth by targeting VEGFR2. Cancer Immunol. Immunother. CII 2023, 72, 3243–3257. [Google Scholar] [CrossRef] [PubMed]
- Wesche, H.; Henzel, W.J.; Shillinglaw, W.; Li, S.; Cao, Z. MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity 1997, 7, 837–847. [Google Scholar] [CrossRef]
- Schenten, D.; Nish, S.A.; Yu, S.; Yan, X.; Lee, H.K.; Brodsky, I.; Pasman, L.; Yordy, B.; Wunderlich, T.; Brüning, J.C.; et al. MyD88 signaling in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity 2014, 40, 78–90. [Google Scholar] [CrossRef]
- Mandraju, R.; Jain, A.; Gao, Y.; Ouyang, Z.; Norgard, M.V.; Pasare, C. MyD88 Signaling in T Cells Is Critical for Effector CD4 T Cell Differentiation following a Transitional T Follicular Helper Cell Stage. Infect. Immun. 2018, 86, e00791-17. [Google Scholar] [CrossRef]
- Foster, A.E.; Mahendravada, A.; Shinners, N.P.; Chang, W.C.; Crisostomo, J.; Lu, A.; Khalil, M.; Morschl, E.; Shaw, J.L.; Saha, S.; et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol. Ther. 2017, 25, 2176–2188. [Google Scholar] [CrossRef]
- Stein, M.N.; Teply, B.A.; Gergis, U.; Strickland, D.; Senesac, J.; Bayle, H.; Chatwal, M.S.; Bilen, M.A.; Stadler, W.M.; Dumbrava, E.E. Early results from a phase 1, multicenter trial of PSCA-specific GoCAR T cells (BPX-601) in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2023, 41, 140. [Google Scholar] [CrossRef]
- Duong, M.T.; Collinson-Pautz, M.R.; Morschl, E.; Lu, A.; Szymanski, S.P.; Zhang, M.; Brandt, M.E.; Chang, W.C.; Sharp, K.L.; Toler, S.M.; et al. Two-Dimensional Regulation of CAR-T Cell Therapy with Orthogonal Switches. Mol. Ther.–Oncolytics 2019, 12, 124–137. [Google Scholar] [CrossRef]
- Prinzing, B.; Schreiner, P.; Bell, M.; Fan, Y.; Krenciute, G.; Gottschalk, S. MyD88/CD40 signaling retains CAR T cells in a less differentiated state. JCI Insight 2020, 5, e136093. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jasinski, D.L.; Medina, J.L.; Spencer, D.M.; Foster, A.E.; Bayle, J.H. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020, 4, 1950–1964. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.-Y.; Tay, J.C.K.; Li, Z.; Wang, J.; Zhu, J.; Wang, S. T Cells Expressing NKG2D CAR with a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ai, K.; Liu, B.; Chen, X.; Huang, C.; Yang, L.; Zhang, W.; Weng, J.; Du, X.; Wu, K.; Lai, P. Optimizing CAR-T cell therapy for solid tumors: Current challenges and potential strategies. J. Hematol. Oncol. 2024, 17, 105. [Google Scholar] [CrossRef]
- Li, S.; Zhao, R.; Zheng, D.; Qin, L.; Cui, Y.; Li, Y.; Jiang, Z.; Zhong, M.; Shi, J.; Li, M.; et al. DAP10 integration in CAR-T cells enhances the killing of heterogeneous tumors by harnessing endogenous NKG2D. Mol. Ther.–Oncolytics 2022, 26, 15–26. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Navarro, M.; Fernández, A.; Escudero, A.; Esteso, G.; Campos-Silva, C.; Navarro-Aguadero, M.Á.; Leivas, A.; Caracuel, B.R.; Rodríguez-Antolín, C.; Ortiz, A.; et al. NKG2D-CAR memory T cells target pediatric T-cell acute lymphoblastic leukemia in vitro and in vivo but fail to eliminate leukemia initiating cells. Front. Immunol. 2023, 14, 1187665. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xie, W.; Song, D.-G.; Powell, D.J. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018, 11, 92. [Google Scholar] [CrossRef]
- Driouk, L.; Gicobi, J.K.; Kamihara, Y.; Rutherford, K.; Dranoff, G.; Ritz, J.; Baumeister, S.H.C. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 11, 580328. [Google Scholar] [CrossRef]
- Solid tumor immunotherapy using NKG2D-based adaptor CAR T cells. Cell Rep. Med. 2024, 5, 101827. [CrossRef]
- Yang, D.; Sun, B.; Li, S.; Wei, W.; Liu, X.; Cui, X.; Zhang, X.; Liu, N.; Yan, L.; Deng, Y.; et al. NKG2D-CAR T cells eliminate senescent cells in aged mice and nonhuman primates. Sci. Transl. Med. 2023, 15, eadd1951. [Google Scholar] [CrossRef]
- Interim Results from the Phase I Deplethink Trial Evaluating the Infusion of a NKG2D CAR T-Cell Therapy Post a Non-Myeloablative Conditioning in Relapse or Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome Patients. Available online: https://colab.ws/articles/10.1182%2Fblood-2019-128267 (accessed on 6 December 2024).
- The Third Affiliated Hospital of Guangzhou Medical University. Hepatic Artery Transfusion of NKG2D CAR-T Cells to Treat Patients with Previously Treated Liver Metastatic Colorectal Cancer: A Prospective, Multicenter Clinical Trial; The Third Affiliated Hospital of Guangzhou Medical University: Guangzhou, China, 2022. [Google Scholar]
- Jiang, G.; Ng, Y.Y.; Tay, J.C.K.; Du, Z.; Xiao, L.; Wang, S.; Zhu, J. Dual CAR-T cells to treat cancers co-expressing NKG2D and PD1 ligands in xenograft models of peritoneal metastasis. Cancer Immunol. Immunother. CII 2023, 72, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, W.; Lv, H.; Gu, D.; Wei, X.; Dai, H. Tandem CAR-T cells targeting CLDN18.2 and NKG2DL for treatment of gastric cancer. J. Clin. Oncol. 2022, 40, 4030. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Paget, C.; Apetoh, L. STING-driven activation of T cells: Relevance for the adoptive cell therapy of cancer. Cell Stress 2023, 7, 95–104. [Google Scholar] [CrossRef]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2020, 218, e20200844. [Google Scholar] [CrossRef]
- Conde, E.; Vercher, E.; Soria-Castellano, M.; Suarez-Olmos, J.; Mancheño, U.; Elizalde, E.; Rodriguez, M.L.; Glez-Vaz, J.; Casares, N.; Rodríguez-García, E.; et al. Epitope spreading driven by the joint action of CART cells and pharmacological STING stimulation counteracts tumor escape via antigen-loss variants. J. Immunother. Cancer 2021, 9, e003351. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; Sun, L.; Castelli, S.; Finck, A.V.; Assenmacher, C.-A.; Young, R.M.; Chen, Z.J.; June, C.H. The STING agonist IMSA101 enhances chimeric antigen receptor T cell function by inducing IL-18 secretion. Nat. Commun. 2024, 15, 3933. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Xiao, Y.; Chen, Z.; Ding, H.; Chen, S.; Jiang, G.; Huang, X. Inhalable nanovesicles loaded with a STING agonist enhance CAR-T cell activity against solid tumors in the lung. Nat. Commun. 2025, 16, 262. [Google Scholar] [CrossRef]
- Johnson, L.R.; Lee, D.Y.; Eacret, J.S.; Ye, D.; June, C.H.; Minn, A.J. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell 2021, 184, 4981–4995.e14. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.H.; Muhammad, N.; Tarique, M.; Usmani, D.; Naz, H.; Sarode, A. The Role of Cancer-Specific Target Antigens in CAR T Cell Therapy in Hematological Malignancies. Curr. Tissue Microenviron. Rep. 2024, 5, 61–67. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Included Domain | Innate Immunity Sensor | Cell Type | Introduction into CAR Cell | Mode of Action | Reference |
|---|---|---|---|---|---|
| TIR domain | TLR2 | T-cells | At the 3’ end of CD3ζ in a 2nd generation CAR construct, targeting CD19 or mesothelin |
| [37,52,53] |
| MyD88/CD40–inducible | TLR | T-cells | Coexpressed with CAR construct |
| [42,59,60] |
| MyD88/CD40–constitutive | TLR | T-cells | At the 3’ end of CD8α TM domain, connected via deliberately inefficient 2A linker system |
| [43] |
| MyD88/CD40 | TLR | T-cells | At the 3’ end of CD8α TM domain, followed by CD3ζ |
| [62] |
| MyD88/CD40 | TLR | T-cells | Expression on a separate polypeptide chain |
| [41] |
| Intracellular segment of TLR4 | TLR4 | T-cells | Intracellular part of a CAR construct targeting VEGFR2 |
| [55] |
| N terminal truncation of TLR4 ectodomain | TLR4 | T-cells | At the 3’ end of CD3ζ in a 2nd generation CAR construct, connected via gt2a linker, targeting CD19 |
| [41] |
| TRIF | TLR4 | T-cells | At the 3’ end of CD3ζ in a 2nd generation CAR construct, connected via gs linker |
| [41] |
| DAP10 | NKG2D | T-cells | ScFv replaced with DAP10 and/or in addition to scFv region |
| [67] |
| NKG2D extracellular domain | NKG2D | T-cells | Paired with a CD3ζ activation domain and either a 4-1BB or CD27 costimulatory domain. |
| [71,74] |
| NKG2D/DAP10-12 | NKG2D | T-cells | Coexpression of NKG2D with DAP10 and DAP12 endodomain |
| [73] |
| NKG2D/DAP12 | NKG2D | T-cells | Coexpression of NKG2D ectodomain with DAP12 endodomain |
| [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakar, T.; Skrbinek, M.; Fink, T.; Lainšček, D. Enhancing CAR T-Cell Function with Domains of Innate Immunity Sensors. Int. J. Mol. Sci. 2025, 26, 1339. https://doi.org/10.3390/ijms26031339
Mlakar T, Skrbinek M, Fink T, Lainšček D. Enhancing CAR T-Cell Function with Domains of Innate Immunity Sensors. International Journal of Molecular Sciences. 2025; 26(3):1339. https://doi.org/10.3390/ijms26031339
Chicago/Turabian StyleMlakar, Tjaša, Mojca Skrbinek, Tina Fink, and Duško Lainšček. 2025. "Enhancing CAR T-Cell Function with Domains of Innate Immunity Sensors" International Journal of Molecular Sciences 26, no. 3: 1339. https://doi.org/10.3390/ijms26031339
APA StyleMlakar, T., Skrbinek, M., Fink, T., & Lainšček, D. (2025). Enhancing CAR T-Cell Function with Domains of Innate Immunity Sensors. International Journal of Molecular Sciences, 26(3), 1339. https://doi.org/10.3390/ijms26031339