
Combining Sulfonylureas with Anticancer Drugs: Evidence of Synergistic Efficacy with Doxorubicin In Vitro and In Vivo

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
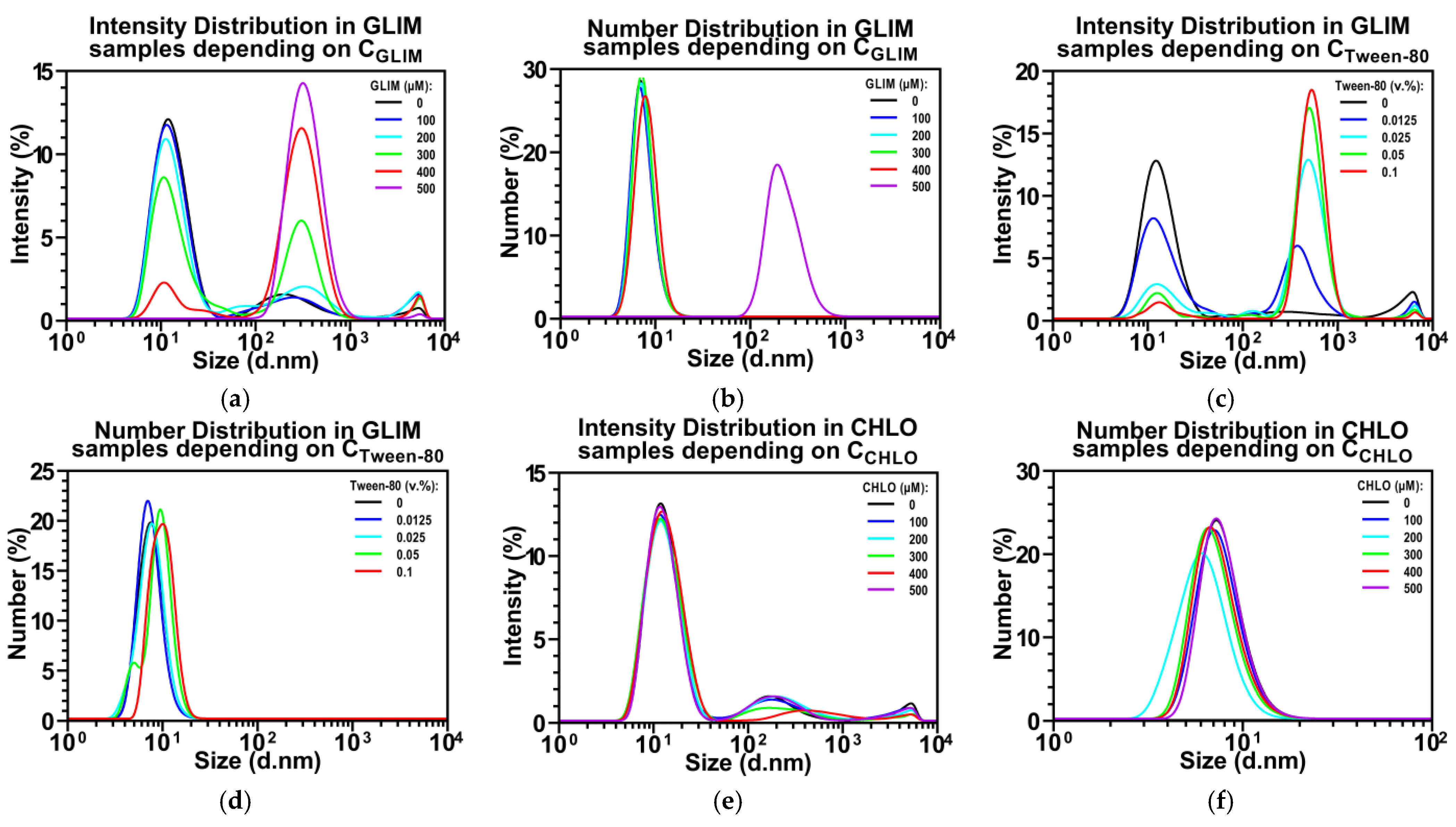
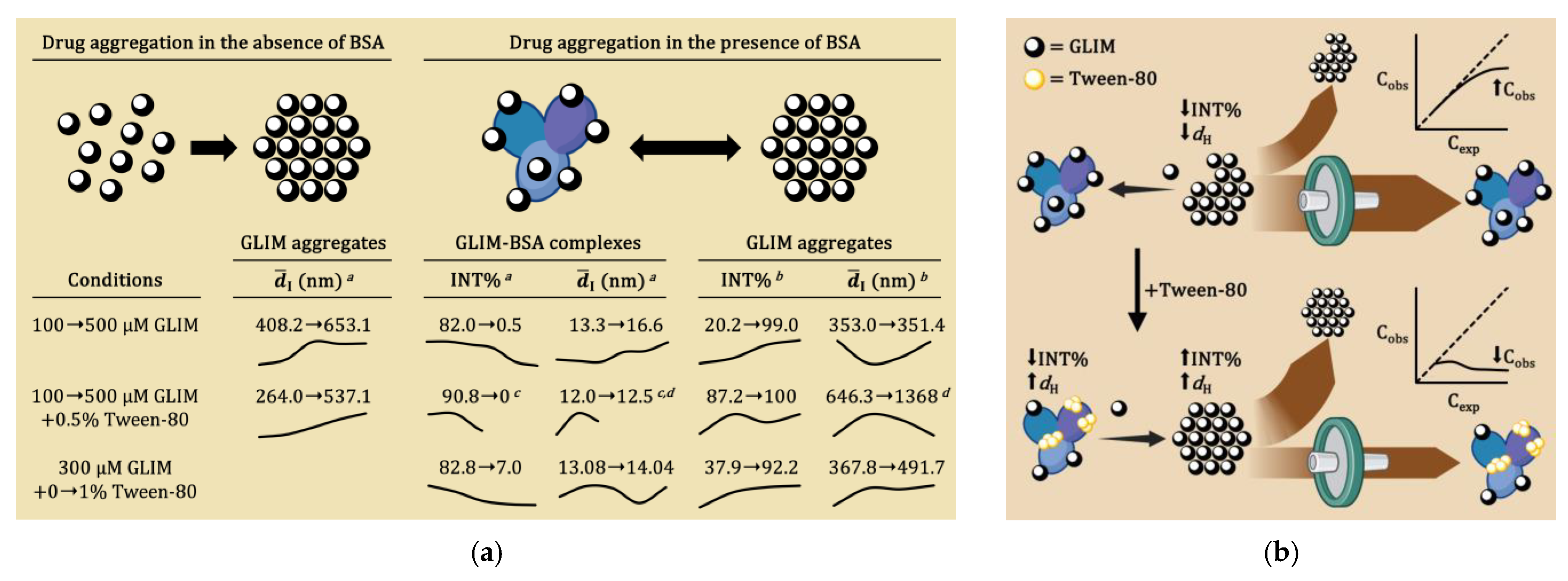
2.1. Effect of Tween-80 on Sulfonylurea Solubility
2.2. Effect of Tween-80 on Drug-Induced Cytotoxicity
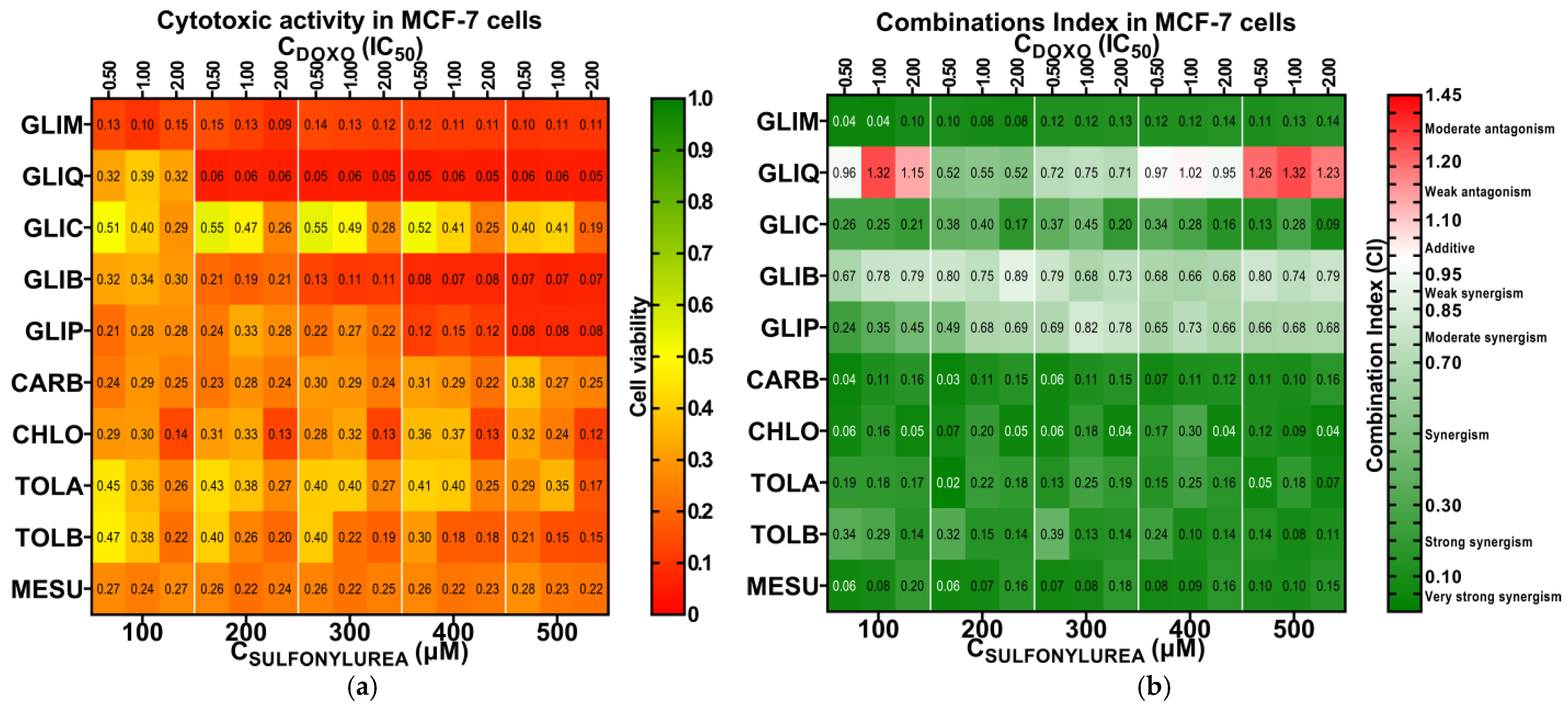
2.3. Preliminary Screening for Cytotoxic Synergy
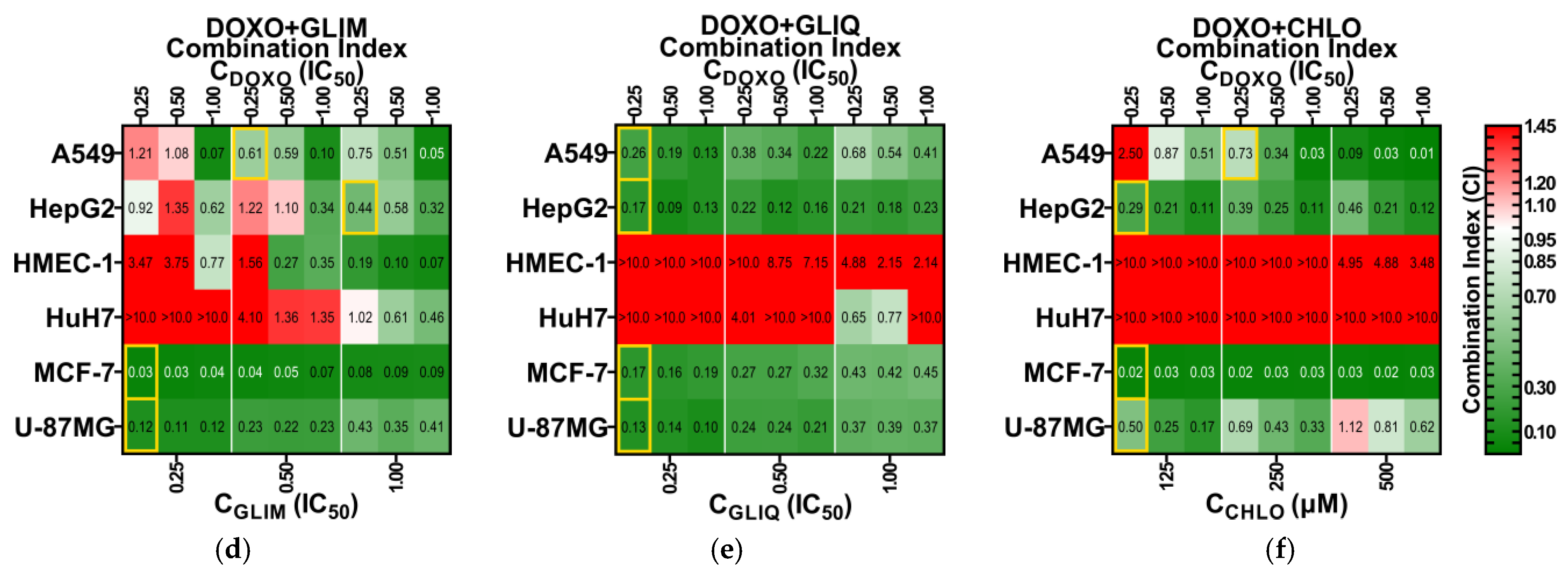
2.4. Expanded Screening of Selected Drug Combinations
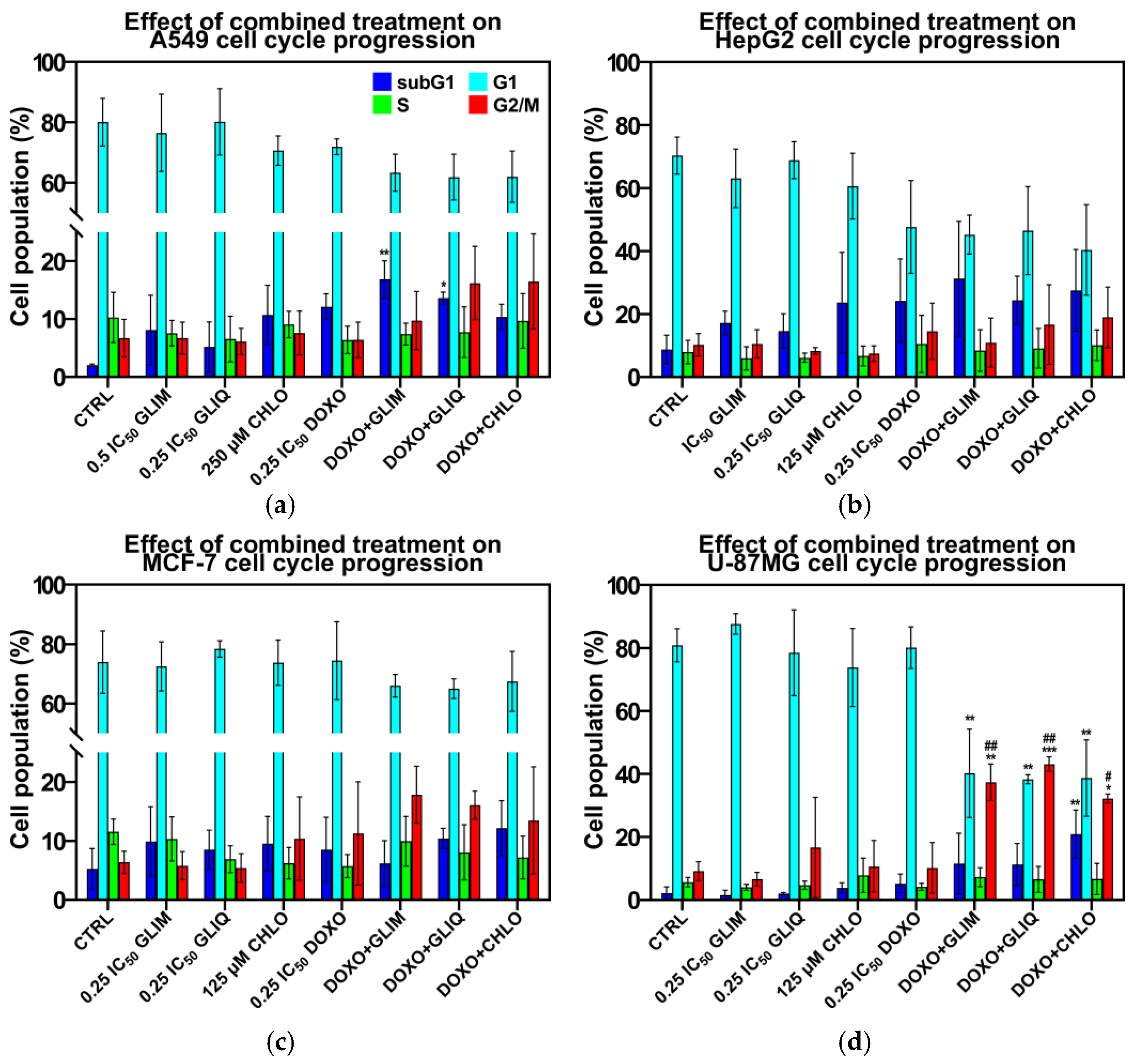
2.5. Effect of Combined Treatment on Cell Cycle Progression
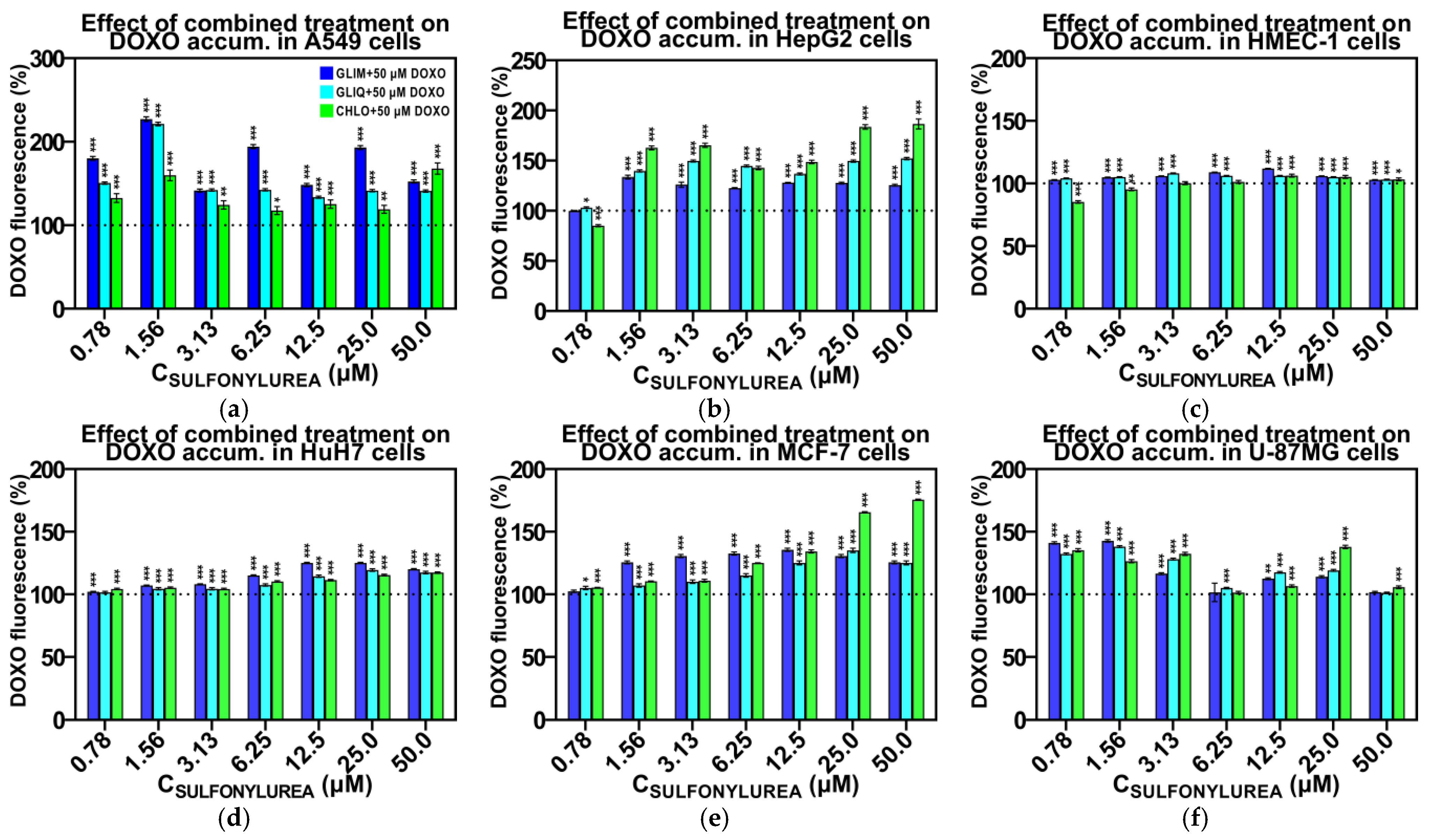
2.6. Effect of Combined Treatment on Doxorubicin Accumulation
2.7. Ex Vivo Toxicity and Microscopy
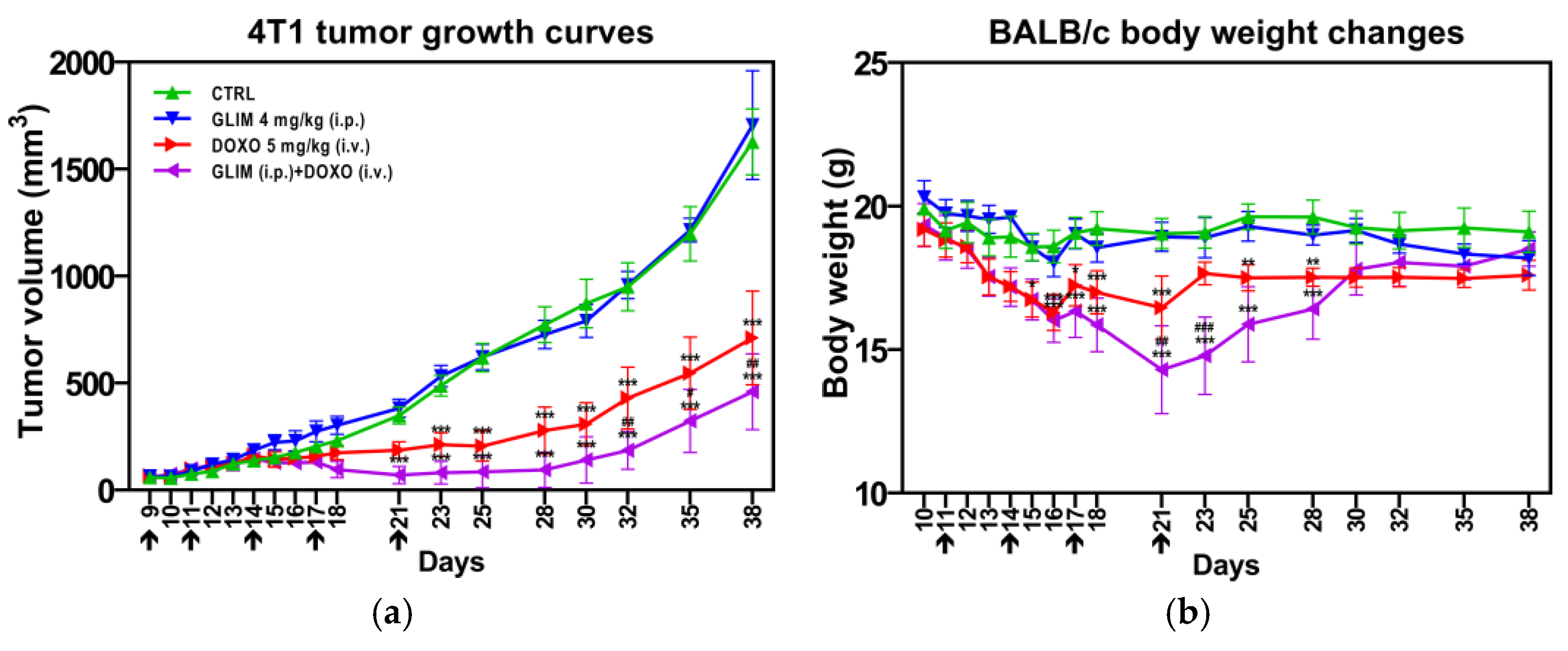
2.8. Findings from In Vivo Studies
3. Materials and Methods
3.1. HPLC Analysis
3.2. DLS Measurements
3.3. Analysis of Drug Solubility
3.4. Analysis of Drug Stability
3.5. Preliminary Cytotoxic Assay
3.6. Synergistic Cytotoxicity Assay
3.7. Flow Cytometric Measurements
3.8. Doxorubicin Accumulation Assay
3.9. Hemolysis and Membrane Integrity Evaluation
3.10. In Vivo Experimental Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sahra, I.B.; Marchand-Brustel, Y.L.; Tanti, J.-F.; Bost, F. Metformin in Cancer Therapy: A New Perspective for an Old Antidiabetic Drug? Mol. Cancer Ther. 2010, 9, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Future Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef] [PubMed]
- Stattin, P.; Björ, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–568. [Google Scholar] [CrossRef]
- Shikata, K.; Ninomiya, T.; Kiyohara, Y. Diabetes mellitus and cancer risk: Review of the epidemiological evidence. Cancer Sci. 2012, 104, 9–14. [Google Scholar] [CrossRef]
- Suh, S.; Kim, K.W. Diabetes and Cancer: Is Diabetes Causally Related to Cancer? Diabetes Metab. J. 2011, 35, 193–198. [Google Scholar] [CrossRef]
- Collins, K.K. The Diabetes-Cancer Link. Diabetes Spectr. 2014, 27, 276–280. [Google Scholar] [CrossRef]
- Kalra, S.; Bahendeka, S.; Sahay, R.; Ghosh, S.; Md, F.; Orabi, A.; Ramaiya, K.; Shammari, S.; Shrestha, D.; Shaikh, K.; et al. Consensus recommendations on sulfonylurea and sulfonylurea combinations in the management of type 2 diabetes mellitus—International Task Force. Indian J. Endocrinol. Metab. 2018, 22, 132–157. [Google Scholar] [CrossRef]
- Aubrey, A.B.; Aguilar-Bryan, L.; Nelson, D.A. Molecular mechanisms of action of glyburide on the beta cell. Am. J. Med. 1990, 89, 3S–10S. [Google Scholar] [CrossRef]
- Alkhalil, M.; Al-Hiari, Y.; Kasabri, V.; Arabiyat, S.; Al-Zweiri, M.; Mamdooh, N.; Telfah, A. Selected pharmacotherapy agents as antiproliferative and anti-inflammatory compounds. Drug Dev. Res. 2020, 81, 470–490. [Google Scholar] [CrossRef]
- Subramaniyam, N.; Arumugam, S.; Ezthupurakkal, P.B.; Ariraman, S.; Biswas, I.; Muthuvel, S.K.; Balakrishnan, A.; Alshammari, G.M.; Chinnasamy, T. Unveiling anticancer potential of glibenclamide: Its synergistic cytotoxicity with doxorubicin on cancer cells. J. Pharm. Biomed. Anal. 2018, 154, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Kmita, H.; Stobienia, O.; Michejda, J. The access of metabolites into yeast mitochondria in the presence and absence of the voltage dependent anion selective channel [YVDAC1]. Acta Biochim. Pol. 1999, 46, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Wonderlin, W.F.; Woodfork, K.A.; Strobl, J.S. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J. Cell. Physiol. 1995, 165, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Núñez, M.; Medina, V.; Cricco, G.; Croci, M.; Cocca, C.; Rivera, E.; Bergoc, R.; Martín, G. Glibenclamide inhibits cell growth by inducing G0/G1 arrest in the human breast cancer cell line MDA-MB-231. BMC Pharmacol Toxicol. 2013, 14, 6. [Google Scholar] [CrossRef]
- Abdul, M.; Hoosein, N. Expression and activity of potassium ion channels in human prostate cancer. Cancer Lett. 2002, 186, 99–105. [Google Scholar] [CrossRef]
- Ru, Q.; Tian, X.; Wu, Y.-X.; Wu, R.-H.; Pi, M.-S.; Li, C.-Y. Voltage-gated and ATP-sensitive K+ channels are associated with cell proliferation and tumorigenesis of human glioma. Oncol. Rep. 2014, 31, 842–848. [Google Scholar] [CrossRef]
- Zúñiga, L.; Cayo, A.; González, W.; Vilos, C.; Zúñiga, R. Potassium Channels as a Target for Cancer Therapy: Current Perspectives. OncoTargets Ther. 2022, 15, 783–797. [Google Scholar] [CrossRef]
- Li, M.; Tian, P.; Zhao, Q.; Ma, X.; Zhang, Y. Potassium channels: Novel targets for tumor diagnosis and chemoresistance. Front. Oncol. 2023, 12, 1074469. [Google Scholar] [CrossRef]
- Villalonga, N.; Ferrere, J.C.; Argiles, J.M.; Condom, E.; Felipe, A. Potassium Channels are a New Target Field in Anticancer Drug Design. Recent Pat. Anticancer Drug Discov. 2007, 2, 212–223. [Google Scholar] [CrossRef]
- Zhou, Q.; Kwan, H.Y.; Chan, H.C.; Jiang, J.L.; Tam, S.C.; Yao, X. Blockage of voltage-gated K+ channels inhibits adhesion and proliferation of hepatocarcinoma cells. Int. J. Mol. Med. 2003, 11, 261–266. [Google Scholar] [CrossRef]
- Suzuki, Y.; Inoue, T.; Murai, M.; Suzuki-Karasaki, M.; Ochiai, T.; Ra, C. Depolarization potentiates TRAIL-induced apoptosis in human melanoma cells: Role for ATP-sensitive K+ channels and endoplasmic reticulum stress. Int. J. Oncol. 2012, 41, 465–475. [Google Scholar] [CrossRef]
- Pasello, G.; Urso, L.; Conte, P.; Favaretto, A. Effects of sulfonylureas on tumor growth: A review of the literature. Oncologist 2013, 18, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.E.; Boom, A.; van Geffel, J.; Jacobs, P.; Masereel, B.; Beauwens, R. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch. 1999, 437, 652–660. [Google Scholar]
- Payen, L.; Delugin, L.; Courtois, A.; Trinquart, Y.; Guillouzo, A.; Fardel, O. The sulphonylurea glibenclamide inhibits multidrug resistance protein (MRP1) activity in human lung cancer cells. Br. J. Pharmacol. 2001, 132, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Cocca, C.; Martín, G.; Núñez, M.; Gutiérrez, A.; Cricco, G.; Mohamad, N.; Medina, V.; Croci, M.; Crescenti, E.; Rivera, E.; et al. Effect of glibenclamide on N-nitroso-N-methylurea-induced mammary tumors in diabetic and nondiabetic rats. Oncol. Res. 2005, 15, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Faridi, U.; Al-Mutairi, F.; Parveen, H.; Khateeb, S. An in-vitro and in Silico Anticancer Study of FDA Approved Antidiabetic Drugs Glimepiride and Empagliflozin: Life Sciences-Biochemistry for better diagnosis and therapy. Int. J. Life Sci. Biotechnol. Pharma Res. 2022, 10, 52–57. [Google Scholar] [CrossRef]
- Szymczak-Pajor, I.; Fleszar, K.; Kasznicki, J.; Gralewska, P.; Śliwińska, A. A potential role of calpains in sulfonylureas (SUs)-mediated death of human pancreatic cancer cells (1.2B4). Toxicol. Vitro 2021, 73, 105128. [Google Scholar] [CrossRef]
- Long, L.; Hu, X.; Li, X.; Zhou, D.; Shi, Y.; Wang, L.; Zeng, H.; Yu, X.; Zhou, W. The Anti-Breast Cancer Effect and Mechanism of Glimepiride-Metformin Adduct. OncoTargets Ther. 2020, 13, 3777–3788. [Google Scholar] [CrossRef]
- Kang, H.; Lee, S.; Kim, K.; Jeon, J.; Kang, S.; Youn, H.; Kim, H.Y.; Youn, B.H. Downregulated CLIP3 induces radioresistance by enhancing stemness and glycolytic flux in glioblastoma. J. Exp. Clin. Cancer Res. 2021, 40, 282. [Google Scholar] [CrossRef]
- Zhan, Z.-T.; Liu, L.; Cheng, M.-Z.; Gao, Y.; Zhou, W.-J. The Effects of 6 Common Antidiabetic Drugs on Anti-PD1 Immune Checkpoint Inhibitor in Tumor Treatment. J. Immunol. Res. 2022, 2022, 2651790. [Google Scholar] [CrossRef]
- Cairns, L.V.; Lappin, K.M.; Mutch, A.; Ali, A.; Matchett, K.B.; Mills, K.I. Multiplex Screening for Interacting Compounds in Paediatric Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2021, 22, 10163. [Google Scholar] [CrossRef]
- Qi, C.; Zhou, Q.; Li, B.; Yang, Y.; Cao, L.; Ye, Y.; Li, J.; Ding, Y.; Wang, H.; Wang, J.; et al. Glipizide, an antidiabetic drug, suppresses tumor growth and metastasis by inhibiting angiogenesis. Oncotarget 2014, 5, 9966–9979. [Google Scholar] [CrossRef]
- Gu, Q.; Wang, C.; Wang, G.; Han, Z.; Li, Y.; Wang, X.; Li, J.; Qi, C.; Xu, T.; Yang, X.; et al. Glipizide suppresses embryonic vasculogenesis and angiogenesis through targeting natriuretic peptide receptor A. Exp. Cell Res. 2015, 333, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Cevik, M.; Caker, S.; Deliorman, G.; Cagatay, P.; Gunduz, M.K.; Susleyici, B. The effects of glipizide on DNA damage and nuclear transport in differentiated 3T3-L1 adipocytes. Mol. Biol. Rep. 2022, 49, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Zheng, S.; Li, J.; Liu, X.; Zhou, Q.; Cao, J.; Zhang, Q.; Zheng, L.; Wang, L.; Qi, C. Glipizide Combined with ANP Suppresses Breast Cancer Growth and Metastasis by Inhibiting Angiogenesis through VEGF/VEGFR2 Signaling. Anticancer Agents Med. Chem. 2022, 22, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Nazim, U.M.D.; Moon, J.-H.; Lee, Y.-J.; Seol, J.-W.; Kim, Y.J.; Park, S.-Y. Glipizide sensitizes lung cancer cells to TRAIL-induced apoptosis via Akt/mTOR/autophagy pathways. Oncotarget 2017, 8, 100021–100033. [Google Scholar] [CrossRef]
- Hendriks, A.M.; Schrijnders, D.; Kleefstra, N.; de Vries, E.; Bilo, H.J.G.; Jalving, M.; Landman, G.W.D. Sulfonylurea derivatives and cancer, friend or foe? Eur. J. Pharmacol. 2019, 861, 172598. [Google Scholar] [CrossRef]
- Owen, S.C.; Doak, A.K.; Wassam, P.; Shoichet, M.S.; Shoichet, B.K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012, 7, 1429–1435. [Google Scholar] [CrossRef]
- Seedher, N.; Kanojia, M. Co-solvent solubilization of some poorly-soluble antidiabetic drugs. Pharm. Dev. Technol. 2009, 14, 185–192. [Google Scholar] [CrossRef]
- Rajesh, S.Y.; Singh, S.K.; Pandey, N.K.; Sharma, P.; Bawa, P.; Kumar, B.; Gulati, M.; Jain, S.K.; Gowthamarajan, K.; Singh, S. Impact of various solid carriers and spray drying on pre/post compression properties of solid SNEDDS loaded with glimepiride: In vitro-ex vivo evaluation and cytotoxicity assessment. Drug Dev. Ind. Pharm. 2018, 44, 1056–1069. [Google Scholar] [CrossRef]
- Pal, A.; Roy, S.; Kumar, A.; Mahmood, S.; Khodapanah, N.; Thomas, S.; Agatemor, C.; Ghosal, K. Physicochemical Characterization, Molecular Docking, and In Vitro Dissolution of Glimepiride-Captisol Inclusion Complexes. ACS Omega 2020, 32, 19968–19977. [Google Scholar] [CrossRef]
- Shukla, U.A.; Chi, E.M.; Lehr, K.-H. Glimepiride Pharmacokinetics in Obese Versus Non-Obese Diabetic Patients. Ann. Pharmacother. 2004, 38, 30–35. [Google Scholar] [CrossRef]
- Badian, M.; Korn, A.; Lehr, K.-H.; Malerczyk, V.; Waldhäusl, W. Absolute bioavailability of glimepiride (Amaryl) after oral administration. Drug Metabol. Drug Interact. 1994, 11, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.-Y.; Park, H.-C.; Kang, W.; Kwon, K.-I. Pharmacokinetic and pharmacodynamic modelling of the effects of glimepiride on insulin secretion and glucose lowering in healthy humans. J. Clin. Pharm. Ther. 2006, 31, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zheng, X.; Hage, D.S. Binding studies based on ultrafast affinity extraction and single- or two-column systems: Interactions of second- and third-generation sulfonylurea drugs with normal or glycated human serum albumin. J. Chromatogr. B 2018, 1102–1103, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Miro, A.; Quaglia, F.; Sorrentino, U.; Rotonda, M.L.; Bianca, R.D.D.V.; Sorrentino, R. Improvement of gliquidone hypoglycaemic effect in rats by cyclodextrin formulations. Eur. J. Pharm. Sci. 2004, 23, 57–64. [Google Scholar] [CrossRef]
- Jia, L.; Yu, Y.; Wang, M.; Ouyang, H.; Gong, J. Preparation and characterization for multicomponent crystals of the antidiabetic drug gliquidone based on crystal engineering. CrystEngComm 2019, 21, 1617–1625. [Google Scholar] [CrossRef]
- Kopitar, Z.; Koss, F.W. Pharmakokinetisches Verhalten von Gliquidone (AR-DF 26), einem neuen Sulfonylharnstoff (Zusammenfassung der bisherigen Untersuchungen) [Pharmacokinetic behaviour of gliquidone (AR-DF 26), a new sulfonyl urea. Summary of the studies so far (author’s transl)]. Arzneimittelforschung 1975, 25, 1933–1938. [Google Scholar]
- Chadha, R.; Rani, D.; Goyal, P. Novel cocrystals of gliclazide: Characterization and evaluation. CrystEngComm 2016, 18, 2275–2283. [Google Scholar] [CrossRef]
- Samie, A.; Desiraju, G.R.; Banik, M. Salts and Cocrystals of the Antidiabetic Drugs Gliclazide, Tolbutamide, an Glipizide: Solubility Enhancements through Drug–Coformer Interactions. Cryst. Growth Des. 2017, 17, 2406–2417. [Google Scholar] [CrossRef]
- Ibrahim, A.Y.; El-Malah, Y.; Abourehab, M.A.S.; Aldawsari, H. Solubility Enhancement of Gliclazide via Co-crystallization with Malonic Acid. Life Sci. J. 2019, 16, 49–53. [Google Scholar] [CrossRef]
- Davis, T.M.E.; Daly, F.; Walsh, J.P.; Ilett, K.F.; Beilby, J.P.; Dusci, L.J.; Barrett, P.H.R. Pharmacokinetics and pharmacodynamics of gliclazide in Caucasians and Australian Aborigines with type 2 diabetes. Br. J. Clin. Pharmacol. 2000, 49, 223–230. [Google Scholar] [CrossRef]
- Campbell, D.B.; Lavielle, R.; Nathan, C. The mode of action and clinical pharmacology of gliclazide: A review. Diabetes Res. Clin. Pract. 1991, 14, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Matsuda, R.; Hage, D.S. Analysis of free drug fractions by ultrafast affinity extraction: Interactions of sulfonylurea drugs with normal or glycated human serum albumin. J. Chromatogr. A 2014, 1371, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Poddar, S.; Woolfork, A.G.; Iftekhar, S.; Ovbude, S.T.; Hage, D.S. Characterization of binding by sulfonylureas with normal or modified human serum albumin using affinity microcolumns prepared by entrapment. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2023, 1226, 123798. [Google Scholar] [CrossRef] [PubMed]
- Goya, P.; Ran, D.; Chadha, R. Crystal Engineering: A Remedy to Tailor the Biopharmaceutical Aspects of Glibenclamide. Cryst. Growth Des. 2018, 18, 105–118. [Google Scholar] [CrossRef]
- Sartor, G.; Melander, A.; Scherstén, B.; Wåhlin-Boll, E. Comparative Single-Dose Kinetics and Effects of Four Sulfonylureas in Healthy Volunteers. Acta Med. Scand. 1980, 208, 301–307. [Google Scholar] [CrossRef]
- Jaber, L.A.; Antal, E.J.; Welshman, I.R. Pharmacokinetics and Pharmacodynamics of Glyburide in Young and Elderly Patients with Non-Insulin-Dependent Diabetes Mellitus. Ann. Pharmacother. 1996, 30, 472–475. [Google Scholar] [CrossRef]
- Shaheen, O.; Othman, S.; Jalal, I.; Awidi, A.; Al-Turk, W. Comparison of pharmacokinetics and pharmacodynamics of a conventional and a new rapidly dissolving glibenclamide preparation. Int. J. Pharm. 1987, 38, 123–131. [Google Scholar] [CrossRef]
- Batisai, E. Solubility Enhancement of Antidiabetic Drugs Using a Co-Crystallization Approach. ChemistryOpen 2021, 10, 1260–1268. [Google Scholar] [CrossRef]
- Pandey, N.K.; Sehal, H.R.; Garg, V.; Gaur, T.; Kumar, B.; Singh, S.K.; Gulati, M.; Gowthamarajan, K.; Bawa, P.; Rajesh, S.Y.; et al. Stable Co-Crystals of Glipizide with Enhanced Dissolution Profiles: Preparation and Characterization. AAPS PharmSciTech 2017, 18, 2454–2465. [Google Scholar] [CrossRef]
- Kradjan, W.A.; Kobayashi, K.A.; Bauer, L.A.; Horn, J.R.; Opheim, K.E.; Wood, F.J. Glipizide Pharmacokinetics: Effects of Age, Diabetes, and Multiple Dosing. J. Clin. Pharmacol. 1989, 29, 1121–1127. [Google Scholar] [CrossRef]
- Jaber, L.A.; Antal, E.J.; Slaughter, R.L.; Welshman, I.R. The pharmacokinetics and pharmacodynamics of 12 weeks of glyburide therapy in obese diabetics. Eur. J. Clin. Pharmacol. 1993, 45, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Yalkowsky, S.H.; Dannenfelser, R.M. The AQUASOL dATAbASE of Aqueous Solubility, 5th ed.; University of Arizona, College of Pharmacy: Tucson, AZ, USA, 1992. [Google Scholar]
- Ridolfo, A.S.; Kirtley, W.R. Clinical experiences with carbutamide, an orally given hypoglycemic agent; preliminary report. JAMA 1956, 160, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Sarraguça, M.C.; Ribeiro, P.R.S.; Nunes, C.; Seabra, C.L. Solids Turn into Liquids—Liquid Eutectic Systems of Pharmaceutics to Improve Drug Solubility. Pharmaceuticals 2022, 15, 279. [Google Scholar] [CrossRef] [PubMed]
- Dodge, A.; Gould, P.L. Dissolution of Chlorpropamide Tablets in A Methanol-Water Binary Solvent System. Drug Dev. Ind. Pharm. 1987, 13, 1817–1826. [Google Scholar] [CrossRef]
- Fadda, H.M.; Chen, X.; Aburub, A.; Mishra, D.; Pinal, R. A Novel Method for Determining the Solubility of Small Molecules in Aqueous Media and Polymer Solvent Systems Using Solution Calorimetry. Pharm. Res. 2014, 31, 1735–17453. [Google Scholar] [CrossRef]
- Taylor, J.A. Pharmacokinetics and biotransformation of chlorpropamide in man. Clin. Pharmacol. Ther. 1972, 13, 710–718. [Google Scholar] [CrossRef]
- Kuldipkumar, A.; Tan, Y.T.F.; Goldstein, M.; Nagasaki, Y.; Zhang, G.G.Z.; Kwon, G.S. Amphiphilic Block Copolymer as a Crystal Habit Modifier. Cryst. Growth Des. 2005, 5, 1781–1785. [Google Scholar] [CrossRef]
- Welling, P.G.; Patel, R.; Patel, U.R.; Gillespie, W.R.; Craig, W.A.; Albert, K.S. Bioavailability of tolazamide from tablets: Comparison of in vitro and in vivo results. J. Pharm. Sci. 1982, 71, 1259–1263. [Google Scholar] [CrossRef]
- Proks, P.; Kramer, H.; Haythorne, E.; Ashcroft, F.M. Binding of sulphonylureas to plasma proteins—A KATP channel perspective. PLoS ONE 2018, 13, e0197634. [Google Scholar] [CrossRef]
- Pawar, S.; Joshi, R.; Ottoor, D. Spectroscopic and molecular docking study to understand the binding interaction of rosiglitazone with bovine serum albumin in presence of valsartan. J. Lumin. 2018, 197, 200–210. [Google Scholar] [CrossRef]
- Ma, Y.; Acosta, D.M.; Whitney, J.R.; Podgornik, R.; Steinmetz, N.F.; French, R.H.; Parsegian, V.A. Determination of the second virial coefficient of bovine serum albumin under varying pH and ionic strength by composition-gradient multi-angle static light scattering. J. Biol. Phys. 2015, 41, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Rout, D.; Sharma, S.; Agarwala, P.; Upadhyaya, A.K.; Sharma, A.; Sasmal, D.K. Interaction of Ibuprofen with Partially Unfolded Bovine Serum Albumin in the Presence of Ionic Micelles and Oligosaccharides at Different λex and pH: A Spectroscopic Analysis. ACS Omega 2023, 8, 3114–3128. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.F.; Hsiao, C.L. Qualitative study of the competition of drugs in binding to serum albumin. Chromatographia 1993, 37, 329–335. [Google Scholar] [CrossRef]
- Shingada, S.R.; Ali, P.S.; Gandhare, N.V.; Pathan, N.B.; Ansari, N.H. Investigation of mechanistic interactions between Rifampicin and bovine serum albumin in the presence of different surfactants. J. Dispers. Sci. Technol. 2021, 44, 1075–1084. [Google Scholar] [CrossRef]
- Tsujino, I.; Yamazaki, T.; Masutani, M.; Sawada, U.; Horie, T. Effect of Tween-80 on cell killing by etoposide in human lung adenocarcinoma cells. Cancer Chemother. Pharmacol. 1999, 43, 29–34. [Google Scholar] [CrossRef]
- Pokhrel, D.R.; Sah, M.K.; Gautam, B.; Basak, H.K.; Bhattarai, A.; Chatterjee, A. A recent overview of surfactant–drug interactions and their importance. RSC Adv. 2023, 13, 17685–17704. [Google Scholar] [CrossRef]
- Jwalapuram, R.; Ahad, H.A.; Haranath, C.; Thadipatri, R.; Varshitha, C.V.H.; Kumar, Y.B. Solubility Enhancement of Drugs with Aid of Surfactants: Research Done Since Last Two Decades. Int. J. Life Sci. Biotechnol. Pharma Res. 2020, 5, 11–16. [Google Scholar] [CrossRef]
- Obata, T.; Suzuki, Y.; Ogawa, N.; Kurimoto, I.; Yamamoto, H.; Furuno, T.; Sasaki, T.; Tanaka, M. Improvement of the Antitumor Activity of Poorly Soluble Sapacitabine (CS-682) by Using Soluplus® as a Surfactant. Biol. Pharm. Bull. 2014, 37, 802–807. [Google Scholar] [CrossRef]
- Lo, Y.L.; Hsu, C.Y.; Huang, J.D. Comparison of effects of surfactants with other MDR reversing agents on intracellular uptake of epirubicin in Caco-2 cell line. Anticancer Res. 1998, 18, 3005–3009. [Google Scholar]
- Bansal, G.; Singh, M.; Jindal, K.C.; Singh, S. Characterization of Mass Ionizable Degradation Products of Gliclazide by LC/ESI-MS. J. Liq. Chromatogr. Relat. 2008, 31, 2174–2193. [Google Scholar] [CrossRef]
- Vargas, F.; Méndez, H.; Tropper, E.; Velázquez, M.; Fraile, G. Studies on the in vitro phototoxicity of the antidiabetes drug glipizide. In Vitr. Mol. Toxicol. 2000, 13, 17–24. [Google Scholar] [PubMed]
- Chou, T.-C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma. 2008, 49, 2059–2080. [Google Scholar] [CrossRef] [PubMed]
- Piao, W.; Yoo, H.; Lee, D.K.; Hwang, H.J.; Kim, J.H. Induction of G(2)/M phase arrest and apoptosis by a new synthetic anti-cancer agent, DW2282, in promyelocytic leukemia (HL-60) cells. Biochem. Pharmacol. 2001, 62, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Drabkin, D.L. The Standardization of Hemoglobin Measurement. Am. J. Med. Sci. 1949, 217, 710–711. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, S.V. Erythrocyte morphological states, phases, transitions and trajectories. Biochim. Biophys. Acta 2010, 1798, 1767–1778. [Google Scholar] [CrossRef]
- Zemlianskykh, N.G. The Effect of Cryoprotective Agents on Proteins of the Erythrocyte Membrane-Cytoskeleton Complex. Biochem. (Mosc.) Suppl. B Biomed. Chem. 2021, 15, 132–138. [Google Scholar] [CrossRef]
- Yi, X.; Liu, M.; Luo, Q.; Zhuo, H.; Cao, H.; Wang, J.; Han, Y. Toxic effects of dimethyl sulfoxide on red blood cells, platelets, and vascular endothelial cells in vitro. FEBS Open Bio 2017, 7, 485–494. [Google Scholar] [CrossRef]
- Ansel, H.C.; Leake, W.F. Hemolysis of erythrocytes by antibacterial preservatives III: Influence of dimethyl sulfoxide on the hemolytic activity of phenol. J. Pharm. Sci. 1966, 55, 685–688. [Google Scholar] [CrossRef]
- Drvenica, I.; Mojsilović, S.; Stančić, A.; Marković, D.; Kovačić, M.; Maslovarić, I.; Rapajić, I.; Vučetić, D.; Ilić, V. The effects of incubation media on the assessment of the shape of human erythrocytes by flow cytometry: A contribution to mathematical data interpretation to enable wider application of the method. Eur. Biophys. J. 2021, 50, 829–846. [Google Scholar] [CrossRef]
- Khan, M.A.; Aljarbou, N.A.; Aldebasi, H.Y.; Alorainy, S.M.; Khan, A. Combination of glycosphingosomes and liposomal doxorubicin shows increased activity against dimethyl-α-benzanthracene-induced fibrosarcoma in mice. Int. J. Nanotechnol. 2015, 2015, 6331–6338. [Google Scholar] [CrossRef]
- Khan, I.; Joshi, G.; Nakhate, K.T.; Ajazuddin Kumar, R.; Gupta, U. Nano-Co-Delivery of Berberine and Anticancer Drug Using PLGA Nanoparticles: Exploration of Better Anticancer Activity and In Vivo Kinetics. Pharm. Res. 2019, 36, 149. [Google Scholar] [CrossRef] [PubMed]
- Meloni, G.; Meloni, T. Glyburide-induced acute haemolysis in a G6PD-deficient patient with NIDDM. Br. J. Haematol. 1996, 92, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Kopicky, J.A.; Packman, C.H. The mechanisms of sulfonylurea-induced immune hemolysis: Case report and review of the literature. Am. J. Hematol. 1986, 23, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Burden, A.C.; Samanta, A.K. Relationship of Maternal and Fetal Outcome to Glucose Tolerance During Pregnancy in Whites and Indian Asians. Diabetes Care 1990, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.W.; Eeles, G.H.; Litchfield, J.A.; Rahman, M.; Wingham, J. Haemolytic anaemia associated with antibodies to tolbutamide and phenacetin. Br. Med. J. 1972, 1, 728–729. [Google Scholar] [CrossRef]
- Mori, Y.; Komiya, H.; Kurokawa, N.; Tajima, N. Comparison of the effects of glimepiride and glibenclamide on adipose tissue tumour necrosis factor-α mRNA expression and cellularity. Diabetes Obes. Metab. 2003, 6, 28–34. [Google Scholar] [CrossRef]
- Ishola, I.O.; Akataobi, O.E.; Alade, A.A.; Adeyemi, O.O. Glimepiride prevents paraquat-induced Parkinsonism in mice: Involvement of oxidative stress and neuroinflammation. Fundam. Clin. Pharmacol. 2018, 33, 277–285. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Palagiano, F.; Saija, A.; Nava, F.; Trombetta, D.; de Caprariis, P. Synthesis, stability, and pharmacological evaluation of nipecotic acid prodrugs. J. Pharm. Sci. 1999, 88, 561–567. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J.; Paradowska, B.; Olejniczak, A.B.; Studzinska, M.; Seekamp, P.; Schüssler, U.; Gabel, D.; Shinazi, R.F.; Plesek, J. Towards new boron carriers for boron neutron capture therapy: Metallacarboranes and their nucleoside conjugates. Bioorg. Med. Chem. 2005, 13, 4168–4175. [Google Scholar] [CrossRef]
- Chou, T.C.; Martin, N. CompuSyn for Drug Combinations: PC Software and User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values; ComboSyn: Paramus, NJ, USA, 2005. [Google Scholar]
- Kauffman, M.K.; Kauffman, M.E.; Zhu, H.; Jia, Z.; Li, Y.R. Fluorescence-Based Assays for Measuring Doxorubicin in Biological Systems. React. Oxyg. Species 2016, 2, 432–439. [Google Scholar] [CrossRef]
- Seedher, N.; Kanojia, M. Mechanism of interaction of hypoglycemic agents glimepiride and glipizide with human serum albumin. Open Chem. 2009, 7, 96–104. [Google Scholar] [CrossRef]
- Rosenkranz, B.; Profozic, V.; Metelko, Z.; Mrzljak, V.; Lange, C.; Malerczyk, V. Pharmacokinetics and safety of glimepiride at clinically effective doses in diabetic patients with renal impairment. Diabetologia 1996, 39, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Dills, D.G.; Schneider, J. Clinical evaluation of glimepiride versus glyburide in NIDDM in a double-blind comparative study. Glimepiride/Glyburide Research Group. Horm. Metab. Res. 1996, 28, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Draeger, K.E.; Wernicke-Panten, K.; Lomp, H.J.; Schüler, E.; Rosskamp, R. Long-term treatment of type 2 diabetic patients with the new oral antidiabetic agent glimepiride (Amaryl): A double-blind comparison with glibenclamide. Horm. Metab. Res. 1996, 28, 419–425. [Google Scholar] [CrossRef]
- Joy, N.G.; Tate, D.B.; Davis, S.N. Counterregulatory responses to hypoglycemia differ between glimepiride and glyburide in non diabetic individuals. Metabolism 2015, 64, 729–737. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Maddock, H.L.; Baxter, G.F.; Lawrence, C.L.; Standen, N.B.; Yellon, D.M. Glimepiride, a novel sulfonylurea, does not abolish myocardial protection afforded by either ischemic preconditioning or diazoxide. Circulation 2001, 103, 3111–3116. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | IC50 Value in Specific Cell Line (µM) | |||||
|---|---|---|---|---|---|---|
| A549 | HepG2 | HMEC-1 | HuH7 | MCF-7 | U-87MG | |
| 2nd-generation SUs | ||||||
| GLIM | 367.7 ± 16.4 | 179.6 ± 21.6 | 135.5 ± 5.1 | 475.8 ± 7.0 | 180.3 ± 7.6 | 189.4 ± 14.8 |
| GLIQ | 259.1 ± 16.3 | 39.6 ± 2.6 | 156.6 ± 2.2 | 400.7 ± 2.2 | 68.8 ± 2.5 | 57.1 ± 1.8 |
| GLIC | 419.3 ± 53.2 | 447.7 ± 2.3 | n.d. | n.d. | ||
| GLIB | 46.1 ± 1.0 | 248.0 ± 3.6 | 493.2 ± 1.3 | 86.6 ± 3.2 | ||
| GLIP | 228.8 ± 10.9 | 454.1 ± 1.8 | 421.9 ± 0.5 | 339.0 ± 18.8 | ||
| 1st-generation SUs | ||||||
| CARB | 70.7 ± 1.4 | n.d. | n.d. | n.d. | ||
| CHLO | n.d. | 480.3 ± 54.1 | 218.2 ± 1.6 | n.d. | n.d. | n.d. |
| TOLA | 469.8 ± 12.4 | 468.6 ± 0.5 | n.d. | n.d. | ||
| TOLB | 232.2 ± 21.6 | 88.2 ± 4.1 | n.d. | 460.9 ± 53.9 | ||
| Other compounds | ||||||
| MESU | 63.1 ± 0.3 | 78.3 ± 2.4 | n.d. | 495.9 ± 0.8 | ||
| DOXO | 0.27 ± 0.02 | 0.30 ± 0.01 | 0.08 ± 0.01 | 0.28 ± 0.02 | 0.46 ± 0.06 | 0.57 ± 0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomczyk, M.D.; Matczak, K.; Denel-Bobrowska, M.; Dzido, G.; Kubicka, A.; Gendosz de Carrillo, D.; Cichoń, T.; Golec, M.; Powieczko, B.; Rzetelny, W.; et al. Combining Sulfonylureas with Anticancer Drugs: Evidence of Synergistic Efficacy with Doxorubicin In Vitro and In Vivo. Int. J. Mol. Sci. 2025, 26, 1429. https://doi.org/10.3390/ijms26041429
Tomczyk MD, Matczak K, Denel-Bobrowska M, Dzido G, Kubicka A, Gendosz de Carrillo D, Cichoń T, Golec M, Powieczko B, Rzetelny W, et al. Combining Sulfonylureas with Anticancer Drugs: Evidence of Synergistic Efficacy with Doxorubicin In Vitro and In Vivo. International Journal of Molecular Sciences. 2025; 26(4):1429. https://doi.org/10.3390/ijms26041429
Chicago/Turabian StyleTomczyk, Mateusz D., Karolina Matczak, Marta Denel-Bobrowska, Grzegorz Dzido, Anna Kubicka, Daria Gendosz de Carrillo, Tomasz Cichoń, Marlena Golec, Beata Powieczko, Waldemar Rzetelny, and et al. 2025. "Combining Sulfonylureas with Anticancer Drugs: Evidence of Synergistic Efficacy with Doxorubicin In Vitro and In Vivo" International Journal of Molecular Sciences 26, no. 4: 1429. https://doi.org/10.3390/ijms26041429
APA StyleTomczyk, M. D., Matczak, K., Denel-Bobrowska, M., Dzido, G., Kubicka, A., Gendosz de Carrillo, D., Cichoń, T., Golec, M., Powieczko, B., Rzetelny, W., Olejniczak, A. B., & Pérez-Sánchez, H. (2025). Combining Sulfonylureas with Anticancer Drugs: Evidence of Synergistic Efficacy with Doxorubicin In Vitro and In Vivo. International Journal of Molecular Sciences, 26(4), 1429. https://doi.org/10.3390/ijms26041429