
Development of Genome-Wide SSR Markers in Leymus chinensis with Genetic Diversity Analysis and DNA Fingerprints
 , ,
, ,
Abstract
:1. Introduction
2. Results
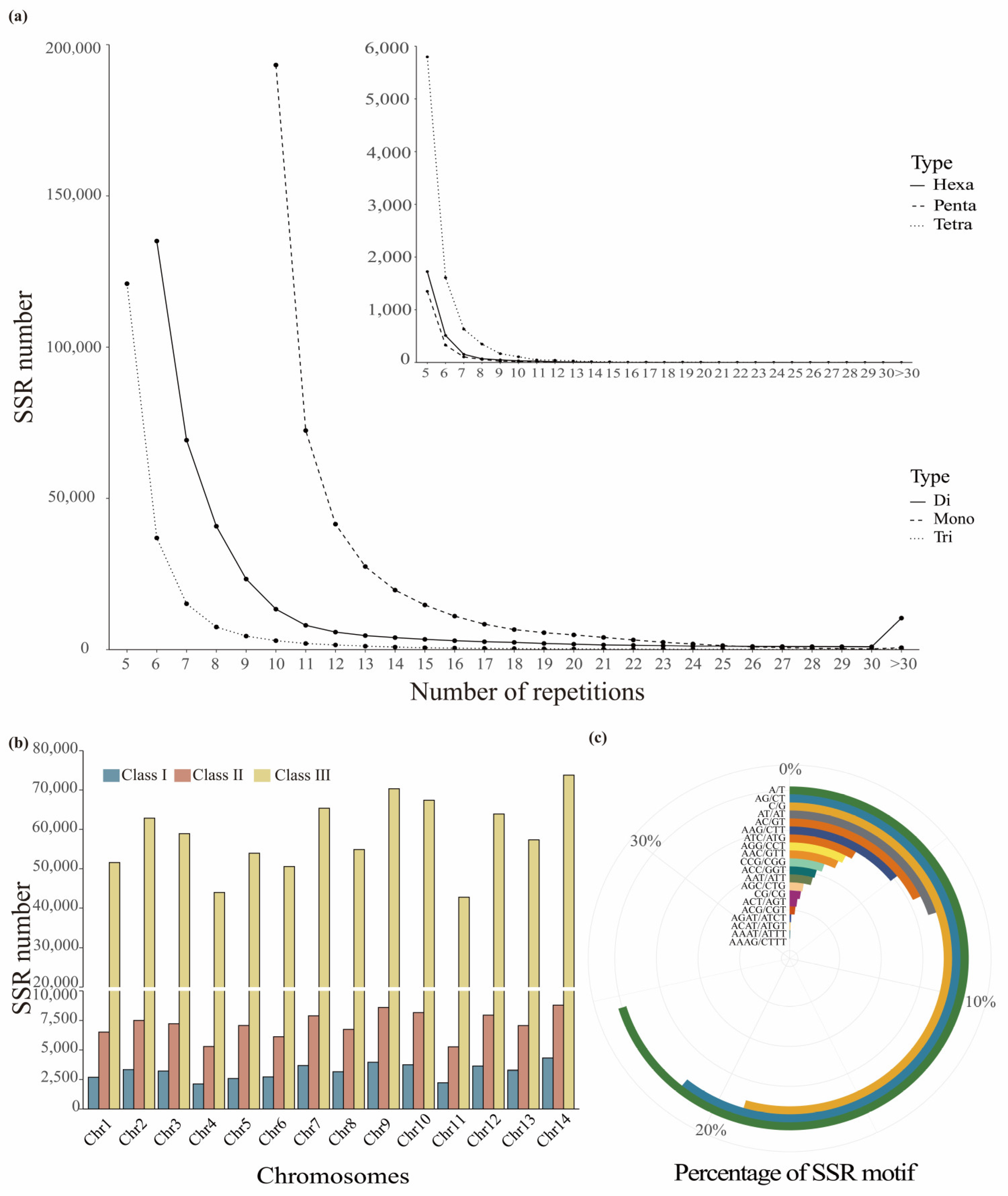
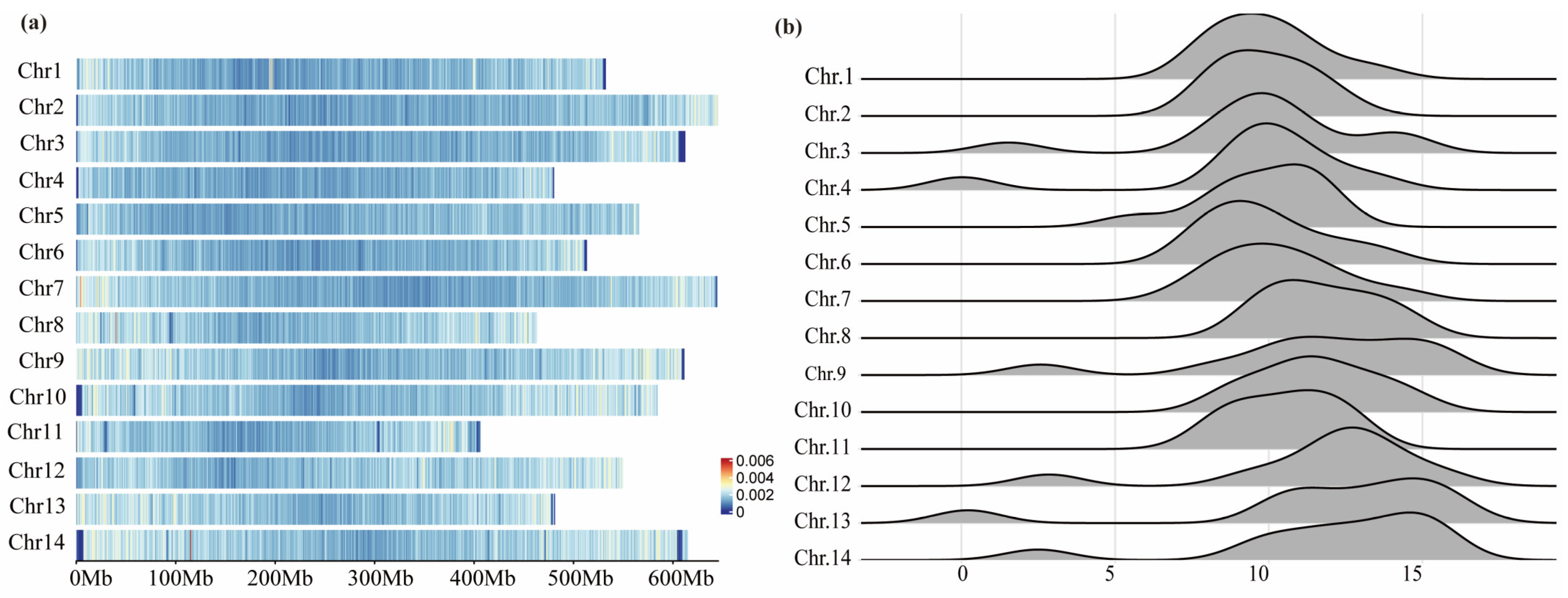
2.1. Distribution and Development of SSRs in Leymus Chinensis Genome
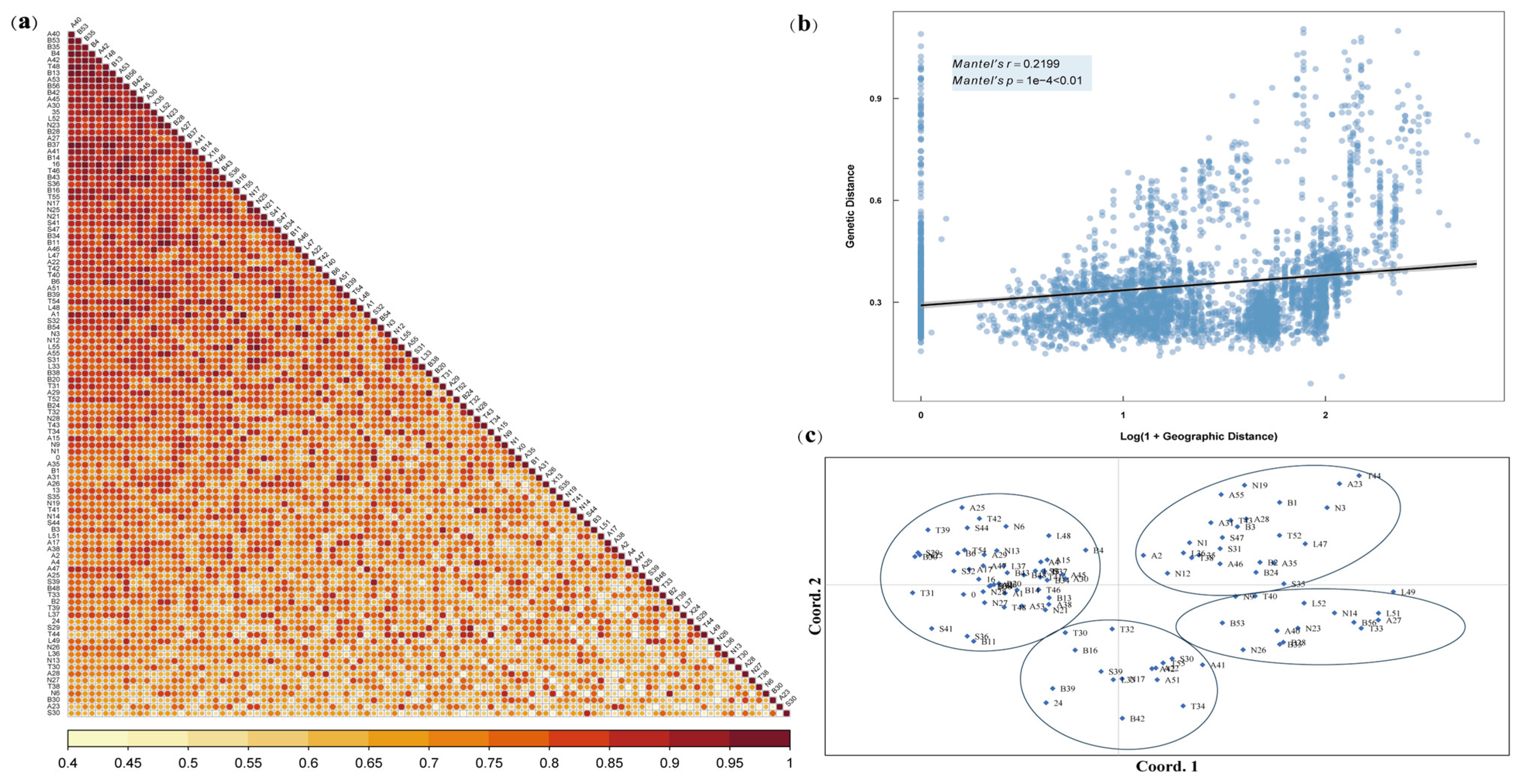
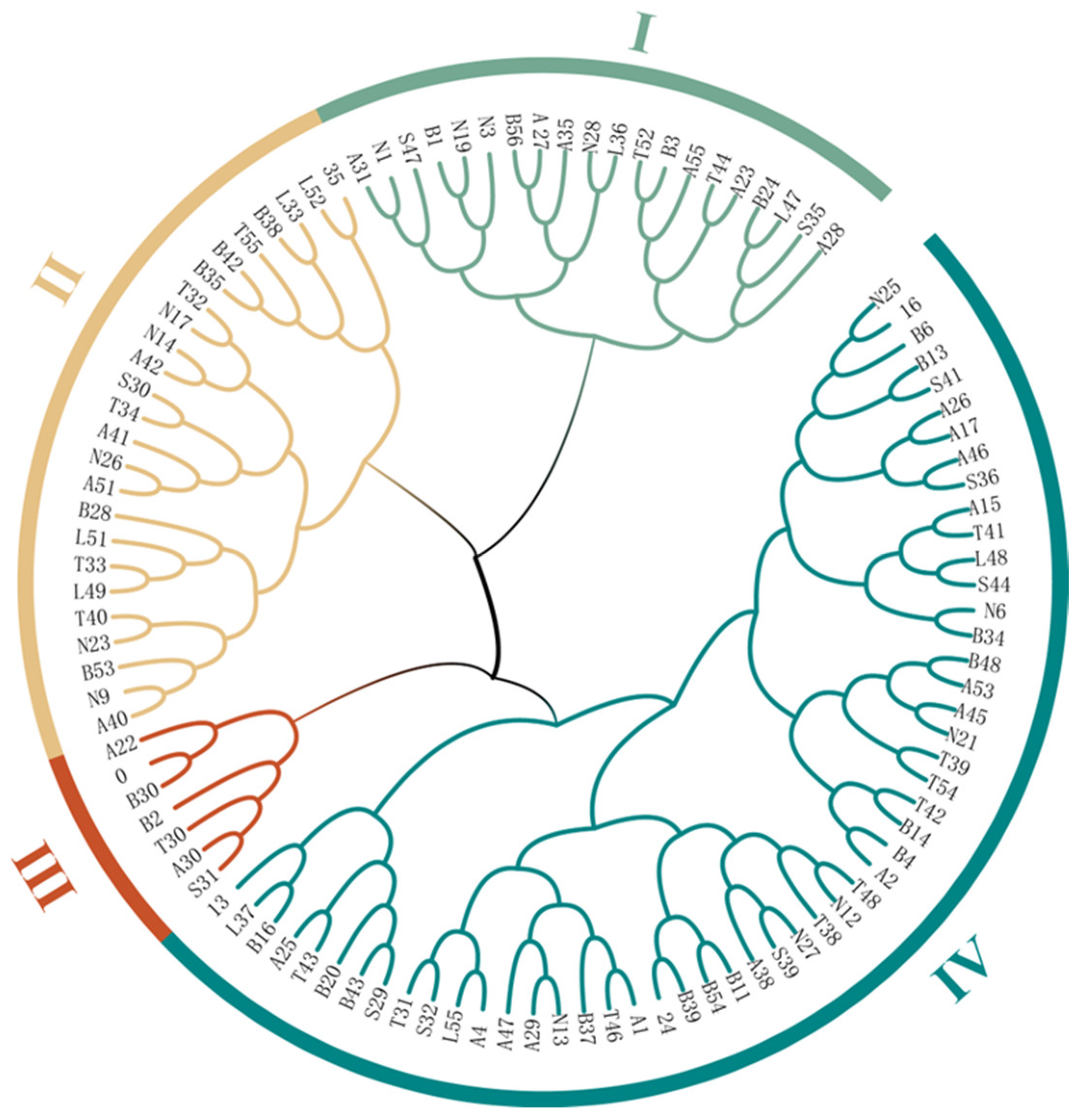
2.2. Genetic Diversity and Population Structure Analysis
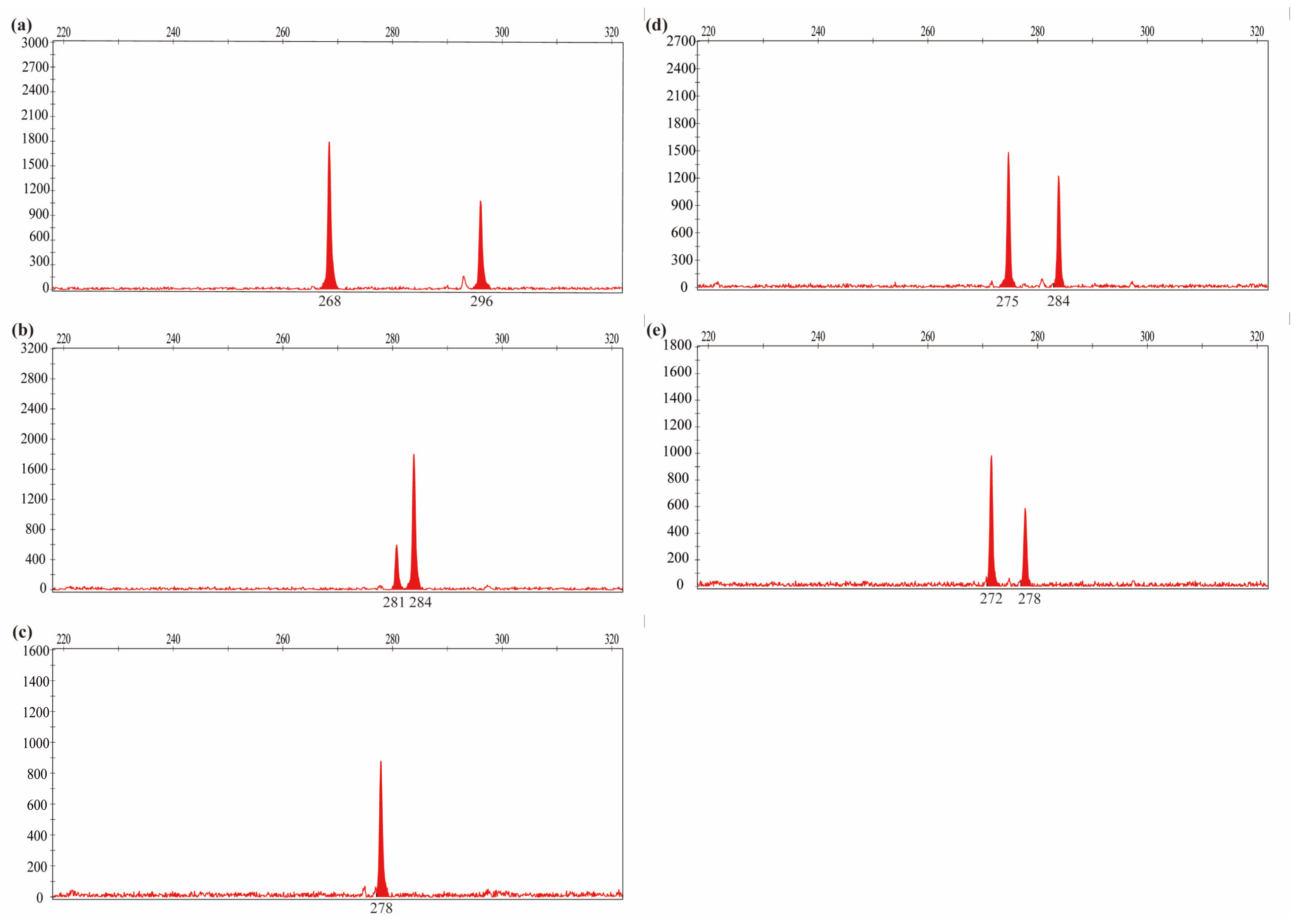
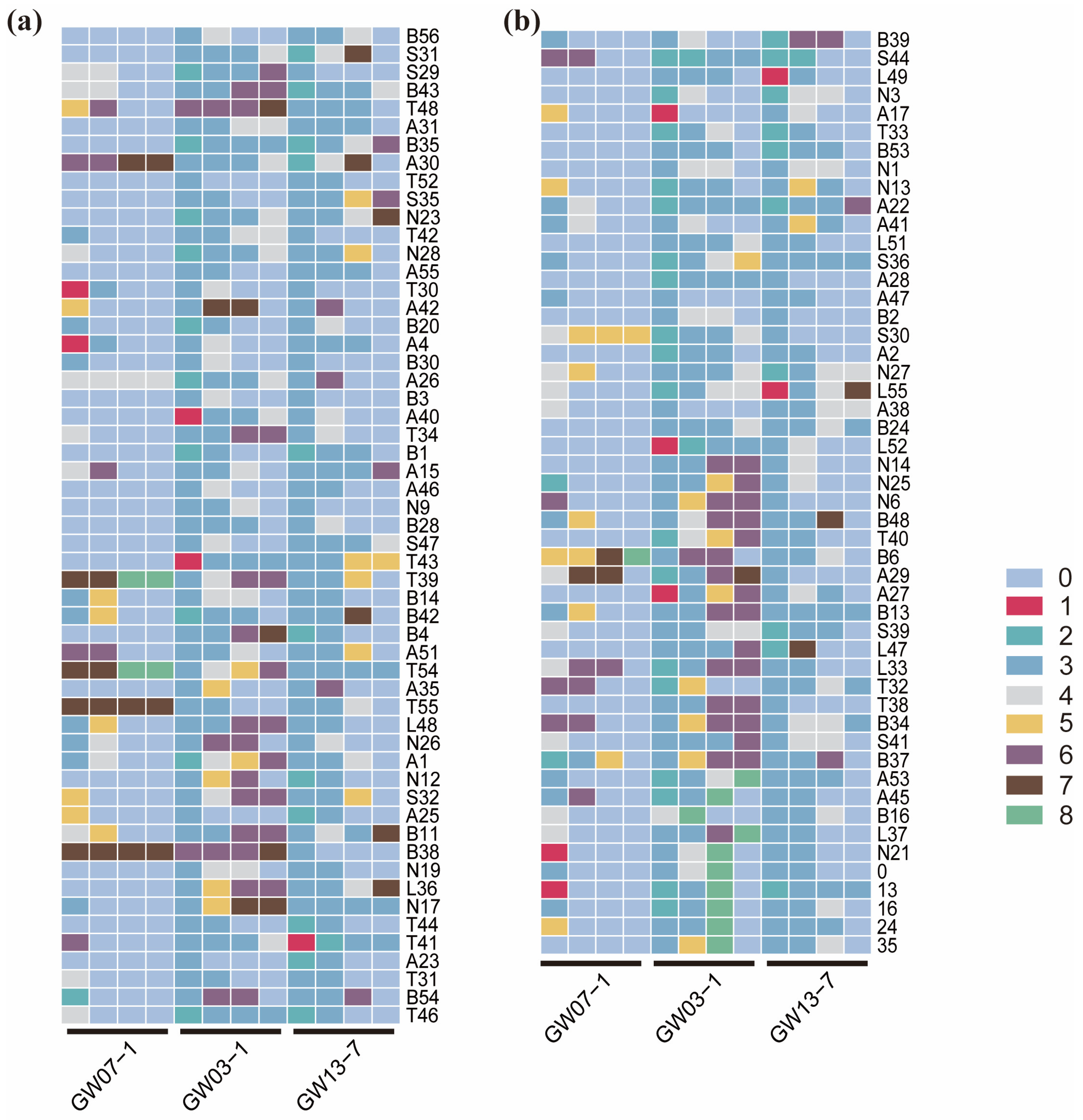
2.3. Fingerprinting of L. chinensis Accessions
3. Discussion
3.1. Distribution of SSRs in L. chinensis Genome
3.2. Genetic Diversity of L. chinensis
3.3. SSR Marker Fingerprint
4. Materials and Methods
4.1. Plant Materials
4.2. Identification and Development of SSR in the L. chinensis Genome
4.3. Primer Screening and PCR Amplification
4.4. Data Analysis
4.5. Construction of DNA Fingerprint
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, S.; Huang, X.; Yan, X.; Liang, Y.; Wang, Y.; Li, X.; Peng, X.; Ma, X.; Zhang, L.; Cai, Y.; et al. Transcriptome analysis in sheepgrass (Leymus chinensis): A dominant perennial grass of the Eurasian Steppe. PLoS ONE 2013, 8, e67974. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, X.; Qi, D.; Chen, S.; Cheng, L. Evaluation and utilization of Leymus chinensis germplasm resources. Chin. Sci. Bull. 2016, 61, 271–281. [Google Scholar] [CrossRef]
- Wu, Z.; Hou, X.; Ren, W.; Chang, C.; Yang, Y.; Yang, Y. Prediction of the potential geographic distribution of Levmus chinensis based on MaxEnt and collection and protection of germplasm. Acta Pratacult. Sin. 2018, 27, 125–135. [Google Scholar] [CrossRef]
- Chen, S.; Jia, J.; Cheng, L.; Zhao, P.; Qi, D.; Yang, W.; Liu, H.; Dong, X.; Li, X.; Liu, G. Transcriptomic Analysis Reveals a Comprehensive Calcium- and Phytohormone-Dominated Signaling Response in Leymus chinensis Self-Incompatibility. Int. J. Mol. Sci. 2019, 20, 2356. [Google Scholar] [CrossRef] [PubMed]
- Tadato, O.; Yang, L. Japanese import of Leymus chinensis will lead to the destruction of grassland vegetation in lnner Mongolia. Foreign. Zootech. (Grassl. Turf) 1997, 2, 47. [Google Scholar] [CrossRef]
- Cao, B. Overview of forage import trade in Japan. Anim. Sci. Vet. Med. 2001, 18, 62–64. [Google Scholar]
- Li, T.; Tang, S.; Li, W.; Zhang, S.; Wang, J.; Pan, D.; Lin, Z.; Ma, X.; Chang, Y.; Liu, B.; et al. Genome evolution and initial breeding of the Triticeae grass Leymus chinensis dominating the Eurasian Steppe. Proc. Natl. Acad. Sci. USA 2023, 120, e2308984120. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, Q.; Liang, Y.; Ma, T.; Cheng, L.; Qi, D.; Liu, H.; Xu, X.; Chen, S.; Liu, G. A novel salt-induced gene from sheepgrass, LcSAIN2, enhances salt tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2013, 64, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Li, M.; Zhao, A.; Xu, X.; Liu, G.; Cheng, L. LcWRKY5: An unknown function gene from sheepgrass improves drought tolerance in transgenic Arabidopsis. Plant Cell Rep. 2014, 33, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Chen, L.; Tang, S.; Zhao, M.; Li, T.; You, J.; You, C.; Li, B.; Zhao, Q.; Zhang, D.; et al. Efficient CRISPR/Cas9-mediated genome editing in sheepgrass (Leymus chinensis). J. Integr. Plant Biol. 2023, 65, 2416–2420. [Google Scholar] [CrossRef]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Temnykh, S.; DeClerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; McCouch, S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef]
- Wang, R.; Zhong, Y.; Hong, W.; Luo, H.; Li, D.; Zhao, L.; Zhang, H.; Wang, J. Genetic diversity evaluation and core collection construction of pomegranate (Punica granatum L.) using genomic SSR markers. Sci. Hortic. 2023, 319, 112192. [Google Scholar] [CrossRef]
- Cheng, S.; Su, L.; Guo, X.; Shao, D.; Qin, Y.; Liu, X.; Chu, Q.; Zhou, X.; He, Z. Genome-wide development of simple sequence repeats markers and genetic diversity analysis of chayote. BMC Plant Biol. 2024, 24, 603. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Li, H.; Shi, W. Application of DNA molecular marker technology in research on forage germplasm resources. Grassl. Turf 2010, 30, 91–96. [Google Scholar] [CrossRef]
- Wu, F.; Cai, G.; Xi, P.; Guo, Y.; Xu, M.; Li, A. Genetic Diversity Analysis and Fingerprint Construction for 87 Passionfruit (Passiflora spp.) Germplasm Accessions on the Basis of SSR Fluorescence Markers. Int. J. Mol. Sci. 2024, 25, 10815. [Google Scholar] [CrossRef] [PubMed]
- Bryan, G.J.; Collins, A.J.; Stephenson, P.; Orry, A.; Smith, J.B.; Gale, M.D. Isolation and characterisation of microsatellites from hexaploid bread wheat. Theor. Appl. Genet. 1997, 94, 557–563. [Google Scholar] [CrossRef]
- Deng, P.; Wang, M.; Feng, K.; Cui, L.; Tong, W.; Song, W.; Nie, X. Genome-wide characterization of microsatellites in Triticeae species: Abundance, distribution and evolution. Sci. Rep. 2016, 6, 32224. [Google Scholar] [CrossRef]
- Biswas, M.K.; Xu, Q.; Mayer, C.; Deng, X. Genome wide characterization of short tandem repeat markers in sweet orange (Citrus sinensis). PLoS ONE 2014, 9, e104182. [Google Scholar] [CrossRef] [PubMed]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 2010, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Martina, M.; Acquadro, A.; Barchi, L.; Gulino, D.; Brusco, F.; Rabaglio, M.; Portis, F.; Portis, E.; Lanteri, S. Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L. Int. J. Mol. Sci. 2022, 23, 3126. [Google Scholar] [CrossRef]
- He, B.; Geng, R.; Cheng, L.; Yang, X.; Ge, H.; Ren, M. Genetic diversity and fingerprinting of 33 standard flue-cured tobacco varieties for use in distinctness, uniformity, and stability testing. BMC Plant Biol. 2020, 20, 378. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, W.; Zhang, C.; Yang, L.; Chang, R.; Gaut, B.; Qiu, L. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol. 2010, 188, 242–253. [Google Scholar] [CrossRef]
- Liu, J.; Liu, G.; Qi, D.; Li, F. Construction of Genetic Fingerprints of Aneurolepidium chinensis Using Microsatellite Sequences. Acta Bot. Sin. 2000, 42, 985–987. [Google Scholar]
- Yang, Y.; Hou, X.; Wei, Z.; Wei, Z.; Qiao, Z.; Chang, C.; Ren, W.; Wu, Z. Screening and genetic diversity analysis of chloroplast non-coding regions in Leymus chinensis. Acta Pratacult. Sin. 2018, 27, 147–157. [Google Scholar] [CrossRef]
- Ahmed, N.; Hou, X. Genetic variation and population structure analysis of Leymus chinensis (Trin.) Tzvelev from Eurasian steppes using SSR makers. Genet. Resour. Crop Evol. 2022, 69, 2425–2436. [Google Scholar] [CrossRef]
- Kong, W.; Wang, Y.; Zhang, S.; Yu, J.; Zhang, X. Recent Advances in Assembly of Complex Plant Genomes. Genom. Proteom. Bioinform. 2023, 21, 427–439. [Google Scholar] [CrossRef]
- Sun, M.; Yan, H.; Zhang, A.; Jin, Y.; Lin, C.; Luo, L.; Wu, B.; Fan, Y.; Tian, S.; Cao, X.; et al. Milletdb: A multi-omics database to accelerate the research of functional genomics and molecular breeding of millets. Plant Biotechnol. J. 2023, 21, 2348–2357. [Google Scholar] [CrossRef]
- Yan, H.; Jin, Y.; Yu, H.; Wang, C.; Wu, B.; Jones, C.S.; Wang, X.; Xie, Z.; Huang, L. Genomic selection for agronomical phenotypes using genome-wide SNPs and SVs in pearl millet. Theor. Appl. Genet. 2024, 137, 244. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J.; Wilson, V.; Thein, S.L. Individual-specific ’fingerprints’ of human DNA. Nature 1985, 316, 76–79. [Google Scholar] [CrossRef]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, B.; Wang, S.; Du, P.; Qi, F.; Fang, Y.; Sun, Z.; Zheng, Z.; Dong, W.; Zhang, X. Development and characterization of whole genome SSR in tetraploid wild Peanut (Arachis monticola). Sci. Agric. Sin. 2019, 52, 2567–2580. [Google Scholar] [CrossRef]
- Tong, Z.; Jiao, F.; Xiao, B. Analysis of SSR loci in Nicotina tabacum genome and its two ancestral species genome. Sci. Agric. Sin. 2015, 48, 2108–2117. [Google Scholar] [CrossRef]
- Patil, P.G.; Singh, N.V.; Bohra, A.; Raghavendra, K.P.; Mane, R.; Mundewadikar, D.M.; Babu, K.D.; Sharma, J. Comprehensive Characterization and Validation of Chromosome-Specific Highly Polymorphic SSR Markers From Pomegranate (Punica granatum L.) cv. Tunisia Genome. Front. Plant Sci. 2021, 12, 645055. [Google Scholar] [CrossRef]
- Sonah, H.; Deshmukh, R.K.; Sharma, A.; Singh, V.P.; Gupta, D.K.; Gacche, R.N.; Rana, J.C.; Singh, N.K.; Sharma, T.R. Genome-wide distribution and organization of microsatellites in plants: An insight into marker development in Brachypodium. PLoS ONE 2011, 6, e21298. [Google Scholar] [CrossRef]
- Song, X.; Yang, Q.; Bai, Y.; Gong, K.; Wu, T.; Yu, T.; Pei, Q.; Duan, W.; Huang, Z.; Wang, Z.; et al. Comprehensive analysis of SSRs and database construction using all complete gene-coding sequences in major horticultural and representative plants. Hortic. Res. 2021, 8, 122. [Google Scholar] [CrossRef]
- Han, B.; Wang, C.; Tang, Z.; Ren, Y.; Li, Y.; Zhang, D.; Dong, Y.; Zhao, X. Genome-Wide Analysis of Microsatellite Markers Based on Sequenced Database in Chinese Spring Wheat (Triticum aestivum L.). PLoS ONE 2015, 10, e0141540. [Google Scholar] [CrossRef] [PubMed]
- Serrote, C.M.L.; Reiniger, L.R.S.; Silva, K.B.; Rabaiolli, S.M.D.S.; Stefanel, C.M. Determining the Polymorphism Information Content of a molecular marker. Gene 2020, 726, 144175. [Google Scholar] [CrossRef]
- Wang, Y. Genotyping of Leymus chinensis Based on Microsatellite Markers. Master’s Thesis, Liaoning University, Shenyang, China, 2019. [Google Scholar]
- Mutegi, E.; Snow, A.A.; Rajkumar, M.; Pasquet, R.; Ponniah, H.; Daunay, M.C.; Davidar, P. Genetic diversity and population structure of wild/weedy eggplant (Solanum insanum, Solanaceae) in southern India: Implications for conservation. Am. J. Bot. 2015, 102, 140–148. [Google Scholar] [CrossRef]
- Ivandic, V.; Hackett, C.A.; Nevo, E.; Keith, R.; Thomas, W.T.; Forster, B.P. Analysis of simple sequence repeats (SSRs) in wild barley from the Fertile Crescent: Associations with ecology, geography and flowering time. Plant Mol. Biol. 2002, 48, 511–527. [Google Scholar] [CrossRef]
- Pagnotta, M.A.; Annicchiarico, P.; Farina, A.; Proietti, S. Characterizing the molecular and morphophysiological diversity of Italian red clover. Euphytica 2011, 179, 393–404. [Google Scholar] [CrossRef]
- Linhart, Y.B.; Grant, M.C. Evolutionary significance of local genetic differentiation in plants. Annu. Rev. Ecol. Syst. 1996, 27, 237–277. [Google Scholar] [CrossRef]
- Pagnotta, M.A.; Mondini, L.; Codianni, P.; Fares, C. Agronomical, quality and molecular characterization of twenty Italian emmer wheat genotypes. Genet. Resour. Crop Evol. 2009, 56, 299–310. [Google Scholar] [CrossRef]
- Zhu, Y. Research on Cultivar Identification and DNA Fingerprinting of Crops Based on Molecular Markers. Ph.D. Thesis, Zhejiang University, Hangzhou, China, 2013. [Google Scholar]
- Liu, L.; Zhao, L.; Gong, Y.; Wang, M.; Chen, L.; Yang, J.; Wang, Y.; Yu, F.; Wang, L. DNA fingerprinting and genetic diversity analysis of late-bolting radish cultivars with RAPD, ISSR and SRAP markers. Sci. Hortic. 2008, 116, 240–247. [Google Scholar] [CrossRef]
- Gade, P.; Alam, M.A.; Barma, N.C.; Majumder, R.; Garapaty, R.; Paranjape, V.D.; Killian, A.; Vijayaraghavan, K.; Kabir, R.; Hakim, A.; et al. Assessment of wheat variety adoption in Bangladesh through DNA fingerprinting. Crop Sci. 2021, 61, 3564–3577. [Google Scholar] [CrossRef]
- Rahman, M.S.; Sohag, M.K.H.; Rahman, L.; Alam, M.S.E.; Nath, U.K.; Bashar, M.K. Genetic fingerprinting for the protection of local rice (Oryza sativa L.) cultivars of Bangladesh. J. Adv. Biotechnol. Exp. Ther. 2020, 3, 182–193. [Google Scholar] [CrossRef]
- Lu, Y.; Yan, J.; Guimarães, C.T.; Taba, S.; Hao, Z.; Gao, S.; Chen, S.; Li, J.; Zhang, S.; Vivek, B.S.; et al. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor. Appl. Genet. 2009, 120, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System; Version 2.1.; Exeter Software: Setauket, NY, USA, 2000. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSR Researching | Number | |||
|---|---|---|---|---|
| Examined sequence size (bp) | 7849381805 | |||
| Total number of SSRs | 973129 | |||
| Total length of perfect SSRs (bp) | 15431847 | |||
| The percentage of sequence covered by SSRs | 0.2 | |||
| Relative abundance of SSRs (loci/Mb) | 123.98 | |||
| Relative density of SSRs (bp/Mb) | 1966.01 | |||
| Type | Length | No. of repeats/ [mean(range)] | Relative abundance | Relative density |
| Mononucleotide | 5,107,083 | 13.1 (10–68) | 53.73 | 650.64 |
| Dinucleotide | 6,375,336 | 9.3 (6–152) | 43.47 | 812.21 |
| Trinucleotide | 3,604,197 | 6.1 (5–343) | 25.09 | 459.17 |
| Tetranucleotide | 201,908 | 5.8 (5–28) | 1.12 | 25.72 |
| Pentanucleotide | 55,195 | 6.1 (5–24) | 0.25 | 7.03 |
| Hexanucleotide | 88,128 | 5.7 (5–24) | 0.33 | 11.23 |
| Locus | Na | Ne | Ho | He | I | PIC |
|---|---|---|---|---|---|---|
| GW03-1 | 36 | 20.027 | 0.943 | 0.950 | 3.211 | 0.948 |
| GW03-7 | 39 | 11.526 | 0.800 | 0.913 | 2.941 | 0.908 |
| GW07-1 | 46 | 30.071 | 0.508 | 0.967 | 3.608 | 0.966 |
| GW07-4 | 41 | 15.989 | 0.689 | 0.937 | 3.119 | 0.934 |
| GW02-3 | 15 | 6.229 | 1.000 | 0.839 | 2.136 | 0.823 |
| GW06-6 | 32 | 9.492 | 1.000 | 0.895 | 2.796 | 0.889 |
| GW13-5 | 24 | 13.445 | 1.000 | 0.926 | 2.779 | 0.921 |
| GW13-7 | 38 | 8.086 | 0.876 | 0.876 | 2.765 | 0.869 |
| GW01-3 | 25 | 4.736 | 0.933 | 0.789 | 2.275 | 0.778 |
| GW05-2 | 44 | 23.339 | 0.816 | 0.957 | 3.440 | 0.956 |
| GW06-3 | 22 | 11.575 | 0.990 | 0.914 | 2.652 | 0.907 |
| GW11-7 | 24 | 8.136 | 0.541 | 0.877 | 2.460 | 0.866 |
| GW06-2 | 64 | 31.729 | 0.901 | 0.968 | 3.786 | 0.968 |
| GW06-7 | 42 | 25.057 | 0.771 | 0.960 | 3.443 | 0.959 |
| GW09-6 | 37 | 15.320 | 0.750 | 0.935 | 3.083 | 0.931 |
| GW12-4 | 33 | 14.300 | 0.924 | 0.930 | 2.998 | 0.926 |
| GW01-5 | 27 | 13.554 | 0.240 | 0.926 | 2.876 | 0.922 |
| GW01-6 | 18 | 7.172 | 0.500 | 0.861 | 2.185 | 0.846 |
| GW02-6 | 39 | 21.000 | 0.724 | 0.952 | 3.296 | 0.950 |
| GW13-3 | 25 | 16.185 | 0.699 | 0.938 | 2.933 | 0.935 |
| Mean | 33.55 | 15.348 | 0.780 | 0.916 | 2.939 | 0.910 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, T.; Wu, Z.; Tian, C.; Yang, Y.; Gong, W.; Niu, J.; Li, Z. Development of Genome-Wide SSR Markers in Leymus chinensis with Genetic Diversity Analysis and DNA Fingerprints. Int. J. Mol. Sci. 2025, 26, 918. https://doi.org/10.3390/ijms26030918
Ou T, Wu Z, Tian C, Yang Y, Gong W, Niu J, Li Z. Development of Genome-Wide SSR Markers in Leymus chinensis with Genetic Diversity Analysis and DNA Fingerprints. International Journal of Molecular Sciences. 2025; 26(3):918. https://doi.org/10.3390/ijms26030918
Chicago/Turabian StyleOu, Taiyou, Zinian Wu, Chunyu Tian, Yanting Yang, Wenlong Gong, Jianjiang Niu, and Zhiyong Li. 2025. "Development of Genome-Wide SSR Markers in Leymus chinensis with Genetic Diversity Analysis and DNA Fingerprints" International Journal of Molecular Sciences 26, no. 3: 918. https://doi.org/10.3390/ijms26030918
APA StyleOu, T., Wu, Z., Tian, C., Yang, Y., Gong, W., Niu, J., & Li, Z. (2025). Development of Genome-Wide SSR Markers in Leymus chinensis with Genetic Diversity Analysis and DNA Fingerprints. International Journal of Molecular Sciences, 26(3), 918. https://doi.org/10.3390/ijms26030918