
Unraveling the Ancient Introgression History of Xanthoceras (Sapindaceae): Insights from Phylogenomic Analysis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
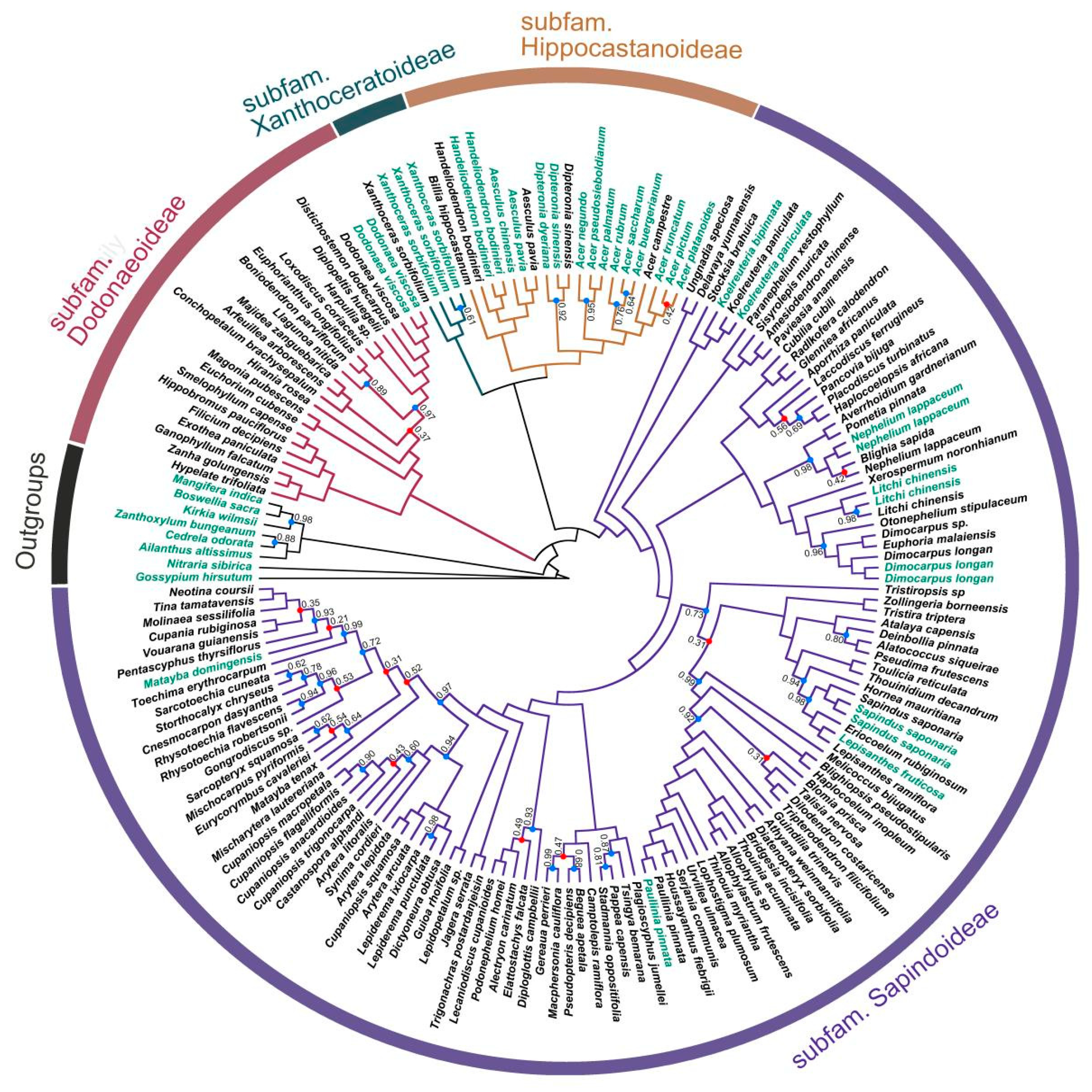
2.1. Phylogenetic Inference
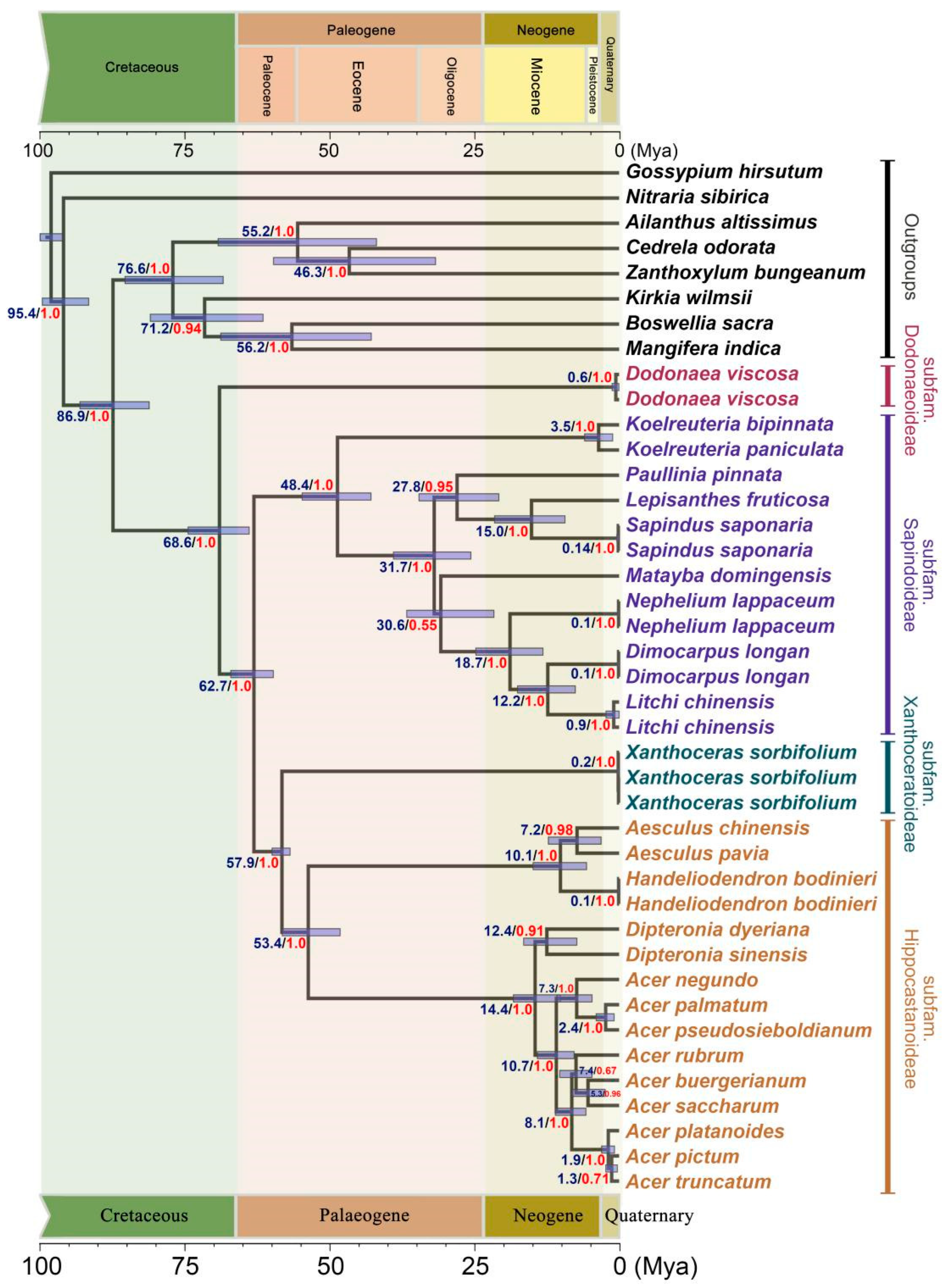
2.2. Divergence Time Estimation
2.3. Analysis of Gene Tree Discordance
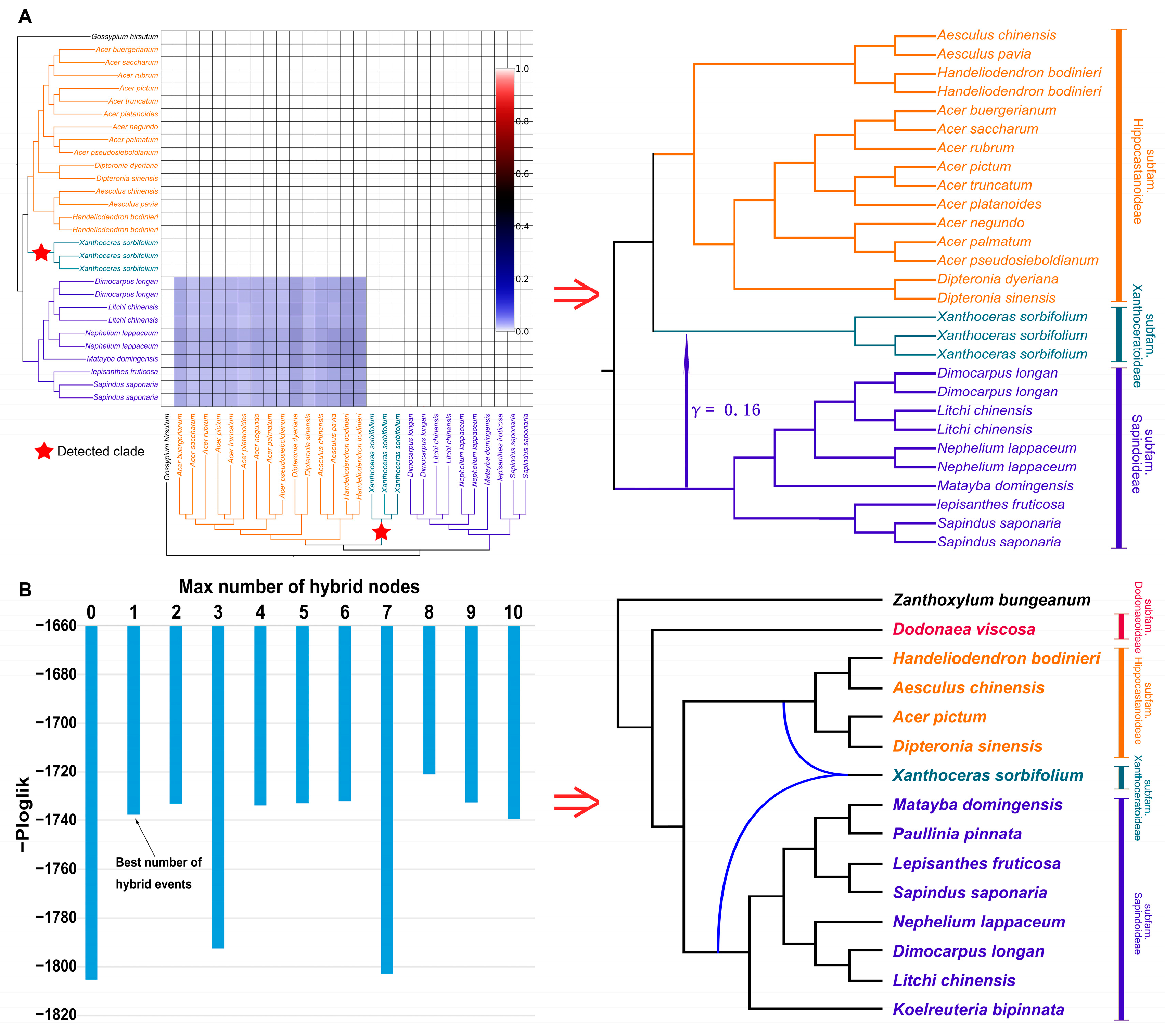
2.4. Assessment of Interspecific Gene Flow
3. Discussion
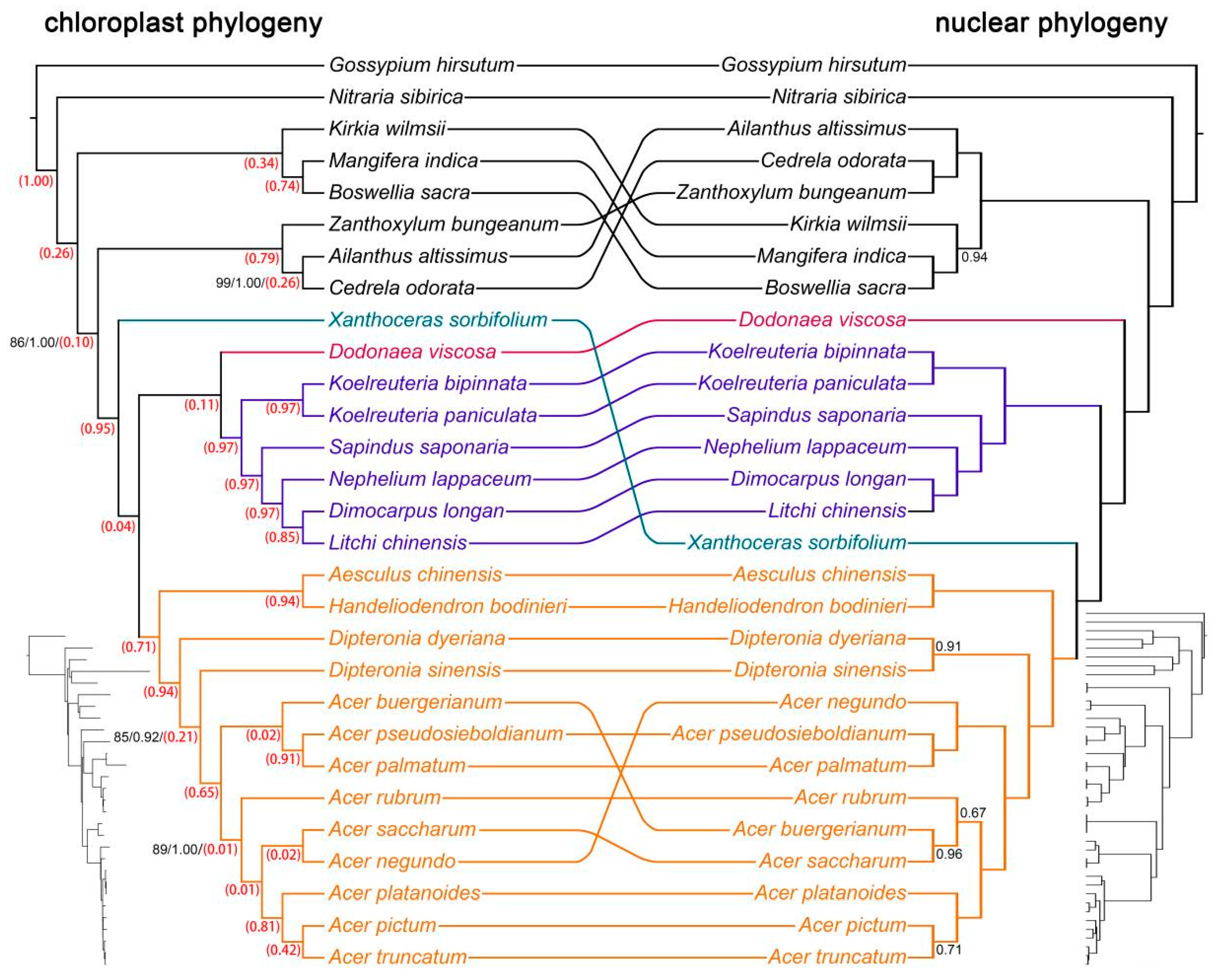
3.1. Deciphering Cyto-Nuclear Discordance in Sapindaceae Phylogeny
3.2. Detecting Ancient Interspecific Gene Flow Events in the Presence of Strong ILS
3.3. Ancient Introgression of Subfam. Xanthoceroideae
4. Materials and Methods
4.1. Data Sources
4.2. Sequence Processing
4.3. Phylogenetic Analysis
4.4. Molecular Dating
4.5. Detecting Gene Tree-Species Tree Discordance
4.6. Coalescent Simulation for Tree Discordance
4.7. Introgression Events Detection
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rieseberg, L.H.; Raymond, O.; Rosenthal, D.M.; Lai, Z.; Livingstone, K.; Nakazato, T.; Durphy, J.L.; Schwarzbach, A.E.; Donovan, L.A.; Lexer, C. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 2003, 301, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Soltis, D.E. The role of hybridization in plant speciation. Ann. Rev. Plant Biol. 2009, 60, 561–588. [Google Scholar] [CrossRef]
- Goulet, B.E.; Roda, F.; Hopkins, R. Hybridization in plants: Old ideas, new techniques. Plant Physiol. 2017, 173, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Alix, K.; Gérard, P.R.; Schwarzacher, T. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. AoB Plants 2017, 120, 183–194. [Google Scholar] [CrossRef]
- Meier, J.I.; Marques, D.A.; Mwaiko, S.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 2017, 8, 14363. [Google Scholar] [CrossRef]
- Meier, J.I.; Stelkens, R.B.; Joyce, D.A.; Mwaiko, S.; Phiri, N.; Schliewen, U.K.; Selz, O.M.; Wagner, C.E.; Katongo, C.; Seehausen, O. The coincidence of ecological opportunity with hybridization explains rapid adaptive radiation in Lake Mweru cichlid fishes. Nat. Commun. 2019, 10, 5391. [Google Scholar] [CrossRef]
- Stankowski, S.; Streisfeld, M.A. Introgressive hybridization facilitates adaptive divergence in a recent radiation of monkeyflowers. Proc. R. Soc. B: Biol. Sci. 2015, 282, 20151666. [Google Scholar] [CrossRef]
- Marques, D.A.; Meier, J.I.; Seehausen, O. A combinatorial view on speciation and adaptive radiation. Trends Ecol. Evol. 2019, 34, 531–544. [Google Scholar] [CrossRef]
- McGee, M.D.; Borstein, S.R.; Meier, J.I.; Marques, D.A.; Mwaiko, S.; Taabu, A.; Seehausen, O. The ecological and genomic basis of explosive adaptive radiation. Nature 2020, 586, 75–79. [Google Scholar] [CrossRef]
- Rosser, N.; Seixas, F.; Queste, L.M.; Cama, B.; Mori-Pezo, R.; Kryvokhyzha, D.; Nelson, M.; Waite-Hudson, R.; Goringe, M.; Costa, M. Hybrid speciation driven by multilocus introgression of ecological traits. Nature 2024, 628, 811–817. [Google Scholar] [CrossRef]
- Taylor, S.A.; Larson, E.L. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 2019, 3, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Mavárez, J.; Salazar, C.A.; Bermingham, E.; Salcedo, C.; Jiggins, C.D.; Linares, M. Speciation by hybridization in Heliconius butterflies. Nature 2006, 441, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Lamichhaney, S.; Han, F.; Webster, M.T.; Andersson, L.; Grant, B.R.; Grant, P.R. Rapid hybrid speciation in Darwin’s finches. Science 2018, 359, 224–228. [Google Scholar] [CrossRef]
- Zhang, B.W.; Xu, L.L.; Li, N.; Yan, P.C.; Jiang, X.H.; Woeste, K.E.; Bai, W.N. Phylogenomics reveals an ancient hybrid origin of the Persian walnut. Mol. Biol. Evol. 2019, 36, 2451–2461. [Google Scholar] [CrossRef]
- Guo, X.; Thomas, D.C.; Saunders, R.M. Gene tree discordance and coalescent methods support ancient intergeneric hybridisation between Dasymaschalon and Friesodielsia (Annonaceae). Mol. Phylogenetics Evol. 2018, 127, 14–29. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, M.; Li, J.; Zhang, Z.; Wang, Y.; Chen, C.; Liu, J. Genomic evidence for homoploid hybrid speciation between ancestors of two different genera. Nat. Commun. 2022, 13, 1987. [Google Scholar] [CrossRef]
- Acosta, M.C.; Premoli, A.C. Evidence of chloroplast capture in south American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogenet. Evol. 2010, 54, 235–242. [Google Scholar] [CrossRef]
- Rose, J.P.; Toledo, C.A.P.; Lemmon, E.M.; Lemmon, A.R.; Sytsma, K.J. Out of sight, out of mind: Widespread nuclear and plastid-nuclear discordance in the flowering plant genus Polemonium (Polemoniaceae) suggests widespread historical gene flow despite limited nuclear signal. Syst. Biol. 2021, 70, 162–180. [Google Scholar] [CrossRef]
- Liu, B.; Ren, C.; Kwak, M.; Hodel, R.G.J.; Xu, C.; He, J.; Zhou, W.; Huang, C.; Ma, H.; Qian, G.; et al. Phylogenomic conflict analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving diversification, with insights into the complex biogeographic history in the Northern Hemisphere. J. Integr. Plant Biol. 2022, 64, 1020–1043. [Google Scholar] [CrossRef]
- He, J.; Lyu, R.; Luo, Y.; Xiao, J.; Xie, L.; Wen, J.; Li, W.; Pei, L.; Cheng, J. A phylotranscriptome study using silica gel-dried leaf tissues produces an updated robust phylogeny of Ranunculaceae. Mol. Phylogenet. Evol. 2022, 174, 107545. [Google Scholar] [CrossRef]
- Zhou, Q.Y.; Liu, G.S. The embryology of Xanthoceras and its phylogenetic implications. Plant Syst. Evol. 2012, 298, 457–468. [Google Scholar] [CrossRef]
- Harrington, M.G.; Edwards, K.J.; Johnson, S.A.; Chase, M.W.; Gadek, P.A. Phylogenetic inference in Sapindaceae sensu lato using plastid matK and rbcL DNA sequences. Syst. Bot. 2005, 30, 366–382. [Google Scholar] [CrossRef]
- Buerki, S.; Forest, F.; Acevedo-Rodríguez, P.; Callmander, M.W.; Nylander, J.A.; Harrington, M.; Sanmartín, I.; Küpfer, P.; Alvarez, N. Plastid and nuclear DNA markers reveal intricate relationships at subfamilial and tribal levels in the soapberry family (Sapindaceae). Mol. Phylogenet. Evol. 2009, 51, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Buerki, S.; Forest, F.; Alvarez, N.; Nylander, J.A.; Arrigo, N.; Sanmartín, I. An evaluation of new parsimony-based versus parametric inference methods in biogeography: A case study using the globally distributed plant family Sapindaceae. J. Biogeogr. 2011, 38, 531–550. [Google Scholar] [CrossRef]
- Buerki, S.; Lowry, P.P., II; Alvarez, N.; Razafimandimbison, S.G.; Küpfer, P.; Callmander, M.W. Phylogeny and circumscription of Sapindaceae revisited: Molecular sequence data, morphology and biogeography support recognition of a new family, Xanthoceraceae. Plant Ecol. Evol. 2012, 143, 148–159. [Google Scholar] [CrossRef]
- Buerki, S.; Forest, F.; Callmander, M.W.; Lowry, P.P., II; Devey, D.S.; Munzinger, J. Phylogenetic inference of New Caledonian lineages of Sapindaceae: Molecular evidence requires a reassessment of generic circumscriptions. Taxon 2012, 61, 109–119. [Google Scholar] [CrossRef]
- Buerki, S.; Forest, F.; Stadler, T.; Alvarez, N. The abrupt climate change at the Eocene–Oligocene boundary and the emergence of South-East Asia triggered the spread of sapindaceous lineages. AoB Plants 2013, 112, 151–160. [Google Scholar] [CrossRef]
- Muellner-Riehl, A.N.; Weeks, A.; Clayton, J.W.; Buerki, S.; Nauheimer, L.; Chiang, Y.C.; Cody, S.; Pell, S.K. Molecular phylogenetics and molecular clock dating of Sapindales based on plastid rbcL, atpB and trnL-trnF DNA sequences. Taxon 2016, 65, 1019–1036. [Google Scholar] [CrossRef]
- Xiang, Q.; He, J.; Wang, X.; Sun, K.; Xu, J.; Zhang, D.; Guan, W. Two complete plastid genome sequences of Sapindales: Zanthoxylum nitidum and Xanthoceras sorbifolium, and phylogenetic analyses in Sapindales. Mitochondrial DNA B 2019, 4, 1716–1717. [Google Scholar] [CrossRef]
- Buerki, S.; Callmander, M.W.; Acevedo-Rodriguez, P.; Lowry, P.P.; Munzinger, J.; Bailey, P.; Maurin, O.; Brewer, G.E.; Epitawalage, N.; Baker, W.J.; et al. An updated infra-familial classification of Sapindaceae based on targeted enrichment data. Am. J. Bot. 2021, 108, 1234–1251. [Google Scholar] [CrossRef]
- Baker, W.J.; Bailey, P.; Barber, V. A comprehensive phylogenomic platform for exploring the angiosperm tree of life. Syst. Biol. 2022, 71, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Joyce, E.M.; Appelhans, M.S.; Buerki, S.; Cheek, M.; De Vos, J.M.; Pirani, J.R.; Zuntini, A.R.; Bachelier, J.B.; Bayly, M.J.; Callmander, M.W.; et al. Phylogenomic analyses of Sapindales support new family relationships, rapid Mid-Cretaceous Hothouse diversification, and heterogeneous histories of gene duplication. Front. Plant Sci. 2023, 14, 1063174. [Google Scholar] [CrossRef]
- Folk, R.A.; Mandel, J.R.; Freudenstein, J.V. Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 2017, 66, 320–337. [Google Scholar] [CrossRef]
- Bruun-Lund, S.; Clement, W.L.; Kjellberg, F.; Rønsted, N. First plastid phylogenomic study reveals potential cyto-nuclear discordance in the evolutionary history of Ficus L.(Moraceae). Mol. Phylogenet. Evol. 2017, 109, 93–104. [Google Scholar] [CrossRef]
- Smith, S.A.; Moore, M.J.; Brown, J.W.; Yang, Y. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 2015, 15, 1–15. [Google Scholar] [CrossRef]
- Blischak, P.D.; Chifman, J.; Wolfe, A.D.; Kubatko, L.S. HyDe: A Python package for genome-scale hybridization detection. Syst. Biol. 2018, 67, 821–829. [Google Scholar] [CrossRef]
- Wen, D.; Yu, Y.; Zhu, J.; Nakhleh, L. Inferring phylogenetic networks using PhyloNet. Syst. Biol. 2018, 67, 735–740. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group (APG). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Wang, Z.; Shrestha, N.; Liu, J. Species divergence with gene flow and hybrid speciation on the Qinghai-Tibet Plateau. New Phytol. 2022, 234, 392–404. [Google Scholar] [CrossRef]
- Kong, S.; Kubatko, L.S. Comparative performance of popular methods for hybrid detection using genomic data. Syst. Biol. 2021, 70, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Z.; Zhang, Y.; Frantz, L.; Roos, C.; Irwin, D.M.; Zhang, C.; Liu, X.; Wu, D.; Huang, S.; et al. Hybrid origin of a primate, the gray snub-nosed monkey. Science 2023, 380, eabl4997. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, L.; Ma, J. Genomic introgression through interspecific hybridization counteracts genetic bottleneck during soybean domestication. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.A.; Maere, S.; Van de Peer, Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA 2009, 106, 5737–5742. [Google Scholar] [CrossRef]
- Vanneste, K.; Maere, S.; Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Res. 2014, 24, 1334–1347. [Google Scholar] [CrossRef]
- One Thousand Plant Transcriptomes Initiative (One KP). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef]
- Koenen, E.J.M.; Ojeda, D.I.; Bakker, F.T.; Wieringa, J.J.; Kidner, C.; Hardy, O.J.; Pennington, R.T.; Herendeen, P.S.; Bruneau, A.; Hughes, C.E. The origin of the legumes is a complex paleopolyploid phylogenomic tangle closely associated with the Cretaceous–Paleogene (K–Pg) mass extinction event. Syst. Biol. 2021, 70, 508–526. [Google Scholar] [CrossRef]
- Hu, G.; Feng, J.; Xiang, X.; Wang, J.; Salojärvi, J.; Liu, C.; Wu, Z.; Zhang, J.; Liang, X.; Jiang, Z.; et al. Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat. Genet. 2022, 54, 73–83. [Google Scholar] [CrossRef]
- Zheng, J.; Meinhardt, L.W.; Goenaga, R.; Matsumoto, T.; Zhang, D.; Yin, Y. The chromosome-level rambutan genome reveals a significant role of segmental duplication in the expansion of resistance genes. Hortic. Res. 2022, 9, uhac014. [Google Scholar] [CrossRef]
- Liang, Q.; Li, H.; Li, S.; Yuan, F.; Sun, J.; Duan, Q.; Li, Q.; Zhang, R.; Sang, Y.L.; Wang, N.; et al. The genome assembly and annotation of yellowhorn (Xanthoceras sorbifolium Bunge). GigaScience 2019, 8, giz071. [Google Scholar] [CrossRef]
- Liu, H.; Yan, X.M.; Wang, X.R.; Zhang, D.X.; Zhou, Q.; Shi, T.L.; Jia, K.H.; Tian, X.C.; Zhou, S.S.; Zhang, R.G.; et al. Centromere-specific retrotransposons and very-long-chain fatty acid biosynthesis in the genome of yellowhorn (Xanthoceras sorbifolium, sapindaceae), an oil-producing tree with significant drought resistance. Front. Plant Sci. 2021, 12, 766389. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Chen, D.; Zhang, T.; Chen, Y.; Fan, H.; Huang, Y.; Li, B. Chromosome-scale assembly and population diversity analyses provide insights into the evolution of Sapindus mukorossi. Hortic. Res. 2022, 9, uhac012. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Zeng, Z.; Wu, F.; Feng, J.; Liu, B.; Mai, Y.; Chu, X.; Wei, W.; Li, X.; et al. SapBase: A central portal for functional and comparative genomics of Sapindaceae species. J. Integr. Plant Biol. 2024, 66, 1561–1570. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-) orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 1–9. [Google Scholar] [CrossRef]
- Duvall, M.R.; Burke, S.V.; Clark, D.C. Plastome phylogenomics of Poaceae: Alternate topologies depend on alignment gaps. Bot. J. Linn. Soc. 2020, 192, 9–20. [Google Scholar] [CrossRef]
- Yang, Y.; Smith, S.A. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: Improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 2014, 31, 3081–3092. [Google Scholar] [CrossRef]
- Mai, U.; Sayyari, E.; Mirarab, S. Minimum variance rooting of phylogenetic trees and implications for species tree reconstruction. PLoS ONE 2017, 12, e0182238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 15–30. [Google Scholar] [CrossRef]
- Chou, J.; Gupta, A.; Yaduvanshi, S.; Davidson, R.; Nute, M.; Mirarab, S.; Warnow, T. A comparative study of SVDquartets and other coalescent-based species tree estimation methods. BMC Genom. 2015, 16, 1–11. [Google Scholar] [CrossRef]
- Yang, Y.; Moore, M.J.; Brockington, S.F.; Soltis, D.E.; Wong, G.K.S.; Carpenter, E.J.; Zhang, Y.; Chen, L.; Yan, Z.; Xie, Y. Dissecting molecular evolution in the highly diverse plant clade Caryophyllales using transcriptome sequencing. Mol. Biol. Evol. 2015, 32, 2001–2014. [Google Scholar] [CrossRef]
- Molloy, E.K.; Warnow, T. TreeMerge: A new method for improving the scalability of species tree estimation methods. Bioinformatics 2019, 35, i417–i426. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Douglas, J.; Jiménez-Silva, C.L.; Bouckaert, R. StarBeast3: Adaptive parallelized Bayesian inference under the multispecies coalescent. Syst. Biol. 2022, 71, 901–916. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef]
- Manchester, S.R. Biogeographical relationships of North American tertiary floras. Ann. Mo. Bot. Gard. 1999, 86, 472–522. [Google Scholar] [CrossRef]
- McClain, A.M.; Manchester, S.R. Dipteronia (Sapindaceae) from the Tertiary of North America and implications for the phytogeographic history of the Aceroideae. Am. J. Bot. 2001, 88, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yi, T.; Gao, L.; Ma, P.; Zhang, T.; Yang, J.; Gitzendanner, M.A.; Fritsch, P.W.; Cai, J.; Luo, Y.; et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 2019, 5, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.A. Toytree: A minimalist tree visualization and manipulation library for Python. Methods Ecol. Evol. 2020, 11, 187–191. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Smith, S.A.; O’Meara, B.C. treePL: Divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef]
- Sukumaran, J.; Holder, M.T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 2010, 26, 1569–1571. [Google Scholar] [CrossRef]
- Sang, T.; Zhong, Y. Testing hybridization hypotheses based on incongruent gene trees. Syst. Biol. 2000, 49, 422–434. [Google Scholar] [CrossRef]
- Edwards, S.V.; Xi, Z.; Janke, A.; Faircloth, B.C.; McCormack, J.E.; Glenn, T.C.; Zhong, B.; Wu, S.; Lemmon, E.M.; Lemmon, A.R. Implementing and testing the multispecies coalescent model: A valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 2016, 94, 447–462. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, P.; Lv, L.; Wang, D.; Ru, D.; Li, Y.; Ma, T.; Zhang, L.; Shen, X.; Meng, F.; et al. Prickly waterlily and rigid hornwort genomes shed light on early angiosperm evolution. Nat. Plants 2020, 6, 215–222. [Google Scholar] [CrossRef]
- Morales-Briones, D.F.; Kadereit, G.; Tefarikis, D.T.; Moore, M.J.; Smith, S.A.; Brockington, S.F.; Timoneda, A.; Yim, W.C.; Cushman, J.C.; Yang, Y. Disentangling sources of gene tree discordance in phylogenomic data sets: Testing ancient hybridizations in Amaranthaceae sl. Syst. Biol. 2021, 70, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.F.; Foulds, L.R. Comparison of phylogenetic trees. Math. Biosci 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L. Phybase: An R package for species tree analysis. Bioinformatics 2010, 26, 962–963. [Google Scholar] [CrossRef]
- Edelman, N.B.; Frandsen, P.B.; Miyagi, M.; Clavijo, B.; Davey, J.; Dikow, R.B.; García-Accinelli, G.; Van Belleghem, S.M.; Patterson, N.; Neafsey, D.E. Genomic architecture and introgression shape a butterfly radiation. Science 2019, 366, 594–599. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.Y. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient admixture in human history. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef]
- McLean, B.S.; Bell, K.C.; Allen, J.M.; Helgen, K.M.; Cook, J.A. Impacts of inference method and data set filtering on phylogenomic resolution in a rapid radiation of ground squirrels (Xerinae: Marmotini). Syst. Biol. 2019, 68, 298–316. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Li, M.; Wu, H.; Cheng, J.; Xie, L. Unraveling the Ancient Introgression History of Xanthoceras (Sapindaceae): Insights from Phylogenomic Analysis. Int. J. Mol. Sci. 2025, 26, 1581. https://doi.org/10.3390/ijms26041581
He J, Li M, Wu H, Cheng J, Xie L. Unraveling the Ancient Introgression History of Xanthoceras (Sapindaceae): Insights from Phylogenomic Analysis. International Journal of Molecular Sciences. 2025; 26(4):1581. https://doi.org/10.3390/ijms26041581
Chicago/Turabian StyleHe, Jian, Mingyang Li, Huanyu Wu, Jin Cheng, and Lei Xie. 2025. "Unraveling the Ancient Introgression History of Xanthoceras (Sapindaceae): Insights from Phylogenomic Analysis" International Journal of Molecular Sciences 26, no. 4: 1581. https://doi.org/10.3390/ijms26041581
APA StyleHe, J., Li, M., Wu, H., Cheng, J., & Xie, L. (2025). Unraveling the Ancient Introgression History of Xanthoceras (Sapindaceae): Insights from Phylogenomic Analysis. International Journal of Molecular Sciences, 26(4), 1581. https://doi.org/10.3390/ijms26041581