
Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology



Abstract
:
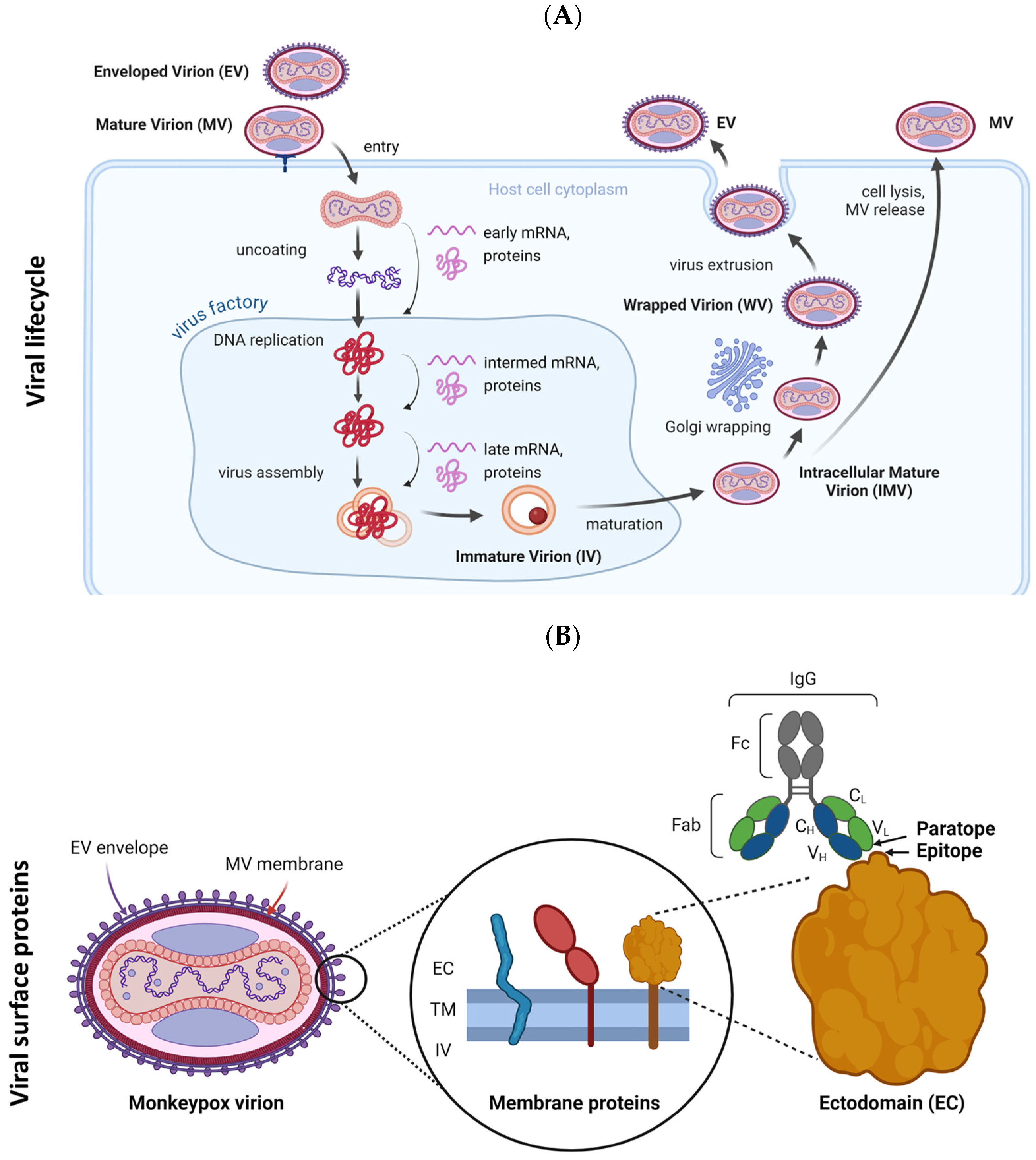
1. Introduction
2. Results
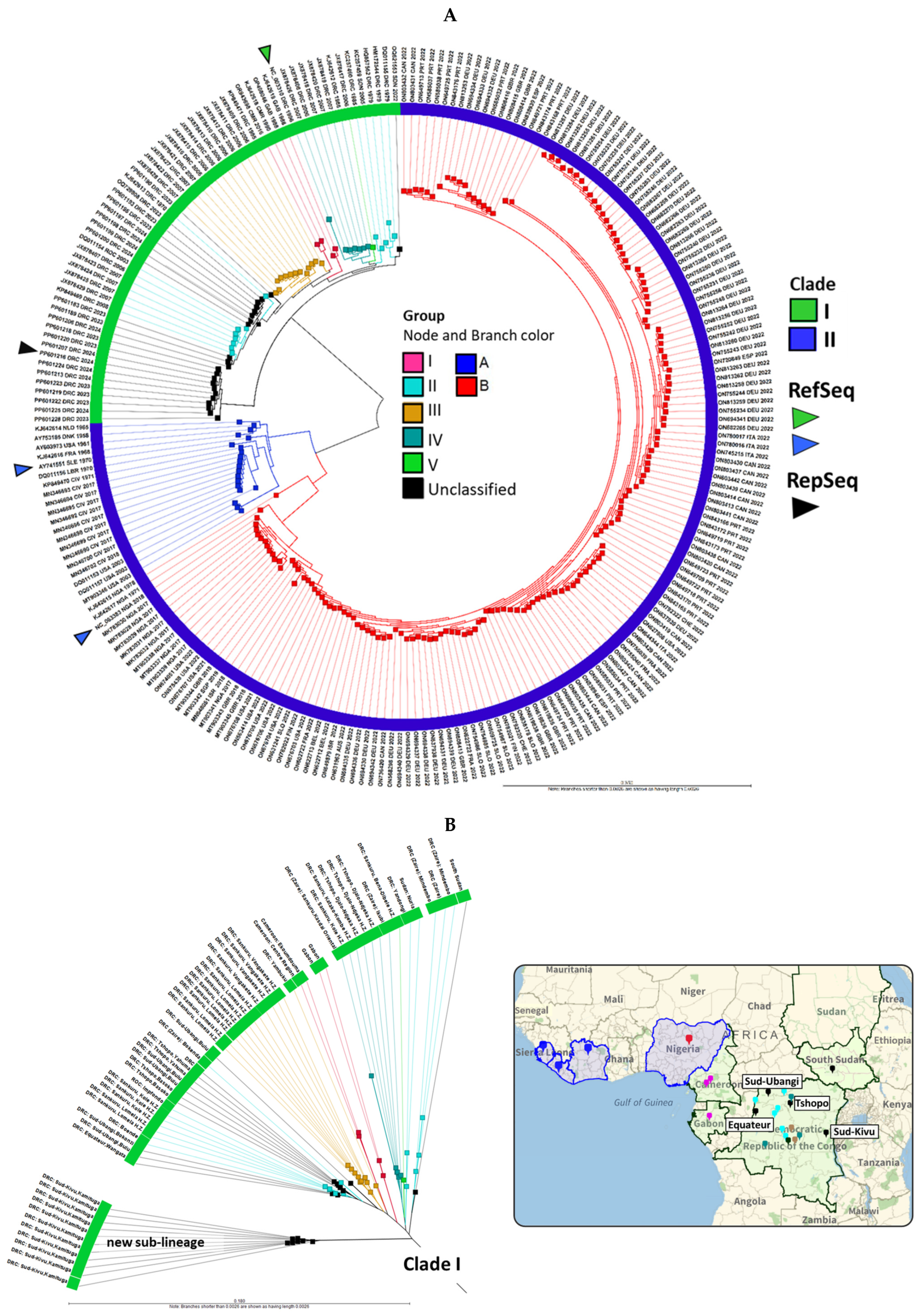
2.1. MPXV Genomes from 2022 to 2024 Clustered with Clades I and IIB, with a New Clade I Sub-Lineage Emerging from Sud-Kivu, DRC
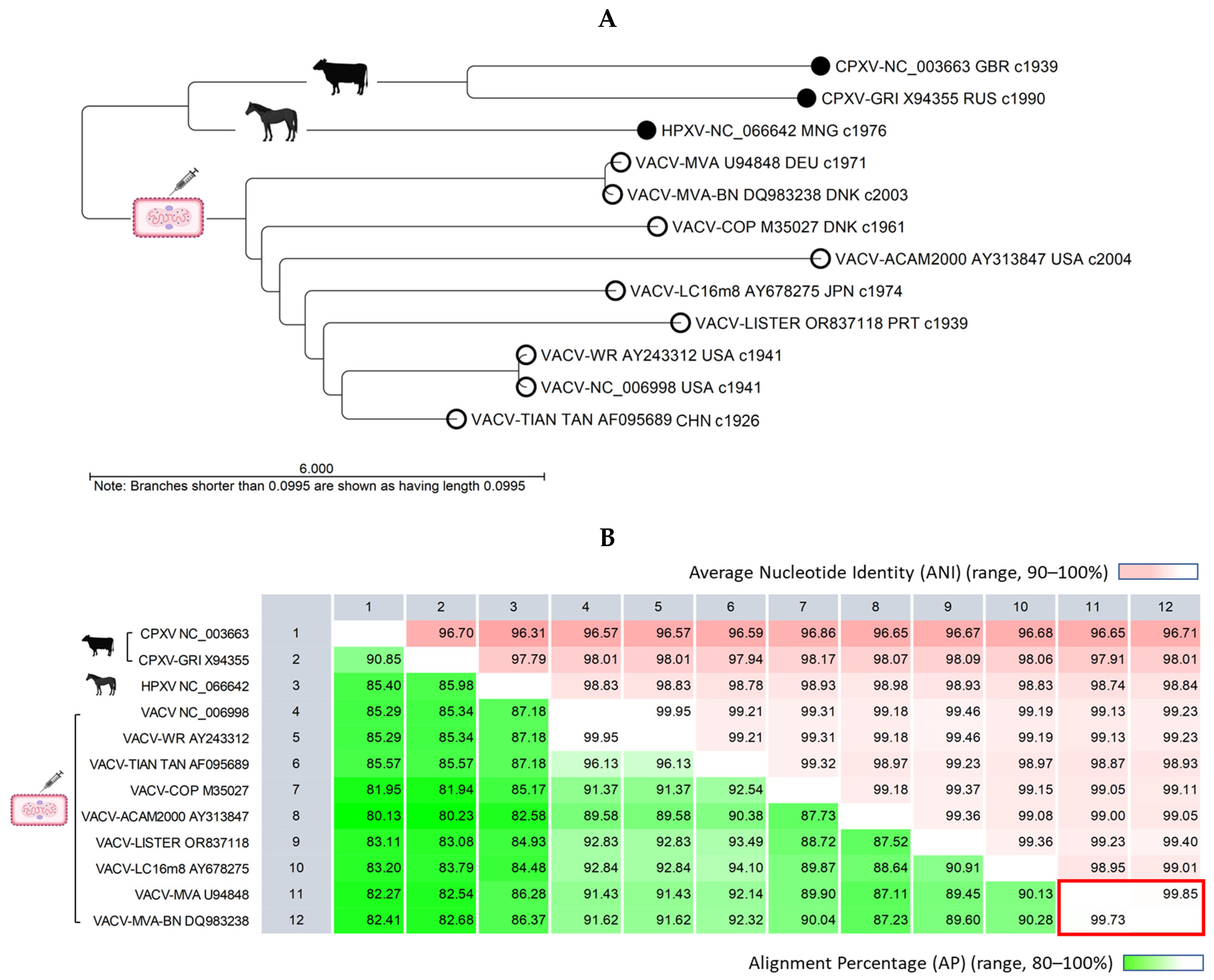
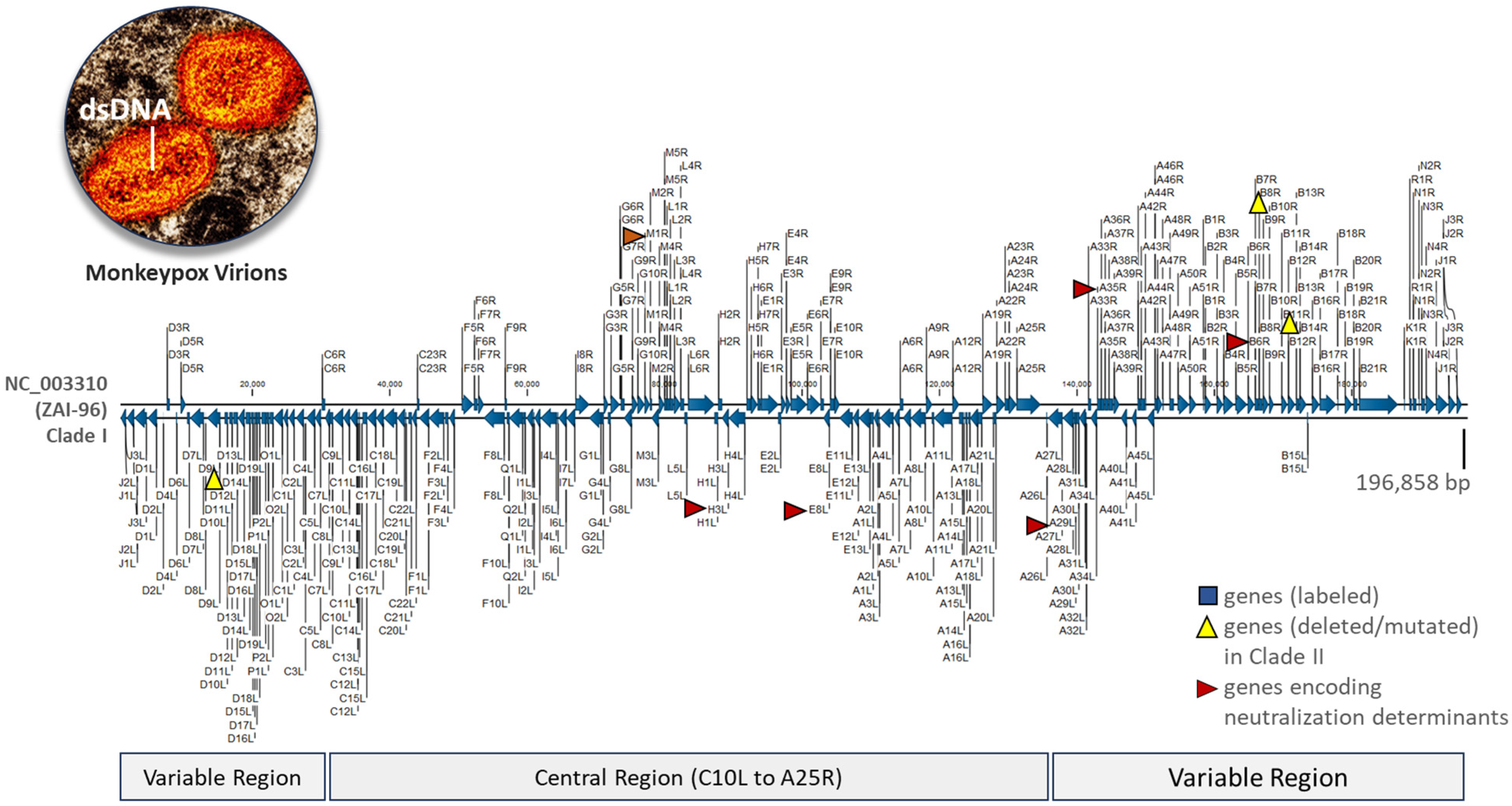
2.2. VACV Genomes Feature a Conserved Central Region and Variable Terminal Regions, Which Exhibit Rearrangements, Truncations, and Deletions in Later Generations of the Vaccine
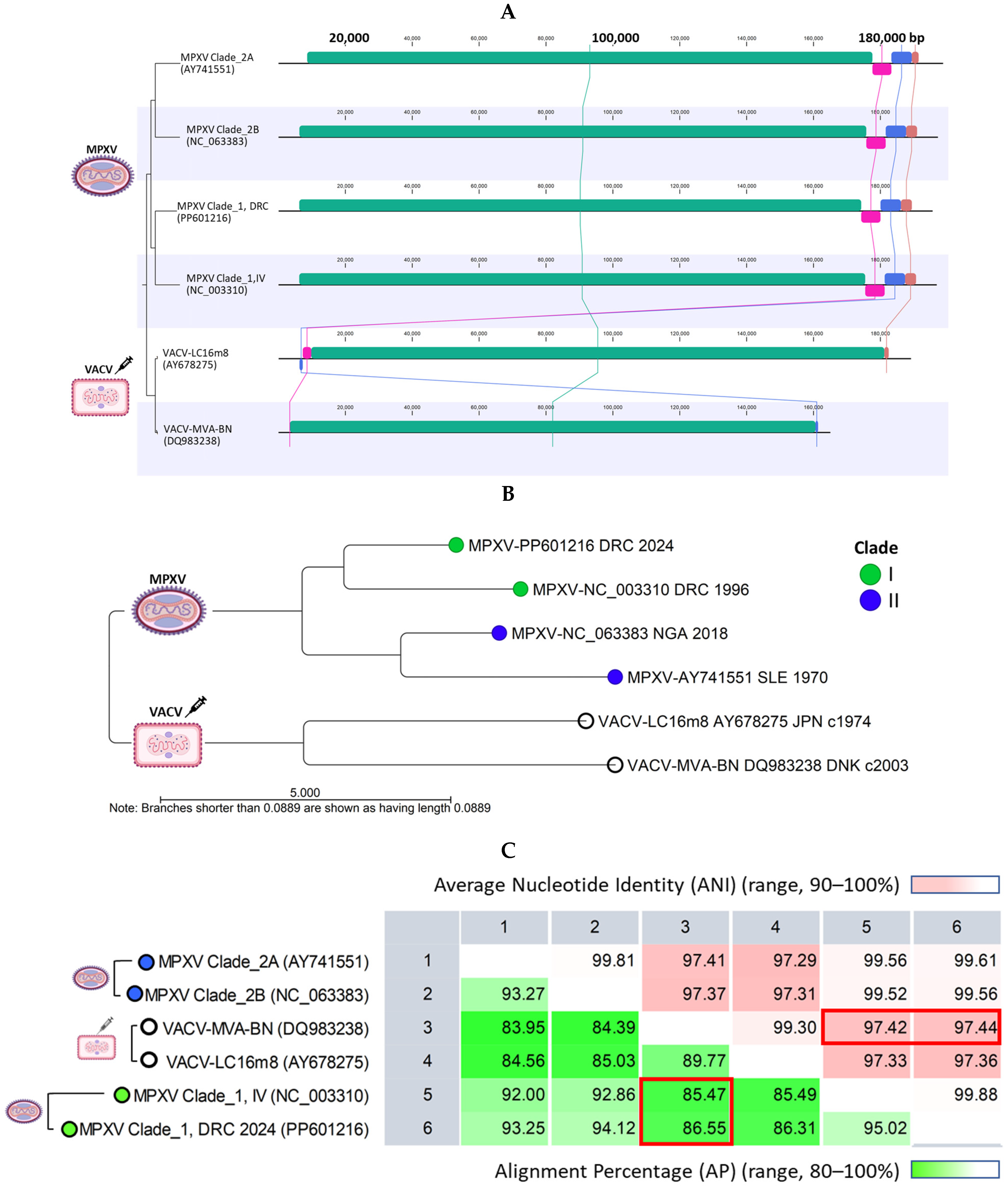
2.3. MPXV Clade I Genomes and Current VACV Vaccines Differ in Genome Lengths and Terminal Regions but Share over 85% Alignment Similarity
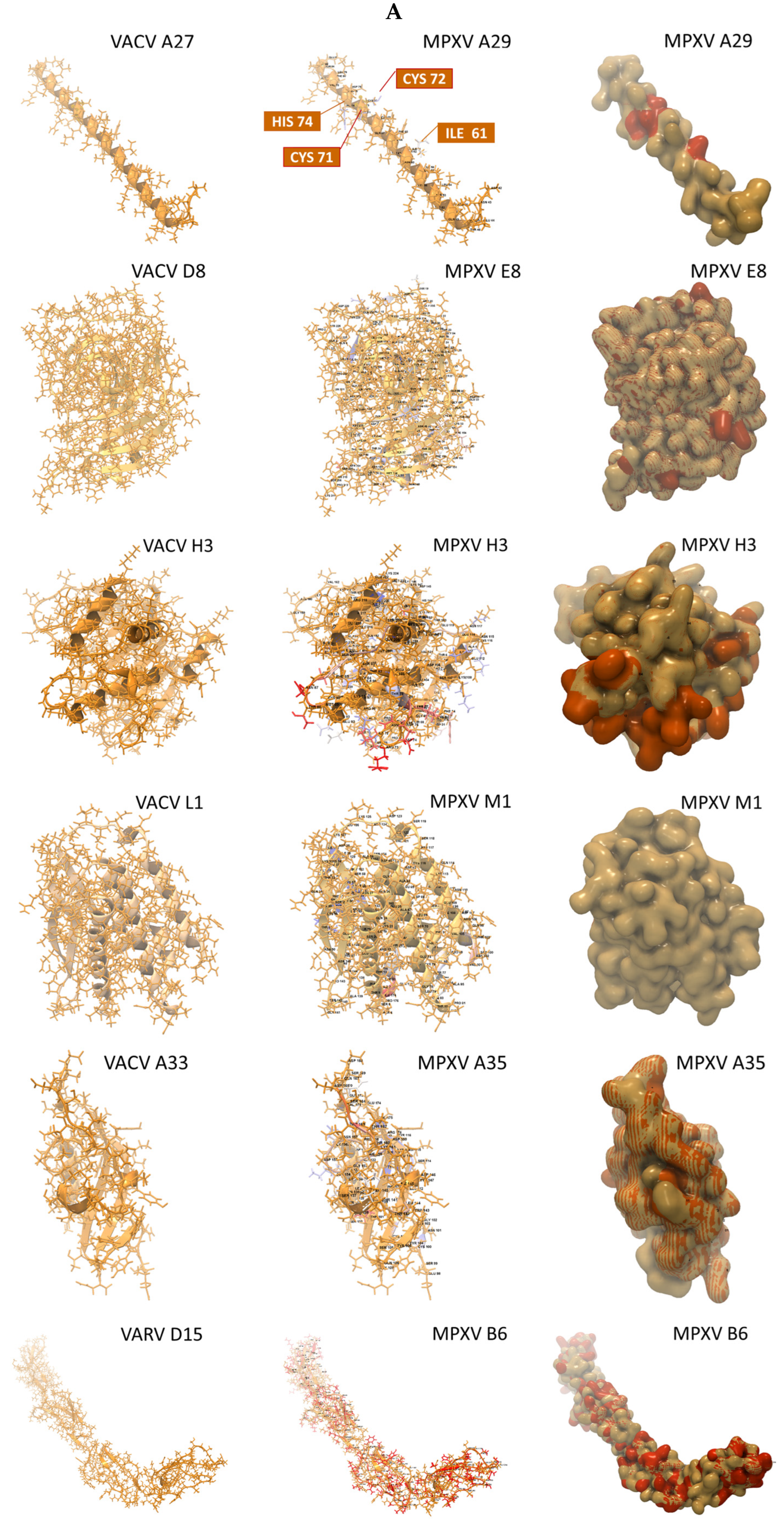
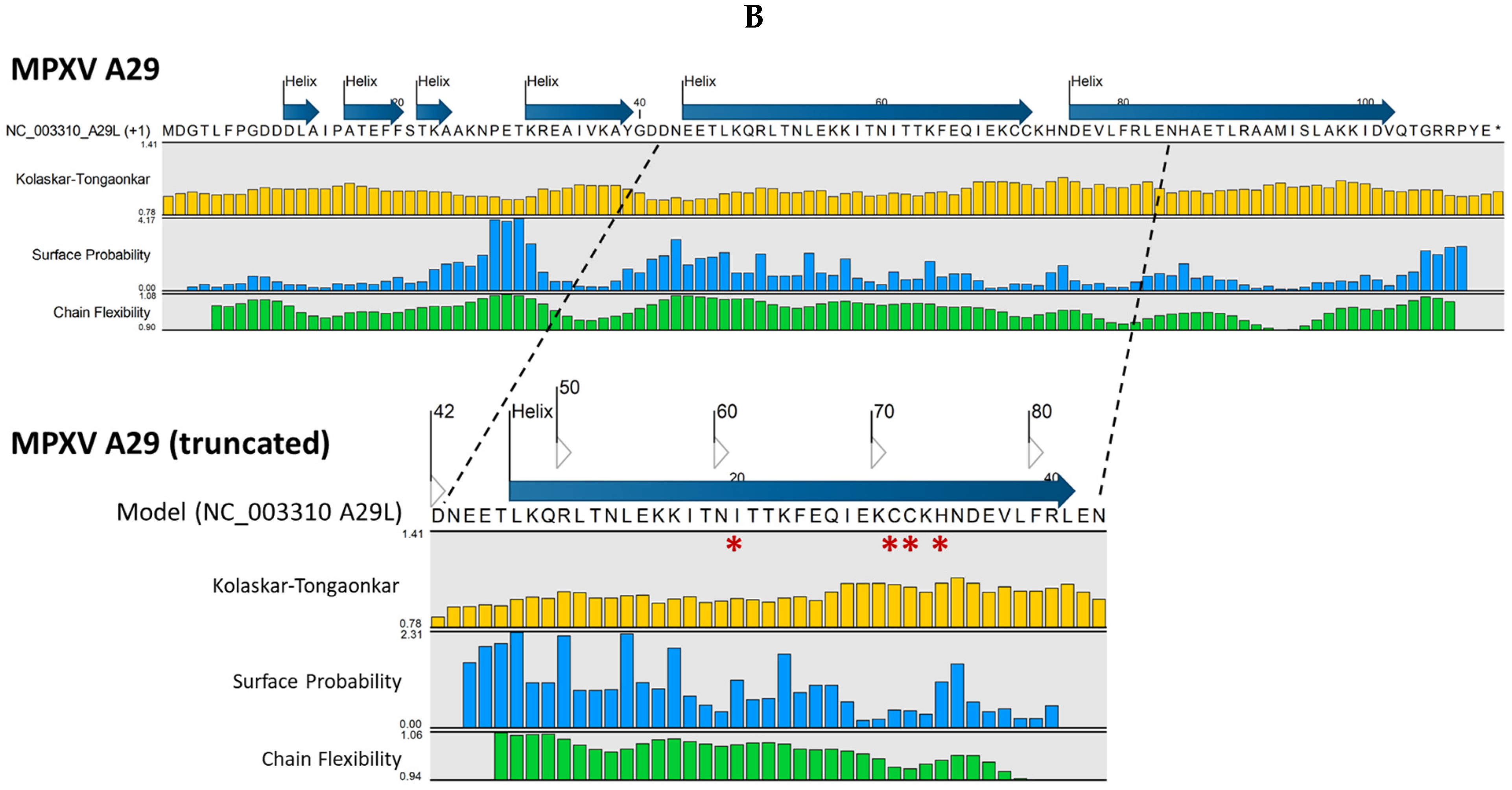
2.4. Variations in Sequence and Structure Between Homologous MPXV and VACV Antigens May Impact Epitope/Paratope Binding
3. Discussion
4. Materials and Methods
4.1. MPXV-Customized Reference Database and Genome Dataset (2022–2024)
4.2. Vaccinia Virus (VACV) Genome Dataset and Major Neutralization Determinants
4.3. Whole-Genome Alignment, Pairwise Comparison, and Phylogenomic Analysis
4.4. Antigenic Protein Structure Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Use of Artificial Intelligence
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization (WHO). Available online: https://www.who.int/news-room/speeches/item/who-director-general-s-opening-remarks-at-the-ihr-emergency-committee-meeting-regarding-the-upsurge-of-mpox-2024---14-august-2024 (accessed on 14 August 2024).
- World Health Organization (WHO). Smallpox and mpox (orthopoxviruses) vaccine position paper. Wkly. Epidemiol. Rec. 2024, 99, 429–456. [Google Scholar]
- Shchelkunov, S.N.; Totmenin, A.V.; Babkin, I.V.; Safronov, P.F.; Ryazankina, O.I.; Petrov, N.A.; Gutorov, V.V.; Uvarova, E.A.; Mikheev, M.V.; Sisler, J.R.; et al. Human monkeypox and smallpox viruses: Genomic comparison. FEBS Lett. 2001, 509, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Li, G.; Liszewski, M.K.; Atkinson, J.P.; Jahrling, P.B.; Feng, Z.; Schriewer, J.; Buck, C.; Wang, C.; Lefkowitz, E.J.; et al. Virulence differences between monkeypox virus isolates from West Africa and the Congo basin. Virology 2005, 340, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Berthet, N.; Descorps-Declère, S.; Besombes, C.; Curaudeau, M.; Meyong, A.A.N.; Selekon, B.; Labouba, I.; Gonofio, E.C.; Ouilibona, R.S.; Tchetgna, H.D.S.; et al. Genomic history of human monkey pox infections in the Central African Republic between 2001 and 2018. Sci. Rep. 2021, 11, 13085. [Google Scholar] [CrossRef]
- Shen-Gunther, J.; Cai, H.; Wang, Y. A Customized Monkeypox Virus Genomic Database (MPXV DB v1.0) for Rapid Sequence Analysis and Phylogenomic Discoveries in CLC Microbial Genomics. Viruses 2022, 15, 40. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-statement-on-the-press-conference-following-IHR-emergency-committee-regarding-the-multi--country-outbreak-of-monkeypox--23-july-2022 (accessed on 10 November 2022).
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef]
- Chen, Y.; Li, M.; Fan, H. The monkeypox outbreak in 2022: Adaptive evolution associated with APOBEC3 may account for. Signal Transduct. Target. Ther. 2022, 7, 1–3. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Pozzoli, U.; Sironi, M. An APOBEC3 Mutational Signature in the Genomes of Human-Infecting Orthopoxviruses. mSphere 2023, 8, e0006223. [Google Scholar] [CrossRef]
- O’toole, Á.; Neher, R.A.; Ndodo, N.; Borges, V.; Gannon, B.; Gomes, J.P.; Groves, N.; King, D.J.; Maloney, D.; Lemey, P.; et al. APOBEC3 deaminase editing in mpox virus as evidence for sustained human transmission since at least 2016. Science 2023, 382, 595–600. [Google Scholar] [CrossRef]
- Vakaniaki, E.H.; Kacita, C.; Kinganda-Lusamaki, E.; O’toole, Á.; Wawina-Bokalanga, T.; Mukadi-Bamuleka, D.; Amuri-Aziza, A.; Malyamungu-Bubala, N.; Mweshi-Kumbana, F.; Mutimbwa-Mambo, L.; et al. Sustained human outbreak of a new MPXV clade I lineage in eastern Democratic Republic of the Congo. Nat. Med. 2024, 30, 2791–2795. [Google Scholar] [CrossRef]
- Masirika, L.M.; Kumar, A.; Dutt, M.; Ostadgavahi, A.T.; Hewins, B.; Nadine, M.B.; Steeven, B.K.; Mweshi, F.K.; Mambo, L.M.; Mbiribindi, J.B.; et al. Complete Genome Sequencing, Annotation, and Mutational Profiling of the Novel Clade I Human Mpox Virus, Kamituga Strain. J. Infect. Dev. Ctries. 2024, 18, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Masirika, L.M.; Udahemuka, J.C.; Schuele, L.; Ndishimye, P.; Otani, S.; Mbiribindi, J.B.; Marekani, J.M.; Mambo, L.M.; Bubala, N.M.; Boter, M.; et al. Ongoing mpox outbreak in Kamituga, South Kivu province, associated with monkeypox virus of a novel Clade I sub-lineage, Democratic Republic of the Congo, 2024. Eurosurveillance 2024, 29, 2400106. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Available online: https://www.who.int/publications/m/item/multi-country-outbreak-of-mpox--external-situation-report-40--13-october-2024 (accessed on 13 October 2024).
- Chaplin, P.; Howley, P.; Meisinger, C. Modified Vaccinia Ankara Virus Variant (US 7,189,536 B2) USPTO. 2007. Available online: https://patentcenter.uspto.gov/applications/10440073/ifw/docs?application= (accessed on 1 August 2024).
- Grabenstein, J.D.; Hacker, A. Vaccines against mpox: MVA-BN and LC16m8. Expert Rev. Vaccines 2024, 23, 796–811. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). Available online: https://www.fda.gov/vaccines-blood-biologics/jynneos (accessed on 1 August 2024).
- Eto, A.; Saito, T.; Yokote, H.; Kurane, I.; Kanatani, Y. Recent advances in the study of live attenuated cell-cultured smallpox vaccine LC16m8. Vaccine 2015, 33, 6106–6111. [Google Scholar] [CrossRef] [PubMed]
- Kenner, J.; Cameron, F.; Empig, C.; Jobes, D.V.; Gurwith, M. LC16m8: An attenuated smallpox vaccine. Vaccine 2006, 24, 7009–7022. [Google Scholar] [CrossRef]
- Morikawa, S.; Sakiyama, T.; Hasegawa, H.; Saijo, M.; Maeda, A.; Kurane, I.; Maeno, G.; Kimura, J.; Hirama, C.; Yoshida, T.; et al. An Attenuated LC16m8 Smallpox Vaccine: Analysis of Full-Genome Sequence and Induction of Immune Protection. J. Virol. 2005, 79, 11873–11891. [Google Scholar] [CrossRef]
- Monath, T.P.; Caldwell, J.R.; Mundt, W.; Fusco, J.; Johnson, C.S.; Buller, M.; Liu, J.; Gardner, B.; Downing, G.; Blum, P.S.; et al. ACAM2000 clonal Vero cell culture vaccinia virus (New York City Board of Health strain)—A second-generation smallpox vaccine for biological defense. Int. J. Infect. Dis. 2004, 8 (Suppl. S2), S31–S44. [Google Scholar] [CrossRef]
- Center for Disease Control and Prevention (CDC). Available online: https://www.cdc.gov/mpox/hcp/vaccine-considerations/ (accessed on 7 February 2025).
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef]
- Davies, D.H.; Schmidt, C.S.; Sheikh, N.A. Concept and Scope of Modern Vaccines. In Vaccinology Principles and Practice; Morrow, W.J.W., Sheikh, N.A., Schmidt, C.S., Davies, D.H., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 3–11. [Google Scholar]
- Carter, D. Sequence-based computational approaches to vaccine discovery and design. In Vaccinology Principles and Practice; Morrow, W.J.W., Sheikh, N.A., Schmidt, C.S., Davies, D.H., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 133–149. [Google Scholar]
- Barranco, C. The first live attenuated vaccines. Nat. Portf. 2020, 284, S7. Available online: https://www.nature.com/articles/d42859-020-00008-5 (accessed on 14 September 2024).
- Wolf, Y.I.; Koonin, E.V. Genome reduction as the dominant mode of evolution. BioEssays 2013, 35, 829–837. [Google Scholar] [CrossRef]
- Tulman, E.R.; Delhon, G.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. Genome of Horsepox Virus. J. Virol. 2006, 80, 9244–9258. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.T.; Klunk, J.; Porter, A.F.; Dhody, A.N.; Hicks, R.; Smith, G.L.; Humphreys, M.; McCollum, A.M.; Davidson, W.B.; Wilkins, K.; et al. The origins and genomic diversity of American Civil War Era smallpox vaccine strains. Genome Biol. 2020, 21, 175. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liang, M.; Evans, D.H. Genomic analysis of vaccinia virus strain TianTan provides new insights into the evolution and evolutionary relationships between Orthopoxviruses. Virology 2013, 442, 59–66. [Google Scholar] [CrossRef]
- Landsberger, M.; Quick, J.; Mercer, J. Coding-Complete Genome Sequences of Copenhagen and Copenhagen-Derived vP811 Strains of Vaccinia Virus Isolated from Cell Culture. Genome Announc. 2023, 12, e0009023. [Google Scholar] [CrossRef]
- Moretti, M.; Meuwissen, A.; Rezende, A.M.; Zange, S.; Van Nedervelde, E.; de Block, T.; Vercauteren, K.; Demuyser, T.; Allard, S.D. Breakthrough Mpox Outbreak Investigation, the Delicate Balance Between Host Immune Response and Viral Immune Escape. Sex. Transm. Dis. 2024, 51, 499–503. [Google Scholar] [CrossRef]
- Nave, L.; Margalit, I.; Tau, N.; Cohen, I.; Yelin, D.; Lienert, F.; Yahav, D. Immunogenicity and Safety of Modified Vaccinia Ankara (MVA) Vaccine—A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Vaccines 2023, 11, 1410. [Google Scholar] [CrossRef]
- Mason, L.M.; Betancur, E.; Riera-Montes, M.; Lienert, F.; Scheele, S. MVA-BN vaccine effectiveness: A systematic review of real-world evidence in outbreak settings. Vaccine 2024, 42, 126409. [Google Scholar] [CrossRef]
- Gilchuk, I.; Gilchuk, P.; Sapparapu, G.; Lampley, R.; Singh, V.; Kose, N.; Blum, D.L.; Hughes, L.J.; Satheshkumar, P.S.; Townsend, M.B.; et al. Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections. Cell 2016, 167, 684–694.e9. [Google Scholar] [CrossRef]
- Riccardo, V.; Pablo, G.-C. Neutralization Determinants on Poxviruses. Viruses 2023, 15, 2396. [Google Scholar] [CrossRef]
- Moss, B.; Smith, G.L. Poxviridae: The Viruses and Their Replication. In Fields Virology. DNA Viruses, 7th ed.; Howley, P.M., Knipe, D.M., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2022; pp. 573–613. [Google Scholar]
- Condit, R.C. Poxviruses. In Fundamental of Molecular Virology, 2nd ed.; Acheson, N.H., Ed.; John Wiley & Son: Hoboken, NJ, USA, 2011; pp. 312–324. [Google Scholar]
- Happi, C.; Adetifa, I.; Mbala, P.; Njouom, R.; Nakoune, E.; Happi, A.; Ndodo, N.; Ayansola, O.; Mboowa, G.; Bedford, T.; et al. Urgent need for a non-discriminatory and non-stigmatizing nomenclature for monkeypox virus. PLOS Biol. 2022, 20, e3001769. [Google Scholar] [CrossRef]
- Brien, S.C.; LeBreton, M.; Doty, J.B.; Mauldin, M.R.; Morgan, C.N.; Pieracci, E.G.; Ritter, J.M.; Matheny, A.; Tafon, B.G.; Tamoufe, U.; et al. Clinical Manifestations of an Outbreak of Monkeypox Virus in Captive Chimpanzees in Cameroon, 2016. J. Infect. Dis. 2024, 229, S275–S284. [Google Scholar] [CrossRef] [PubMed]
- NCBI GenBank. Available online: https://www.ncbi.nlm.nih.gov/nuccore/OP498046.1 (accessed on 26 June 2024).
- Meseda, C.A.; Mayer, A.E.; Kumar, A.; Garcia, A.D.; Campbell, J.; Listrani, P.; Manischewitz, J.; King, L.R.; Golding, H.; Merchlinsky, M.; et al. Comparative Evaluation of the Immune Responses and Protection Engendered by LC16m8 and Dryvax Smallpox Vaccines in a Mouse Model. Clin. Vaccine Immunol. 2009, 16, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank (PDB). Available online: https://www.rcsb.org/ (accessed on 23 August 2024).
- Qiagen Digital Insights: CLC Genomics Workbench 24.0.2 Find and Model Structure. Available online: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Find_Model_Structure.html (accessed on 23 August 2024).
- Qiagen Digital Insights: CLC Genomics Workbench 24.0.2 Antigenicity. Available online: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Antigenicity.html (accessed on 23 August 2024).
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Sci. Nat. 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Sadeghpour, S.; Khodaee, S.; Rahnama, M.; Rahimi, H.; Ebrahimi, D. Human APOBEC3 Variations and Viral Infection. Viruses 2021, 13, 1366. [Google Scholar] [CrossRef]
- Schrick, L.; Tausch, S.H.; Dabrowski, P.W.; Damaso, C.R.; Esparza, J.; Nitsche, A. An Early American Smallpox Vaccine Based on Horsepox. N. Engl. J. Med. 2017, 377, 1491–1492. [Google Scholar] [CrossRef]
- Molteni, C.; Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Genetic ancestry and population structure of vaccinia virus. npj Vaccines 2022, 7, 1–9. [Google Scholar] [CrossRef]
- Downie, A.W. The Immunological Relationship of the Virus of Spontaneous Cowpox to Vaccinia Virus. Br. J. Exp. Pathol. 1939, 20, 158–176. [Google Scholar]
- Marennikova, S.S.; Gashnikov, P.V.; Zhukova, O.A.; Riabchikova, E.I.; Strel’Tsov, V.V.; Riazankina, O.I.; Chekunova, E.V.; Ianova, N.N.; Shchelkunov, S.N. The biotype and genetic characteristics of an isolate of the cowpox virus causing infection in a child. Zh. Mikrobiol. Epidemiol. Immunobiol. 1996, 4, 6–10. (In Russian) [Google Scholar]
- Adebowale, A.; Letebele, P. Phylogenomics for tracking the epidemiology of COVID-19: The genomic data gap for the african continent. In Proceedings of the 2020 2nd International Multidisciplinary Information Technology and Engineering Conference (IMITEC), Kimberley, South Africa, 25–27 November 2020; pp. 1–5. [Google Scholar] [CrossRef]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Congo’s mpox crisis. Science 2024, 386, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, C.; Xu, Y.; Zeng, S.; Zhang, J.; Xu, D. MUFOLD-DB: A processed protein structure database for protein structure prediction and analysis. BMC Genom. 2014, 15, S2. [Google Scholar] [CrossRef]
- van Beusekom, B.; Touw, W.G.; Tatineni, M.; Somani, S.; Rajagopal, G.; Luo, J.; Gilliland, G.L.; Perrakis, A.; Joosten, R.P. Homology-based hydrogen bond information improves crystallographic structures in the PDB. Protein Sci. 2017, 27, 798–808. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Jankauskaitė, J.; Jiménez-García, B.; Dapkūnas, J.; Fernández-Recio, J.; Moal, I.H. SKEMPI 2.0: An updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019, 35, 462–469. [Google Scholar] [CrossRef]
- Structural Database of Kinetics and Energetics of Mutant Protein Interactions (SKEMPI v2.0). Available online: https://life.bsc.es/pid/skempi2/ (accessed on 1 February 2025).
- Islam, S.; Chauhan, V.M.; Pantazes, R.J. Analysis of how antigen mutations disrupt antibody binding interactions toward enabling rapid and reliable antibody repurposing. mAbs 2025, 17, 2440586. [Google Scholar] [CrossRef]
- Delamonica, B.; Davalos, L.; Larijani, M.; Anthony, S.J.; Liu, J.; MacCarthy, T. Evolutionary potential of the monkeypox genome arising from interactions with human APOBEC3 enzymes. Virus Evol. 2023, 9, vead047. [Google Scholar] [CrossRef]
- Dumonteil, E.; Herrera, C.; Sabino-Santos, G. Monkeypox Virus Evolution before 2022 Outbreak. Emerg. Infect. Dis. 2023, 29, 451–453. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). Virus. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ (accessed on 26 June 2024).
- CDC Public Health Image Library (PHIL). Colorized Transmission Electron Microscopic Image of Mpox Virus Particles. 2022. Photograph. Available online: https://phil.cdc.gov/Details.aspx?pid=26503 (accessed on 1 August 2024).
- Qiagen Digital Insights: CLC Genomics Workbench 24.0.2 Whole Genome Alignment. Available online: https://resources.qiagenbioinformatics.com/manuals/wholegenomealignment/current/index.php?manual=Create_Whole_Genome_Alignment.html (accessed on 23 August 2024).
- Hall, B.G. Major methods for estimating phylogenetic trees. In Phylogenetic Trees Made Easy: A How-To Manual, 4th ed.; Hall, B.G., Ed.; Sinauer Associates: Sunderland, MA, USA, 2011; pp. 61–68. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPXV Virion | MPXV Gene (Protein) | Structures Found (n) | Available Structures 1 | Rank 2 | E-Value | Match ID (%) | Coverage (%) |
|---|---|---|---|---|---|---|---|
| MV | A29L (A29) | 1 | 3VOP 3 | 1 | 3.1 × 10−20 | 90.7 | 38.7 |
| MV | E8L (E8) | 1309 | 5USH 4 | 3 | 1.7 × 10−158 | 92.7 | 75.4 |
| MV | H3L (H3) | 1 | 5EJ0 5 | 1 | 1.6 × 10−142 | 84.4 | 65.2 |
| MV | M1R (M1) | 6 | 2I9L 6 | 3 | 4.5 × 10−125 | 99.4 | 68.9 |
| EV | A35R (A35) | 7 | 4LU5 7 | 5 | 1.1 × 10−50 | 90.4 | 45.1 |
| EV | B6R (B6) | 36 | 5FOB 8 | 1 | 1.0 × 10−11 | 29.6 | 67.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen-Gunther, J.; Cai, H.; Wang, Y. Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology. Int. J. Mol. Sci. 2025, 26, 1428. https://doi.org/10.3390/ijms26041428
Shen-Gunther J, Cai H, Wang Y. Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology. International Journal of Molecular Sciences. 2025; 26(4):1428. https://doi.org/10.3390/ijms26041428
Chicago/Turabian StyleShen-Gunther, Jane, Hong Cai, and Yufeng Wang. 2025. "Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology" International Journal of Molecular Sciences 26, no. 4: 1428. https://doi.org/10.3390/ijms26041428
APA StyleShen-Gunther, J., Cai, H., & Wang, Y. (2025). Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology. International Journal of Molecular Sciences, 26(4), 1428. https://doi.org/10.3390/ijms26041428