
Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms
 , and
, and
Abstract
1. Introduction
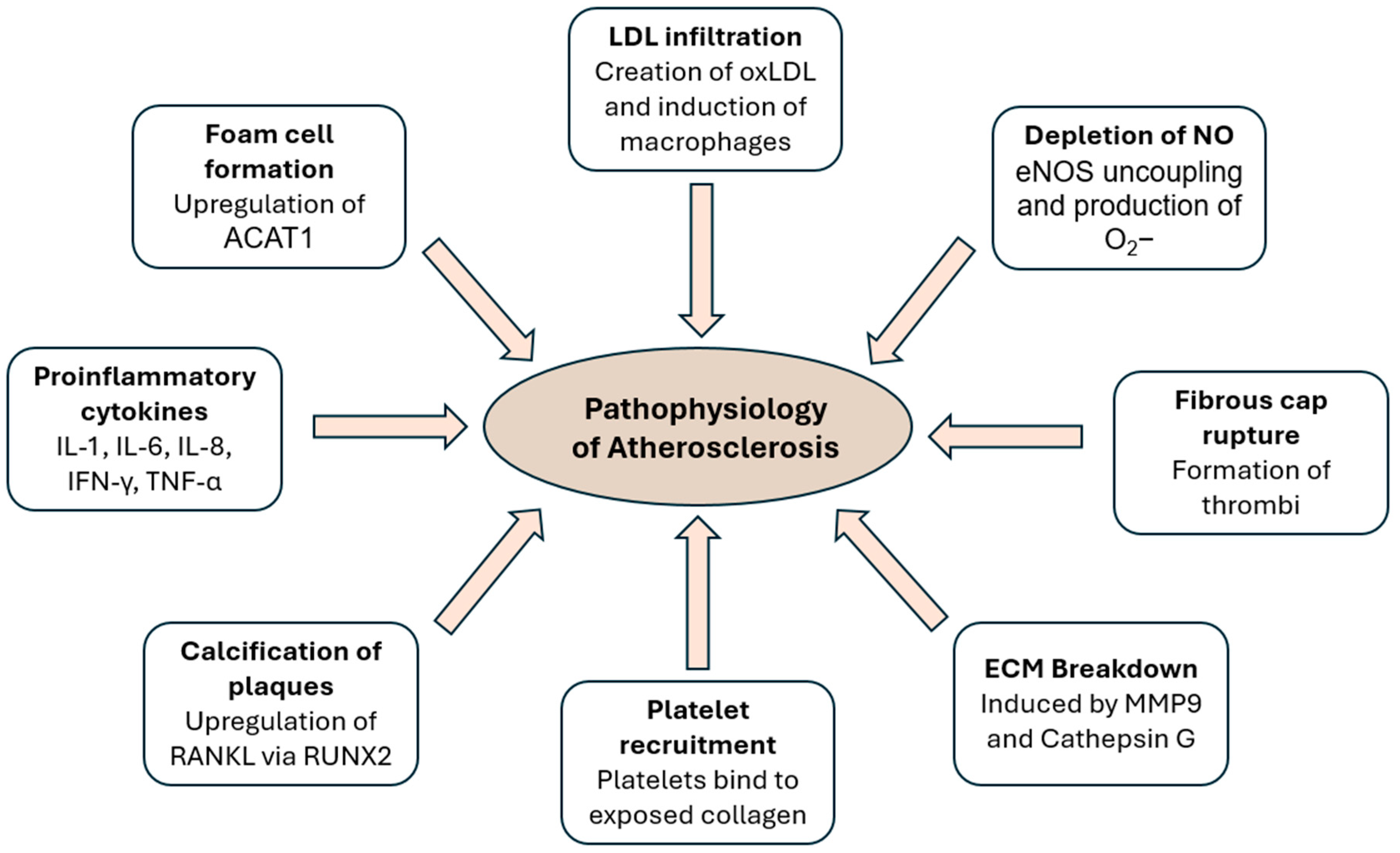
2. Pathophysiology
2.1. Anatomy
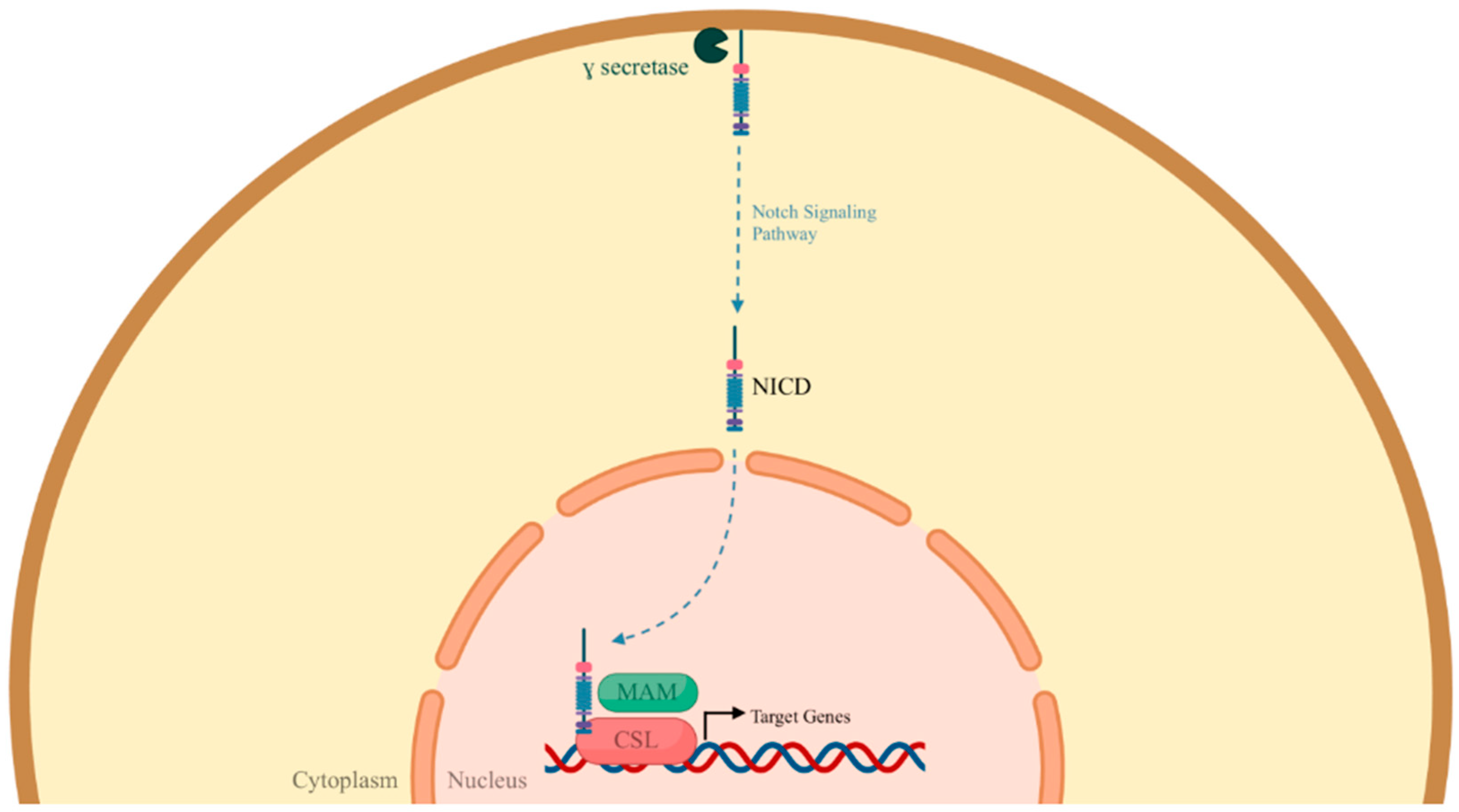
2.2. Important Signaling Pathways
2.3. The Role of Nitric Oxide
2.4. The Role of Connexins in Atherosclerosis
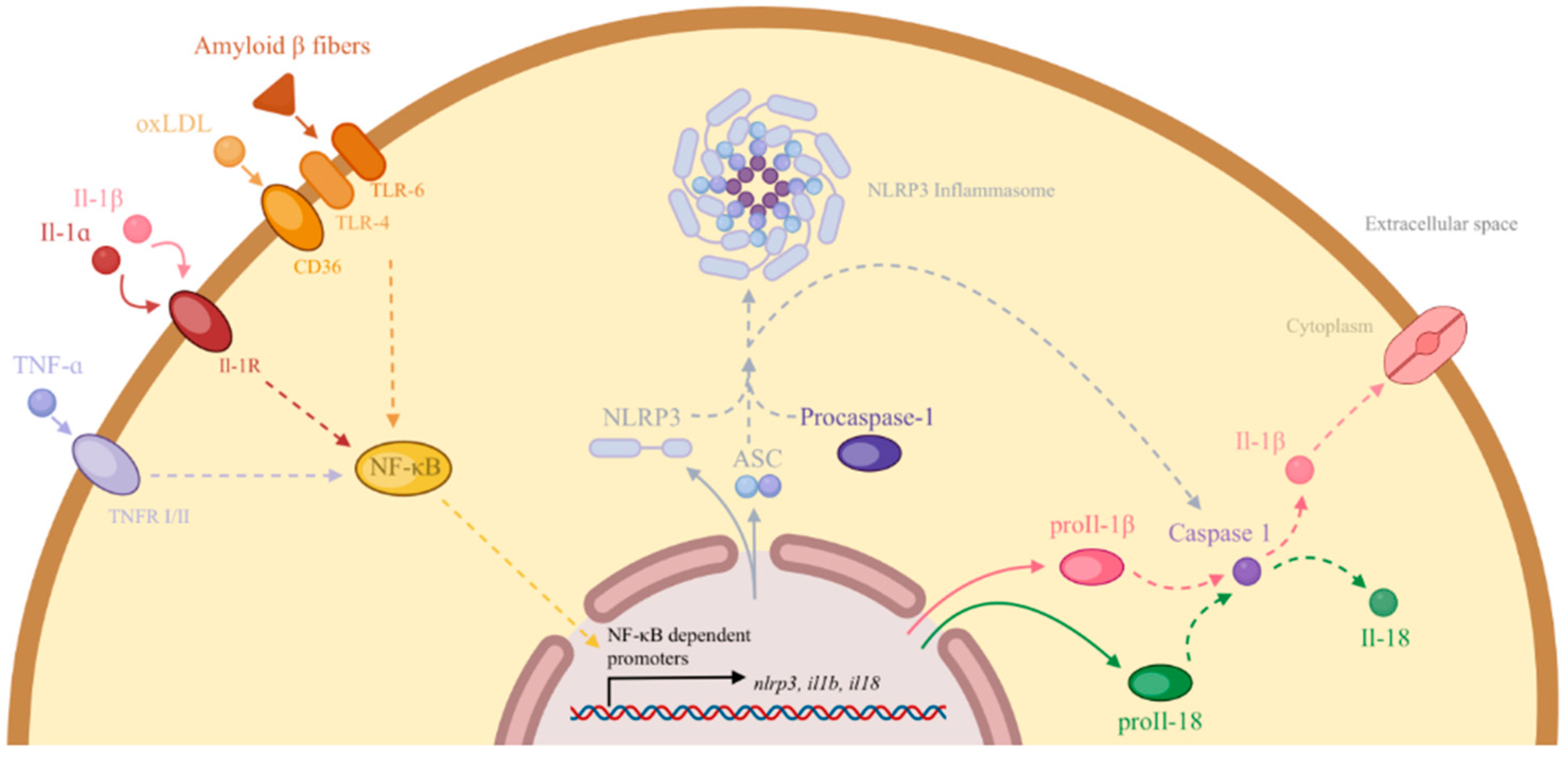
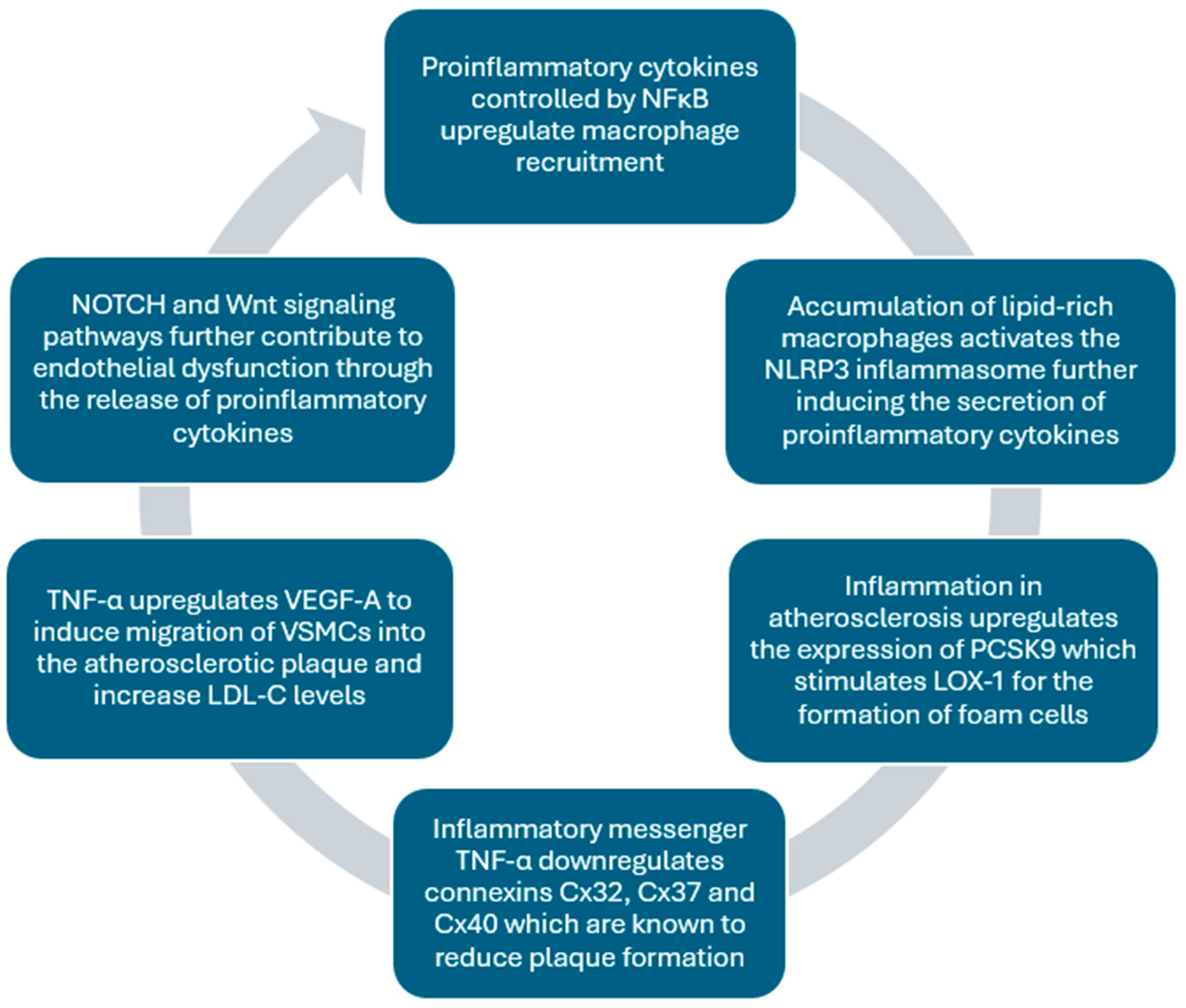
2.5. The Role of Inflammation
2.6. The Role of microRNA and siRNA Therapies
2.7. The Role of VEGFs
3. Risk Factors
4. Chronic Coronary Disease (CCD) Guidelines
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fan, J.; Watanabe, T. Atherosclerosis: Known and Unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, E.; Atzeni, F.; Sarzi-Puttini, P.; Turiel, M.; Lopez, L.R.; Nurmohamed, M.T. Is Atherosclerosis an Autoimmune Disease? BMC Med. 2014, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Glovaci, D.; Fan, W.; Wong, N.D. Epidemiology of Diabetes Mellitus and Cardiovascular Disease. Curr. Cardiol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Citrin, K.M.; Fernández-Hernando, C.; Suárez, Y. MicroRNA Regulation of Cholesterol Metabolism. Ann. N. Y. Acad. Sci. 2021, 1495, 55–77. [Google Scholar] [CrossRef]
- Sarzani, R.; Spannella, F.; Di Pentima, C.; Giulietti, F.; Landolfo, M.; Allevi, M. Molecular Therapies in Cardiovascular Diseases: Small Interfering RNA in Atherosclerosis, Heart Failure, and Hypertension. Int. J. Mol. Sci. 2023, 25, 328. [Google Scholar] [CrossRef]
- Seidelmann, S.B.; Lighthouse, J.K.; Greif, D.M. Development and Pathologies of the Arterial Wall. Cell. Mol. Life Sci. 2014, 71, 1977–1999. [Google Scholar] [CrossRef]
- Mironov, A.A.; Beznoussenko, G.V. Opinion: On the Way towards the New Paradigm of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 2152. [Google Scholar] [CrossRef]
- Nezu, T.; Hosomi, N.; Aoki, S.; Matsumoto, M. Carotid Intima-Media Thickness for Atherosclerosis. J. Atheroscler. Thromb. 2016, 23, 18–31. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Mironov, A.A.; Sesorova, I.S.; Dimov, I.D.; Karelina, N.R.; Beznoussenko, G.V. Intracellular Transports and Atherogenesis. Front. Biosci. 2020, 25, 1230–1258. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Mikhailenko, I.A. Modification of Low Density Lipoprotein by Desialylation Causes Lipid Accumulation in Cultured Cells: Discovery of Desialylated Lipoprotein with Altered Cellular Metabolism in the Blood of Atherosclerotic Patients. Biochem. Biophys. Res. Commun. 1989, 162, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Mezentsev, A.; Bezsonov, E.; Kashirskikh, D.; Baig, M.S.; Eid, A.H.; Orekhov, A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines 2021, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Torres, N.; Guevara-Cruz, M.; Velázquez-Villegas, L.A.; Tovar, A.R. Nutrition and Atherosclerosis. Arch. Med. Res. 2015, 46, 408–426. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, J.; Liu, Y.; Xu, D. Sialic Acid Metabolism as a Potential Therapeutic Target of Atherosclerosis. Lipids Health Dis. 2019, 18, 173. [Google Scholar] [CrossRef]
- Falck-Hansen, M.; Kassiteridi, C.; Monaco, C. Toll-Like Receptors in Atherosclerosis. Int. J. Mol. Sci. 2013, 14, 14008–14023. [Google Scholar] [CrossRef]
- Björkbacka, H. Multiple Roles of Toll-like Receptor Signaling in Atherosclerosis. Curr. Opin. Lipidol. 2006, 17, 527. [Google Scholar] [CrossRef]
- Bayer, A.L.; Alcaide, P. MyD88: At the Heart of Inflammatory Signaling and Cardiovascular Disease. J. Mol. Cell. Cardiol. 2021, 161, 75–85. [Google Scholar] [CrossRef]
- den Dekker, W.K.; Cheng, C.; Pasterkamp, G.; Duckers, H.J. Toll like Receptor 4 in Atherosclerosis and Plaque Destabilization. Atherosclerosis 2010, 209, 314–320. [Google Scholar] [CrossRef]
- Tedgui, A.; Mallat, Z. Cytokines in Atherosclerosis: Pathogenic and Regulatory Pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef]
- Deguine, J.; Barton, G.M. MyD88: A Central Player in Innate Immune Signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-G.; Ryu, S.Y.; Jung, I.-H.; Lee, Y.-H.; Kang, K.J.; Lee, M.-R.; Lee, M.-N.; Sonn, S.K.; Lee, J.H.; Lee, H.; et al. Evaluation of VCAM-1 Antibodies as Therapeutic Agent for Atherosclerosis in Apolipoprotein E-Deficient Mice. Atherosclerosis 2013, 226, 356–363. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of Foam Cell Formation in Atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and Its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Ringor, M. Figure 1. BioRender. 2025. Available online: https://app.biorender.com/citation/678476749924434a21f76dc3 (accessed on 1 February 2025).
- Gutierrez, P.S. Foam Cells in Atherosclerosis. Arq. Bras. Cardiol. 2022, 119, 542–543. [Google Scholar] [CrossRef]
- Matsuo, M. ABCA1 and ABCG1 as Potential Therapeutic Targets for the Prevention of Atherosclerosis. J. Pharmacol. Sci. 2022, 148, 197–203. [Google Scholar] [CrossRef]
- McLaren, J.E.; Michael, D.R.; Ashlin, T.G.; Ramji, D.P. Cytokines, Macrophage Lipid Metabolism and Foam Cells: Implications for Cardiovascular Disease Therapy. Prog. Lipid Res. 2011, 50, 331–347. [Google Scholar] [CrossRef]
- Littlewood, T.D.; Bennett, M.R. Apoptotic Cell Death in Atherosclerosis. Curr. Opin. Lipidol. 2003, 14, 469–475. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Akers, E.J.; Nicholls, S.J.; Di Bartolo, B.A. Plaque Calcification: Do Lipoproteins Have a Role? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; López-Ongil, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL Increases Vascular Smooth Muscle Cell Calcification through a RANK-BMP4-Dependent Pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Heart Assoc. 2015, 4, e002499. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, G.P.; Sykes, M.; Weiss, D.; Platt, M.O.; Saha, A.; Hwang, J.; Boyd, N.; Boo, Y.C.; Vega, J.D.; Taylor, W.R.; et al. Bone Morphogenic Protein 4 Produced in Endothelial Cells by Oscillatory Shear Stress Stimulates an Inflammatory Response. J. Biol. Chem. 2003, 278, 31128–31135. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Huang, A.L.; Kyaw, T.S.; Bobik, A.; Peter, K. Atherosclerotic Plaque Rupture: Identifying the Straw That Breaks the Camel’s Back. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e63–e72. [Google Scholar] [CrossRef]
- Pedrigi, R.M.; de Silva, R.; Bovens, S.M.; Mehta, V.V.; Petretto, E.; Krams, R. Thin-Cap Fibroatheroma Rupture Is Associated with a Fine Interplay of Shear and Wall Stress. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2224–2231. [Google Scholar] [CrossRef]
- Asada, Y.; Yamashita, A.; Sato, Y.; Hatakeyama, K. Pathophysiology of Atherothrombosis: Mechanisms of Thrombus Formation on Disrupted Atherosclerotic Plaques. Pathol. Int. 2020, 70, 309–322. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis Formation on Atherosclerotic Lesions and Plaque Rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, Platelets and Thrombosis in Acute Ischaemic Heart Disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Martinez Bravo, G.; Annarapu, G.; Carmona, E.; Nawarskas, J.; Clark, R.; Novelli, E.; Mota Alvidrez, R.I. Platelets in Thrombosis and Atherosclerosis: A Double-Edged Sword. Am. J. Pathol. 2024, 194, 1608–1621. [Google Scholar] [CrossRef]
- Zaman, A.G.; Helft, G.; Worthley, S.G.; Badimon, J.J. The Role of Plaque Rupture and Thrombosis in Coronary Artery Disease. Atherosclerosis 2000, 149, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Gutstein, D.E.; Fuster, V. Pathophysiology and Clinical Significance of Atherosclerotic Plaque Rupture. Cardiovasc. Res. 1999, 41, 323–333. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, D.; Xu, Y.; Jiang, F.; Zheng, J.; Feng, Y.; Cao, J.; Zhou, X. Nitric Oxide-Releasing Platforms for Treating Cardiovascular Disease. Pharmaceutics 2022, 14, 1345. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial Dysfunction Due to eNOS Uncoupling: Molecular Mechanisms as Potential Therapeutic Targets. Cell. Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Łuczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathway in the Pathogenesis of Autoimmune Rheumatic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1417981. [Google Scholar] [CrossRef]
- Cziráki, A.; Lenkey, Z.; Sulyok, E.; Szokodi, I.; Koller, A. L-Arginine-Nitric Oxide-Asymmetric Dimethylarginine Pathway and the Coronary Circulation: Translation of Basic Science Results to Clinical Practice. Front. Pharmacol. 2020, 11, 569914. [Google Scholar] [CrossRef]
- Willeit, P.; Freitag, D.F.; Laukkanen, J.A.; Chowdhury, S.; Gobin, R.; Mayr, M.; Di Angelantonio, E.; Chowdhury, R. Asymmetric Dimethylarginine and Cardiovascular Risk: Systematic Review and Meta-Analysis of 22 Prospective Studies. J. Am. Heart Assoc. 2015, 4, e001833. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; Ramirez, S.H.; Nenov, M.N. Asymmetric Dimethylarginine Induces Maladaptive Function of the Blood-Brain Barrier. Front. Cell Dev. Biol. 2024, 12, 1476386. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, D.; Quinn, K.M.; Sanders, J.; Plumblee, L.; Morinelli, T.A.; Nadig, S.N. Connexins in Endothelial Cells as a Therapeutic Target for Solid Organ Transplantation. Am. J. Transpl. 2022, 22, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Kiełbowski, K.; Bakinowska, E.; Pawlik, A. The Potential Role of Connexins in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 2600. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Burnier, L.; Kwak, B.R. Connexins Participate in the Initiation and Progression of Atherosclerosis. Semin. Immunopathol. 2009, 31, 49–61. [Google Scholar] [CrossRef]
- Sedovy, M.W.; Leng, X.; Leaf, M.R.; Iqbal, F.; Payne, L.B.; Chappell, J.C.; Johnstone, S.R. Connexin 43 across the Vasculature: Gap Junctions and Beyond. J. Vasc. Res. 2023, 60, 101–113. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and Atherosclerosis: Signaling Pathways and Therapeutic Intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Ma, M.; Hou, C.; Liu, J. Effect of PCSK9 on Atherosclerotic Cardiovascular Diseases and Its Mechanisms: Focus on Immune Regulation. Front. Cardiovasc. Med. 2023, 10, 1148486. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sukhorukov, V.N.; Popov, M.A.; Chegodaev, Y.S.; Postnov, A.Y.; Orekhov, A.N. Mechanisms of the Wnt Pathways as a Potential Target Pathway in Atherosclerosis. J. Lipid Atheroscler. 2023, 12, 223–236. [Google Scholar] [CrossRef]
- Peng, X.; Wang, S.; Chen, H.; Chen, M. Role of the Notch1 Signaling Pathway in Ischemic Heart Disease (Review). Int. J. Mol. Med. 2023, 51, 27. [Google Scholar] [CrossRef]
- Souilhol, C.; Tardajos Ayllon, B.; Li, X.; Diagbouga, M.R.; Zhou, Z.; Canham, L.; Roddie, H.; Pirri, D.; Chambers, E.V.; Dunning, M.J.; et al. JAG1-NOTCH4 Mechanosensing Drives Atherosclerosis. Sci. Adv. 2022, 8, eabo7958. [Google Scholar] [CrossRef]
- Ringor, M. Figure 3. Created in BioRender. 2025. Available online: https://app.biorender.com/citation/678478ab8d3ebba993846ac0 (accessed on 1 February 2025).
- Li, Z.; Zhao, Y.; Suguro, S.; Suguro, R. MicroRNAs Regulate Function in Atherosclerosis and Clinical Implications. Oxid. Med. Cell. Longev. 2023, 2023, 2561509. [Google Scholar] [CrossRef] [PubMed]
- Churov, A.; Summerhill, V.; Grechko, A.; Orekhova, V.; Orekhov, A. MicroRNAs as Potential Biomarkers in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 5547. [Google Scholar] [CrossRef] [PubMed]
- Bruen, R.; Fitzsimons, S.; Belton, O. miR-155 in the Resolution of Atherosclerosis. Front. Pharmacol. 2019, 10, 463. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Thavarajah, T.; Gu, W.; Cai, J.; Xu, Q. Impact of miRNA in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e159–e170. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, P.; Ruotsalainen, A.-K.; Ylä-Herttuala, S. Nucleic Acid–Based Therapies for Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 10. [Google Scholar] [CrossRef]
- Li, Q.; Yin, K.; Ma, H.-P.; Liu, H.-H.; Li, S.; Luo, X.; Hu, R.; Zhang, W.-W.; Lv, Z.-S.; Niu, X.-L.; et al. Application of Improved GalNAc Conjugation in Development of Cost-Effective siRNA Therapies Targeting Cardiovascular Diseases. Mol. Ther. 2024, 32, 637–645. [Google Scholar] [CrossRef]
- Kraaijenhof, J.M.; Tromp, T.R.; Nurmohamed, N.S.; Reeskamp, L.F.; Langenkamp, M.; Levels, J.H.M.; Boekholdt, S.M.; Wareham, N.J.; Hoekstra, M.; Stroes, E.S.G.; et al. ANGPTL3 (Angiopoietin-Like 3) Preferentially Resides on High-Density Lipoprotein in the Human Circulation, Affecting Its Activity. J. Am. Heart Assoc. 2023, 12, e030476. [Google Scholar] [CrossRef]
- Gencer, B.; Kronenberg, F.; Stroes, E.S.; Mach, F. Lipoprotein(a): The Revenant. Eur. Heart J. 2017, 38, 1553–1560. [Google Scholar] [CrossRef]
- Beheshtizadeh, N.; Gharibshahian, M.; Bayati, M.; Maleki, R.; Strachan, H.; Doughty, S.; Tayebi, L. Vascular Endothelial Growth Factor (VEGF) Delivery Approaches in Regenerative Medicine. Biomed. Pharmacother. 2023, 166, 115301. [Google Scholar] [CrossRef]
- Florek, K.; Mendyka, D.; Gomułka, K. Vascular Endothelial Growth Factor (VEGF) and Its Role in the Cardiovascular System. Biomedicines 2024, 12, 1055. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The Effect of HIF on Metabolism and Immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lv, X.; Yang, J.; Chen, Z.; Qiao, W.; Zhou, T.; Zhang, Y. Variation in VEGFA and Risk of Cardiovascular Disease in the UK Biobank. Front. Cardiovasc. Med. 2023, 10, 1240288. [Google Scholar] [CrossRef] [PubMed]
- Mallick, R.; Ylä-Herttuala, S. Therapeutic Potential of VEGF-B in Coronary Heart Disease and Heart Failure: Dream or Vision? Cells 2022, 11, 4134. [Google Scholar] [CrossRef] [PubMed]
- Moessinger, C.; Nilsson, I.; Muhl, L.; Zeitelhofer, M.; Heller Sahlgren, B.; Skogsberg, J.; Eriksson, U. VEGF-B Signaling Impairs Endothelial Glucose Transcytosis by Decreasing Membrane Cholesterol Content. EMBO Rep. 2020, 21, e49343. [Google Scholar] [CrossRef]
- Zuo, B.; Zhu, S.; Zhong, G.; Bu, H.; Chen, H. Causal Association between Placental Growth Factor and Coronary Heart Disease: A Mendelian Randomization Study. Aging 2023, 15, 10117–10132. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, X.; Cui, H.; Shi, J.; Yuan, G.; Shi, S.; Hu, Y. The Role of the VEGF Family in Coronary Heart Disease. Front. Cardiovasc. Med. 2021, 8, 738325. [Google Scholar] [CrossRef]
- Glinton, K.E.; Ma, W.; Lantz, C.; Grigoryeva, L.S.; DeBerge, M.; Liu, X.; Febbraio, M.; Kahn, M.; Oliver, G.; Thorp, E.B. Macrophage-Produced VEGFC Is Induced by Efferocytosis to Ameliorate Cardiac Injury and Inflammation. J. Clin. Investig. 2022, 132, e140685. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sukhorukov, V.N.; Guo, S.; Postnov, A.Y.; Orekhov, A.N. Sex Differences Define the Vulnerability to Atherosclerosis. Clin. Med. Insights Cardiol. 2023, 17, 11795468231189044. [Google Scholar] [CrossRef]
- Man, J.J.; Beckman, J.A.; Jaffe, I.Z. Sex as a Biological Variable in Atherosclerosis. Circ. Res. 2020, 126, 1297–1319. [Google Scholar] [CrossRef]
- Ishida, M.; Sakai, C.; Kobayashi, Y.; Ishida, T. Cigarette Smoking and Atherosclerotic Cardiovascular Disease. J. Atheroscler. Thromb. 2024, 31, 189–200. [Google Scholar] [CrossRef]
- Frohlich, E.D.; Susic, D. Blood Pressure, Large Arteries and Atherosclerosis. Adv. Cardiol. 2007, 44, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Mancusi, C.; Manzi, M.V.; de Simone, G.; Morisco, C.; Lembo, M.; Pilato, E.; Izzo, R.; Trimarco, V.; De Luca, N.; Trimarco, B. Carotid Atherosclerosis Predicts Blood Pressure Control in Patients With Hypertension: The Campania Salute Network Registry. J. Am. Heart Assoc. 2022, 11, e022345. [Google Scholar] [CrossRef] [PubMed]
- Abrahamowicz, A.A.; Ebinger, J.; Whelton, S.P.; Commodore-Mensah, Y.; Yang, E. Racial and Ethnic Disparities in Hypertension: Barriers and Opportunities to Improve Blood Pressure Control. Curr. Cardiol. Rep. 2023, 25, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed]
- Balla, S.; Nusair, M.B.; Alpert, M.A. Risk Factors for Atherosclerosis in Patients with Chronic Kidney Disease: Recognition and Management. Curr. Opin. Pharmacol. 2013, 13, 192–199. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease: More, Less, or Just Different? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Thompson, P.D.; Buchner, D.; Pina, I.L.; Balady, G.J.; Williams, M.A.; Marcus, B.H.; Berra, K.; Blair, S.N.; Costa, F.; Franklin, B.; et al. Exercise and Physical Activity in the Prevention and Treatment of Atherosclerotic Cardiovascular Disease: A Statement from the Council on Clinical Cardiology (Subcommittee on Exercise, Rehabilitation, and Prevention) and the Council on Nutrition, Physical Activity, and Metabolism (Subcommittee on Physical Activity). Circulation 2003, 107, 3109–3116. [Google Scholar] [CrossRef]
- Aengevaeren, V.L.; Mosterd, A.; Bakker, E.A.; Braber, T.L.; Nathoe, H.M.; Sharma, S.; Thompson, P.D.; Velthuis, B.K.; Eijsvogels, T.M.H. Exercise Volume Versus Intensity and the Progression of Coronary Atherosclerosis in Middle-Aged and Older Athletes: Findings From the MARC-2 Study. Circulation 2023, 147, 993–1003. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent Developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Anyfanti, P.; Ainatzoglou, A.; Angeloudi, E.; Michailou, O.; Defteraiou, K.; Bekiari, E.; Kitas, G.D.; Dimitroulas, T. Cardiovascular Risk in Rheumatoid Arthritis: Considerations on Assessment and Management. Mediterr. J. Rheumatol. 2024, 35, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Libby, P. Inflammation and Atherosclerosis: Prospects for Clinical Trials. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1899–1905. [Google Scholar] [CrossRef]
- Wong, N.D.; Zhao, Y.; Quek, R.G.W.; Blumenthal, R.S.; Budoff, M.J.; Cushman, M.; Garg, P.; Sandfort, V.; Tsai, M.; Lopez, J.A.G. Residual Atherosclerotic Cardiovascular Disease Risk in Statin-Treated Adults: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2017, 11, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, E.; van der Laarse, A.; Jukema, J.W.; Cobbaert, C.M. High Residual Cardiovascular Risk after Lipid-Lowering: Prime Time for Predictive, Preventive, Personalized, Participatory, and Psycho-Cognitive Medicine. Front. Cardiovasc. Med. 2023, 10, 1264319. [Google Scholar] [CrossRef]
- Matsuura, Y.; Kanter, J.E.; Bornfeldt, K.E. Highlighting Residual Atherosclerotic Cardiovascular Disease Risk. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e1–e9. [Google Scholar] [CrossRef]
- Virani, S.S.; Newby, L.K.; Arnold, S.V.; Bittner, V.; Brewer, L.C.; Demeter, S.H.; Dixon, D.L.; Fearon, W.F.; Hess, B.; Johnson, H.M.; et al. 2023 AHA/ACC/ACCP/ASPC/NLA/PCNA Guideline for the Management of Patients with Chronic Coronary Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2023, 148, e9–e119. [Google Scholar] [CrossRef]
- Winchester, D.E.; ACC Solution Set Oversight Committee; Cibotti-Sun, M. 2023 Chronic Coronary Disease Guideline-at-a-Glance. J. Am. Coll. Cardiol. 2023, 82, 956–960. [Google Scholar] [CrossRef]
- Vrints, C.; Andreotti, F.; Koskinas, K.C.; Rossello, X.; Adamo, M.; Ainslie, J.; Banning, A.P.; Budaj, A.; Buechel, R.R.; Chiariello, G.A.; et al. 2024 ESC Guidelines for the Management of Chronic Coronary Syndromes. Eur. Heart J. 2024, 45, 3415–3537. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ten Take-Home Messages from the 2023 American Heart Association Guidelines Regarding the Management of CCD |
|---|
| Team-based, patient-centered care |
| Incorporation of nonpharmacologic therapies such as diet and exercise |
| Use of habitual physical activity for CCD and cardiac rehabilitation for eligible patients |
| Administration of sodium–glucose cotransporter two inhibitors and glucagon-like peptide-1 receptor agonists in CCD patients with or without diabetes |
| Use of either a calcium channel blocker or a beta-blocker as first-line therapy for angina while avoiding the use of beta-blockers in CCD patients in the absence of myocardial infarction in the past year and left ventricular ejection fraction ≤50% or another primary indication for beta-blocker therapy |
| Use of statins as first-line therapy for lowering lipids in CCD patients |
| Administration of dual antiplatelet therapy for a shorter duration is safe and effective when the risk of bleeding is high and the ischemic risk is low to moderate |
| The utilization of supplements such as fish oil, vitamins, and omega-3 fatty acids is not recommended due to the lack of benefit in reducing cardiovascular events |
| Routine periodic anatomic or ischemic testing without a change in clinical or functional status is not recommended to guide decision-making in CCD patients |
| The usage of e-cigarettes is not recommended as first-line therapy for smoking cessation due to a lack of long-term data regarding safety and risks |
| European Society of Cardiology Guidelines for the Management of Chronic Coronary Syndromes |
|---|
| The term chronic coronary syndrome (CCS) is used to describe clinical presentations of coronary artery disease (CAD) during stable periods, before or after acute coronary syndrome (ACS) events |
| Implementing a general clinical evaluation, cardiac examination, and diagnostic testing should be emphasized to establish the diagnosis of CCS along with the use of lifestyle modifications and disease-modifying medications |
| The employment of non-invasive anatomic or functional imaging as first-line diagnostic testing of suspected CCS |
| Coronary computed tomography angiography (CCTA) should be preferred as an initial test to rule out obstructive CAD. The use of invasive coronary angiography (ICA) can be considered in patients with a very high clinical likelihood of obstructive CAD, symptoms unresponsive to medical therapy, angina at a low level of exercise, and/or high-event risk |
| The use of a single antiplatelet like aspirin or clopidogrel is recommended as part of the long-term therapy for CCS patients with obstructive atherosclerotic CAD |
| CCS patients with functionally significant multivessel CAD or reduced left ventricular function and ischemic cardiomyopathy would benefit from myocardial and surgical revascularization, respectively, over guideline-directed medical therapy (GDMT) alone when taking into consideration overall survival and reduced mortality |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasouli-Drakou, V.; Ogurek, I.; Shaikh, T.; Ringor, M.; DiCaro, M.V.; Lei, K. Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms. Int. J. Mol. Sci. 2025, 26, 1364. https://doi.org/10.3390/ijms26031364
Tasouli-Drakou V, Ogurek I, Shaikh T, Ringor M, DiCaro MV, Lei K. Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms. International Journal of Molecular Sciences. 2025; 26(3):1364. https://doi.org/10.3390/ijms26031364
Chicago/Turabian StyleTasouli-Drakou, Vasiliki, Ian Ogurek, Taha Shaikh, Marc Ringor, Michael V. DiCaro, and KaChon Lei. 2025. "Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms" International Journal of Molecular Sciences 26, no. 3: 1364. https://doi.org/10.3390/ijms26031364
APA StyleTasouli-Drakou, V., Ogurek, I., Shaikh, T., Ringor, M., DiCaro, M. V., & Lei, K. (2025). Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms. International Journal of Molecular Sciences, 26(3), 1364. https://doi.org/10.3390/ijms26031364