
Integrative Multi-Omics Approaches for Identifying and Characterizing Biological Elements in Crop Traits: Current Progress and Future Prospects


Abstract
1. Introduction
2. Principles and Methods of Multi-Omics Technologies
2.1. Elucidating Gene Function and Genetic Variation in Crops
2.2. Investigating Epigenetic Regulation and Its Influence on Gene Expression
2.3. Characterizing Gene Expression Profiles and Regulatory Networks
2.4. Deciphering Protein Interaction Networks and Functional Proteomes
2.5. Analyzing Plant-Microbiome Interactions and Microbial Diversity
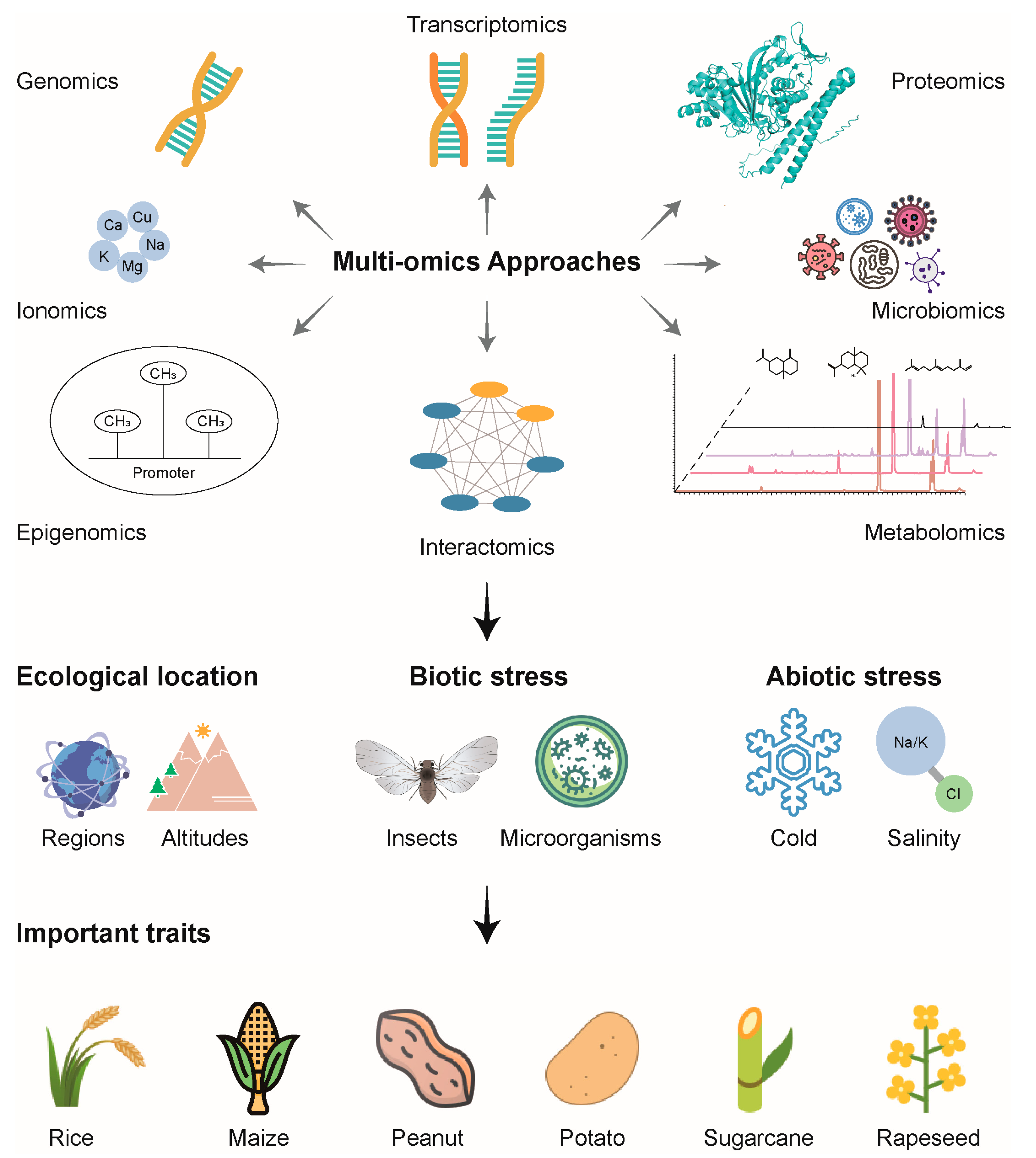
3. Exploring Biological Elements in Crop Research Through Integrative Multi-Omics Approaches
3.1. Exploration of Agronomic Traits
3.2. Understanding Adaptation to Various Environmental Conditions
3.3. Enhancing Resistance to Biological Stresses
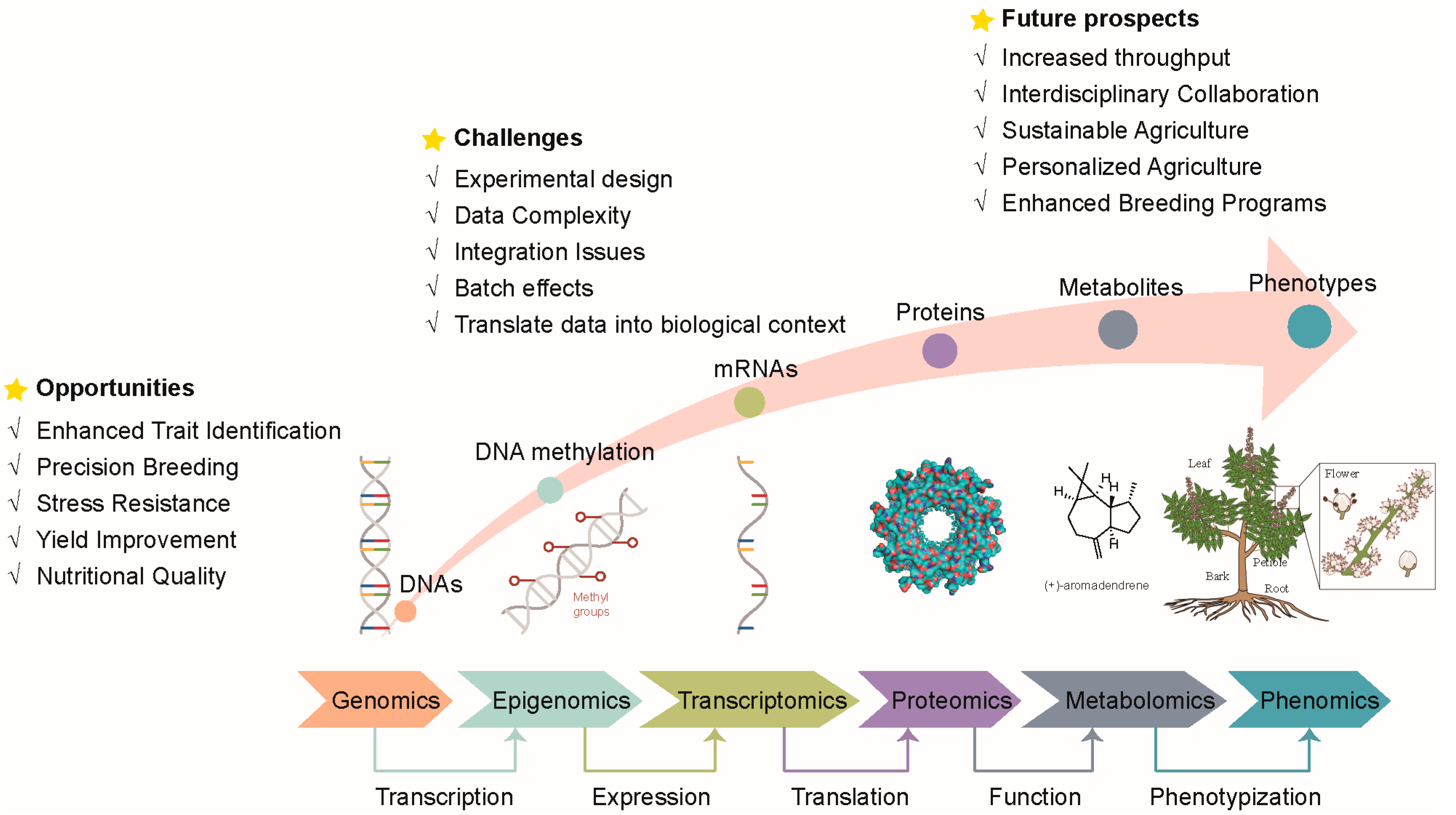
4. Emerging Technologies, Challenges, and Future Prospects
4.1. Advances in Single-Cell and Spatial Omics
4.2. Applications of Plant scRNA-Seq and ST
4.3. Challenges and Future Prospects
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding Crops to Feed 10 Billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-Generation Sequencing Technologies and Their Implications for Crop Genetics and Breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef]
- Wallace, J.G.; Rodgers-Melnick, E.; Buckler, E.S. On the Road to Breeding 4.0: Unraveling the Good, the Bad, and the Boring of Crop Quantitative Genomics. Annu. Rev. Genet. 2018, 52, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Qiu, J.; Yong, K.; Fan, J.; Zhang, Q.; Hua, H.; Liu, J.; Wang, Q.; Olsen, K.M.; Han, B.; et al. A Quantitative Genomics Map of Rice Provides Genetic Insights and Guides Breeding. Nat. Genet. 2021, 53, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Song, J.M.; Xie, W.Z.; Wang, S.; Guo, Y.X.; Koo, D.H.; Kudrna, D.; Gong, C.; Huang, Y.; Feng, J.W.; Zhang, W.; et al. Two Gap-Free Reference Genomes and a Global View of the Centromere Architecture in Rice. Mol. Plant 2021, 14, 1757–1767. [Google Scholar] [CrossRef]
- Shang, L.; Li, X.; He, H.; Yuan, Q.; Song, Y.; Wei, Z.; Lin, H.; Hu, M.; Zhao, F.; Zhang, C.; et al. A Super Pan-Genomic Landscape of Rice. Cell Res. 2022, 32, 878–896. [Google Scholar] [CrossRef]
- Zhu, X.T.; Zhou, R.; Che, J.; Zheng, Y.Y.; Tahir ul Qamar, M.; Feng, J.W.; Zhang, J.; Gao, J.; Chen, L.L. Ribosome Profiling Reveals the Translational Landscape and Allele-Specific Translational Efficiency in Rice. Plant Commun. 2023, 4, 100457. [Google Scholar] [CrossRef]
- Zhao, H.; Li, J.; Yang, L.; Qin, G.; Xia, C.; Xu, X.; Su, Y.; Liu, Y.; Ming, L.; Chen, L.L.; et al. An Inferred Functional Impact Map of Genetic Variants in Rice. Mol. Plant 2021, 14, 1584–1599. [Google Scholar] [CrossRef]
- Zhou, R.; Sanz-Jimenez, P.; Zhu, X.T.; Feng, J.W.; Shao, L.; Song, J.M.; Chen, L.L. Analysis of Rice Transcriptome Reveals the LncRNA/CircRNA Regulation in Tissue Development. Rice 2021, 14, 14. [Google Scholar] [CrossRef]
- Zhan, C.; Lei, L.; Guo, H.; Zhou, S.; Xu, C.; Liu, Z.; Wu, Z.; Deng, Y.; Miao, Y.; Han, Y.; et al. Disease Resistance Conferred by Components of Essential Chrysanthemum Oil and the Epigenetic Regulation of OsTPS1. Sci. China Life Sci. 2023, 66, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Q.; Chen, Y.; Liu, Y.; Lin, W.H.; Wang, J.W. Single-Cell Transcriptome Atlas and Chromatin Accessibility Landscape Reveal Differentiation Trajectories in the Rice Root. Nat. Commun. 2021, 12, 2053. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huan, Q.; Li, K.; Qian, W. Single-Cell Transcriptome Atlas of the Leaf and Root of Rice Seedlings. J. Genet. Genom. 2021, 48, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Muthamilarasan, M.; Singh, N.K.; Prasad, M. Multi-Omics Approaches for Strategic Improvement of Stress Tolerance in Underutilized Crop Species: A Climate Change Perspective. Adv. Genet. 2019, 103, 1–38. [Google Scholar] [CrossRef]
- Bernal-Gallardo, J.J.; de Folter, S. Plant Genome Information Facilitates Plant Functional Genomics. Planta 2024, 259, 117. [Google Scholar] [CrossRef]
- Kajla, M.; Roy, A.; Singh, I.K.; Singh, A. Regulation of the Regulators: Transcription Factors Controlling Biosynthesis of Plant Secondary Metabolites during Biotic Stresses and Their Regulation by MiRNAs. Front. Plant Sci. 2023, 14, 1126567. [Google Scholar] [CrossRef]
- Espinosa, E.; Bautista, R.; Larrosa, R.; Plata, O. Advancements in Long-Read Genome Sequencing Technologies and Algorithms. Genomics 2024, 116, 110842. [Google Scholar] [CrossRef]
- Shi, J.; Tian, Z.; Lai, J.; Huang, X. Plant Pan-Genomics and Its Applications. Mol. Plant 2023, 16, 168–186. [Google Scholar] [CrossRef]
- Kille, B.; Balaji, A.; Sedlazeck, F.J.; Nute, M.; Treangen, T.J. Multiple Genome Alignment in the Telomere-to-Telomere Assembly Era. Genome Biol. 2022, 23, 182. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-W.; Yu, Z.-G.; Huang, X.-M.; Liu, J.-S.; Guo, Y.-X.; Chen, L.-L.; Song, J.-M. GenomeSyn: A Bioinformatics Tool for Visualizing Genome Synteny and Structural Variations. J. Genet. Genom. 2022, 49, 1174–1176. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3,000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Abdellaoui, A.; Yengo, L.; Verweij, K.J.H.; Visscher, P.M. 15 Years of GWAS Discovery: Realizing the Promise. Am. J. Hum. Genet. 2023, 110, 179–194. [Google Scholar] [CrossRef]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and Prospects of Genome-Wide Association Studies in Plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Pantalião, G.F.; Narciso, M.; Guimarães, C.; Castro, A.; Colombari, J.M.; Breseghello, F.; Rodrigues, L.; Vianello, R.P.; Borba, T.O.; Brondani, C. Genome Wide Association Study (GWAS) for Grain Yield in Rice Cultivated under Water Deficit. Genetica 2016, 144, 651–664. [Google Scholar] [CrossRef]
- Chen, S.; Dang, D.; Liu, Y.; Ji, S.; Zheng, H.; Zhao, C.; Dong, X.; Li, C.; Guan, Y.; Zhang, A.; et al. Genome-Wide Association Study Presents Insights into the Genetic Architecture of Drought Tolerance in Maize Seedlings under Field Water-Deficit Conditions. Front. Plant Sci. 2023, 14, 1165582. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Speed, D.; Balding, D.J. Relatedness in the Post-Genomic Era: Is It Still Useful? Nat. Rev. Genet. 2014, 16, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A Unified Mixed-Model Method for Association Mapping That Accounts for Multiple Levels of Relatedness. Nat. Genet. 2005, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Owens, B.F.; Lipka, A.E.; Magallanes-Lundback, M.; Tiede, T.; Diepenbrock, C.H.; Kandianis, C.B.; Kim, E.; Cepela, J.; Mateos-Hernandez, M.; Buell, C.R.; et al. A Foundation for Provitamin A Biofortification of Maize: Genome-Wide Association and Genomic Prediction Models of Carotenoid Levels. Genetics 2014, 198, 1699–1716. [Google Scholar] [CrossRef] [PubMed]
- Jamann, T.M.; Balint-Kurti, P.J.; Holland, J.B. QTL Mapping Using High-Throughput Sequencing. In Plant Functional Genomics: Methods and Protocols; Alonso, J.M., Stepanova, A.N., Eds.; Springer: New York, NY, USA, 2015; pp. 257–285. ISBN 978-1-4939-2444-8. [Google Scholar]
- Li, B. Identification of Genes Conferring Plant Salt Tolerance Using GWAS: Current Success and Perspectives. Plant Cell Physiol. 2020, 61, 1419–1426. [Google Scholar] [CrossRef]
- Khan, S.U.; Saeed, S.; Khan, M.H.U.; Fan, C.; Ahmar, S.; Arriagada, O.; Shahzad, R.; Branca, F.; Mora-Poblete, F. Advances and Challenges for QTL Analysis and GWAS in the Plant-Breeding of High-Yielding: A Focus on Rapeseed. Biomolecules 2021, 11, 1516. [Google Scholar] [CrossRef]
- Masojć, P. The Application of Molecular Markers in the Process of Selection. Cell Mol. Biol. Lett. 2002, 7, 499–509. [Google Scholar]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Novik, K.L.; Nimmrich, I.; Genc, B.; Maier, S.; Piepenbrock, C.; Olek, A.; Beck, S. Epigenomics: Genome-Wide Study of Methylation Phenomena. Curr. Issues Mol. Biol. 2002, 4, 111–128. [Google Scholar] [CrossRef]
- Alabert, C.; Groth, A. Chromatin Replication and Epigenome Maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Fu, Z.; Jiang, S.; Sun, Y.; Zheng, S.; Zong, L.; Li, P. Cut&tag: A Powerful Epigenetic Tool for Chromatin Profiling. Epigenetics 2024, 19, 2293411. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Neumann, D.A.; Schmitz, R.J. Crop Epigenomics: Identifying, Unlocking, and Harnessing Cryptic Variation in Crop Genomes. Mol. Plant 2015, 8, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.-K. Epigenetic Gene Regulation in Plants and Its Potential Applications in Crop Improvement. Nat. Rev. Mol. Cell Biol. 2024, 2024, 51–67. [Google Scholar] [CrossRef]
- Cao, S.; Chen, K.; Lu, K.; Chen, S.; Zhang, X.; Shen, C.; Zhu, S.; Niu, Y.; Fan, L.; Chen, Z.J.; et al. Asymmetric Variation in DNA Methylation during Domestication and De-Domestication of Rice. Plant Cell 2023, 35, 3429–3443. [Google Scholar] [CrossRef]
- Liu, B.; Yang, D.; Wang, D.; Liang, C.; Wang, J.; Lisch, D.; Zhao, M. Heritable Changes of Epialleles near Genes in Maize Can Be Triggered in the Absence of CHH Methylation. Plant Physiol. 2024, 194, 2511–2532. [Google Scholar] [CrossRef]
- Ziegler, D.J.; Khan, D.; Pulgar-Vidal, N.; Parkin, I.A.P.; Robinson, S.J.; Belmonte, M.F. Genomic Asymmetry of the Brassica Napus Seed: Epigenetic Contributions of DNA Methylation and Small RNAs to Subgenome Bias. Plant J. 2023, 115, 690–708. [Google Scholar] [CrossRef]
- Axel, R. Cleavage of DNA in Nuclei and Chromatin with Staphylococcal Nuclease. Biochemistry 1975, 14, 2921–2925. [Google Scholar] [CrossRef]
- Zhou, W.; Sherwood, B.; Ji, Z.; Xue, Y.; Du, F.; Bai, J.; Ying, M.; Ji, H. Genome-Wide Prediction of DNase I Hypersensitivity Using Gene Expression. Nat. Commun. 2017, 8, 1038. [Google Scholar] [CrossRef]
- Tsompana, M.; Buck, M.J. Chromatin Accessibility: A Window into the Genome. Epigenetics Chromatin 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pugh, B.F. High-Resolution Genome-Wide Mapping of the Primary Structure of Chromatin. Cell 2011, 144, 175–186. [Google Scholar] [CrossRef]
- Nelson, N.J. Microarrays Have Arrived: Gene Expression Tool Matures. J. Natl. Cancer Inst. 2001, 93, 492–493. [Google Scholar] [CrossRef]
- McGettigan, P.A. Transcriptomics in the RNA-Seq Era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Wang, N.; Huo, Y.X. Using Genome and Transcriptome Analysis to Elucidate Biosynthetic Pathways. Curr. Opin. Biotechnol. 2022, 75, 102708. [Google Scholar] [CrossRef]
- Ming, L.; Fu, D.; Wu, Z.; Zhao, H.; Xu, X.; Xu, T.; Xiong, X.; Li, M.; Zheng, Y.; Li, G.; et al. Transcriptome-Wide Association Analyses Reveal the Impact of Regulatory Variants on Rice Panicle Architecture and Causal Gene Regulatory Networks. Nat. Commun. 2023, 14, 7501. [Google Scholar] [CrossRef]
- Zhong, Y.; Luo, Y.; Sun, J.; Qin, X.; Gan, P.; Zhou, Z.; Qian, Y.; Zhao, R.; Zhao, Z.; Cai, W.; et al. Pan-Transcriptomic Analysis Reveals Alternative Splicing Control of Cold Tolerance in Rice. Plant Cell 2024, 36, 2117–2139. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Poretsky, E.; Huffaker, A. MutRank: An R Shiny Web-Application for Exploratory Targeted Mutual Rank-Based Coexpression Analyses Integrated with User-Provided Supporting Information. PeerJ 2020, 8, e10264. [Google Scholar] [CrossRef]
- Singh, A. Soil Salinity: A Global Threat to Sustainable Development. Soil. Use Manag. 2022, 38, 39–67. [Google Scholar] [CrossRef]
- Fang, Z.; Cui, X. Design and Validation Issues in RNA-Seq Experiments. Brief. Bioinform. 2011, 12, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Govind, G.; Vokkaliga Thammegowda, H.; Jayaker Kalaiarasi, P.; Iyer, D.R.; Muthappa, S.K.; Nese, S.; Makarla, U.K. Identification and Functional Validation of a Unique Set of Drought Induced Genes Preferentially Expressed in Response to Gradual Water Stress in Peanut. Mol. Genet. Genom. 2009, 281, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Locasale, J.W. Metabolomics: A Primer. Trends Biochem. Sci. 2017, 42, 274–284. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The Link between Genotypes and Phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Jamil, I.N.; Remali, J.; Azizan, K.A.; Nor Muhammad, N.A.; Arita, M.; Goh, H.H.; Aizat, W.M. Systematic Multi-Omics Integration (MOI) Approach in Plant Systems Biology. Front. Plant Sci. 2020, 11, 944. [Google Scholar] [CrossRef]
- Ribay, V.; Praud, C.; Letertre, M.P.M.; Dumez, J.N.; Giraudeau, P. Hyperpolarized NMR Metabolomics. Curr. Opin. Chem. Biol. 2023, 74, 102307. [Google Scholar] [CrossRef]
- Honour, J.W. Gas Chromatography-Mass Spectrometry. Methods Mol. Biol. 2006, 324, 53–74. [Google Scholar] [CrossRef]
- Zhong, P.; Wei, X.; Li, X.; Wei, X.; Wu, S.; Huang, W.; Koidis, A.; Xu, Z.; Lei, H. Untargeted Metabolomics by Liquid Chromatography-Mass Spectrometry for Food Authentication: A Review. Compr. Rev. Food Sci. Food Saf. 2022, 21, 2455–2488. [Google Scholar] [CrossRef]
- Zhang, F.; Huang, J.; Guo, H.; Yang, C.; Li, Y.; Shen, S.; Zhan, C.; Qu, L.; Liu, X.; Wang, S.; et al. OsRLCK160 Contributes to Flavonoid Accumulation and UV-B Tolerance by Regulating OsbZIP48 in Rice. Sci. China Life Sci. 2022, 65, 1380–1394. [Google Scholar] [CrossRef]
- Zhan, C.; Lei, L.; Liu, Z.; Zhou, S.; Yang, C.; Zhu, X.; Guo, H.; Zhang, F.; Peng, M.; Zhang, M.; et al. Selection of a Subspecies-Specific Diterpene Gene Cluster Implicated in Rice Disease Resistance. Nat. Plants 2020, 6, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xing, F.; He, Q.; Qamar, M.T.U.; Chen, L.L.; Xing, Y. Conserved Imprinted Genes between Intra-Subspecies and Inter-Subspecies Are Involved in Energy Metabolism and Seed Development in Rice. Int. J. Mol. Sci. 2020, 21, 9618. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lyv, Y.; Zheng, W.; Yang, C.; Li, Y.; Wang, X.; Chen, R.; Wang, C.; Luo, J.; Qu, L. Comparative Metabolomics Reveals Two Metabolic Modules Affecting Seed Germination in Rice (Oryza Sativa). Metabolites 2021, 11, 880. [Google Scholar] [CrossRef]
- Li, K.; Wang, D.; Gong, L.; Lyu, Y.; Guo, H.; Chen, W.; Jin, C.; Liu, X.; Fang, C.; Luo, J. Comparative Analysis of Metabolome of Rice Seeds at Three Developmental Stages Using a Recombinant Inbred Line Population. Plant J. 2019, 100, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.R.; James, E.K.; Poole, P.S. The Plant Microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The Rhizosphere Microbiome and Plant Health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Dastogeer, K.M.G.; Tumpa, F.H.; Sultana, A.; Akter, M.A.; Chakraborty, A. Plant Microbiome–an Account of the Factors That Shape Community Composition and Diversity. Curr. Plant Biol. 2020, 23, 100161. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, R. Nitrogen and Phosphorus Signaling and Transport During Legume–Rhizobium Symbiosis. Front. Plant Sci. 2021, 12, 683601. [Google Scholar] [CrossRef]
- Compant, S.; Cassan, F.; Kostić, T.; Johnson, L.; Brader, G.; Trognitz, F.; Sessitsch, A. Harnessing the Plant Microbiome for Sustainable Crop Production. Nat. Rev. Microbiol. 2025, 23, 9–23. [Google Scholar] [CrossRef]
- Koza, N.A.; Adedayo, A.A.; Babalola, O.O.; Kappo, A.P. Microorganisms in Plant Growth and Development: Roles in Abiotic Stress Tolerance and Secondary Metabolites Secretion. Microorganisms 2022, 10, 1528. [Google Scholar] [CrossRef]
- Malusá, E.; Sas-Paszt, L.; Ciesielska, J. Technologies for Beneficial Microorganisms Inocula Used as Biofertilizers. Sci. World J. 2012, 2012, 491206. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Diksha; Sindhu, S.S.; Kumar, R. Biofertilizers: An Ecofriendly Technology for Nutrient Recycling and Environmental Sustainability. Curr. Res. Microb. Sci. 2021, 3, 100094. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Ramlal, A.; Mallick, D.; Mishra, V. An Overview of Some Biopesticides and Their Importance in Plant Protection for Commercial Acceptance. Plants 2021, 10, 1185. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.-S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A. Opportunities and Challenges of Whole-Genome and -Exome Sequencing. BMC Genet. 2017, 18, 14. [Google Scholar] [CrossRef]
- Jelin, A.C.; Vora, N. Whole Exome Sequencing: Applications in Prenatal Genetics. Obstet. Gynecol. Clin. North. Am. 2018, 45, 69–81. [Google Scholar] [CrossRef]
- Dellino, G.I.; Pelicci, P.G. Next-Generation Sequencing and DNA Replication in Human Cells: The Future Has Arrived. Future Oncol. 2014, 10, 683–693. [Google Scholar] [CrossRef]
- Furey, T.S. ChIP-Seq and beyond: New and Improved Methodologies to Detect and Characterize Protein-DNA Interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Capurso, D.; Tang, Z.; Ruan, Y. Methods for Comparative ChIA-PET and Hi-C Data Analysis. Methods 2020, 170, 69–74. [Google Scholar] [CrossRef]
- Chu, C.; Quinn, J.; Chang, H.Y. Chromatin Isolation by RNA Purification (ChIRP). J. Vis. Exp. 2012, 3912. [Google Scholar] [CrossRef]
- Gu, H.; Raman, A.T.; Wang, X.; Gaiti, F.; Chaligne, R.; Mohammad, A.W.; Arczewska, A.; Smith, Z.D.; Landau, D.A.; Aryee, M.J.; et al. Smart-RRBS for Single-Cell Methylome and Transcriptome Analysis. Nat. Protoc. 2021, 16, 4004–4030. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Xu, W.; Park, K.H.; Ryu, K.H.; Xiang, R. Comprehensive Evaluation of Differential Methylation Analysis Methods for Bisulfite Sequencing Data. Int. J. Environ. Res. Public. Health 2021, 18, 7975. [Google Scholar] [CrossRef]
- Aberg, K.A.; Chan, R.F.; Xie, L.; Shabalin, A.A.; van den Oord, E.J.C.G. Methyl-CpG-Binding Domain Sequencing: MBD-Seq. Methods Mol. Biol. 2018, 1708, 171–189. [Google Scholar] [CrossRef]
- Crawford, G.E.; Holt, I.E.; Whittle, J.; Webb, B.D.; Tai, D.; Davis, S.; Margulies, E.H.; Chen, Y.D.; Bernat, J.A.; Ginsburg, D.; et al. Genome-Wide Mapping of DNase Hypersensitive Sites Using Massively Parallel Signature Sequencing (MPSS). Genome Res. 2006, 16, 123–131. [Google Scholar] [CrossRef]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-Wide Measurement of RNA Secondary Structure in Yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In Vivo Genome-Wide Profiling of RNA Secondary Structure Reveals Novel Regulatory Features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global Identification of MicroRNA-Target RNA Pairs by Parallel Analysis of RNA Ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef]
- Patwardhan, R.P.; Hiatt, J.B.; Witten, D.M.; Kim, M.J.; Smith, R.P.; May, D.; Lee, C.; Andrie, J.M.; Lee, S.I.; Cooper, G.M.; et al. Massively Parallel Functional Dissection of Mammalian Enhancers in Vivo. Nat. Biotechnol. 2012, 30, 265–270. [Google Scholar] [CrossRef]
- Guo, Z.; Shafik, A.M.; Jin, P.; Wu, Z.; Wu, H. Detecting M6A Methylation Regions from Methylated RNA Immunoprecipitation Sequencing. Bioinformatics 2021, 37, 2818–2824. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Beaulaurier, J.; Deikus, G.; Wu, T.P.; Strahl, M.; Hao, Z.; Luo, G.; Gregory, J.A.; Chess, A.; He, C.; et al. Mapping and Characterizing N6-Methyladenine in Eukaryotic Genomes Using Single-Molecule Real-Time Sequencing. Genome Res. 2018, 28, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.S.K.; Gu, L.; Reddy, A.S.N. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Mercer, T.R.; Gerhardt, D.J.; Dinger, M.E.; Crawford, J.; Trapnell, C.; Jeddeloh, J.A.; Mattick, J.S.; Rinn, J.L. Targeted RNA Sequencing Reveals the Deep Complexity of the Human Transcriptome. Nat. Biotechnol. 2011, 30, 99–104. [Google Scholar] [CrossRef]
- Calviello, L.; Ohler, U. Beyond Read-Counts: Ribo-Seq Data Analysis to Understand the Functions of the Transcriptome. Trends Genet. 2017, 33, 728–744. [Google Scholar] [CrossRef]
- Cardiello, J.F.; Sanchez, G.J.; Allen, M.A.; Dowell, R.D. Lessons from ERNAs: Understanding Transcriptional Regulation through the Lens of Nascent RNAs. Transcription 2020, 11, 3–18. [Google Scholar] [CrossRef]
- Chen, F.X.; Marshall, S.A.; Deng, Y.; Tianjiao, S. Measuring Nascent Transcripts by Nascent-Seq. Methods Mol. Biol. 2018, 1712, 19–26. [Google Scholar] [CrossRef]
- Churchman, L.S.; Weissman, J.S. Native Elongating Transcript Sequencing (NET-Seq). Curr. Protoc. Mol. Biol. 2012, 4.14.1–4.14.17. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, Y.; Zhou, Y.; Shi, K.; Hu, X.; Li, D.; Ye, H.; Zhou, Y.; Wang, K. Full-Length Annotation with Multistrategy RNA-Seq Uncovers Transcriptional Regulation of LncRNAs in Cotton. Plant Physiol. 2021, 185, 179–195. [Google Scholar] [CrossRef]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, T.; Qian, W.J.; Petyuk, V.A.; Smith, R.D. Liquid Chromatography-Mass Spectrometry-Based Quantitative Proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, A.M.; Smith, H.; Labrie, M.; Mills, G.B. Characterization of Anticancer Drug Resistance by Reverse-Phase Protein Array: New Targets and Strategies. Expert. Rev. Proteom. 2022, 19, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Jorrin-Novo, J.V.; Komatsu, S.; Sanchez-Lucas, R.; Rodríguez de Francisco, L.E. Gel Electrophoresis-Based Plant Proteomics: Past, Present, and Future. Happy 10th Anniversary Journal of Proteomics! J. Proteom. 2019, 198, 1–10. [Google Scholar] [CrossRef]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein Labeling by ITRAQ: A New Tool for Quantitative Mass Spectrometry in Proteome Research. Proteomics 2007, 7, 340–350. [Google Scholar] [CrossRef]
- Hoedt, E.; Zhang, G.; Neubert, T.A. Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) for Quantitative Proteomics. Adv. Exp. Med. Biol. 2019, 1140, 531–539. [Google Scholar] [CrossRef]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass Spectrometry-Based Metabolomics: A Guide for Annotation, Quantification and Best Reporting Practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Araya, C.L.; Chircus, L.M.; Layton, C.J.; Chang, H.Y.; Snyder, M.P.; Greenleaf, W.J. Quantitative Analysis of RNA-Protein Interactions on a Massively Parallel Array Reveals Biophysical and Evolutionary Landscapes. Nat. Biotechnol. 2014, 32, 562–568. [Google Scholar] [CrossRef]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.H.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of Neuronal RNA Targets of TDP-43-Containing Ribonucleoprotein Complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef]
- Baker, M. The Interaction Map. Nature 2012, 484, 271–275. [Google Scholar] [CrossRef]
- Kodama, Y.; Hu, C.D. Bimolecular Fluorescence Complementation (BiFC): A 5-Year Update and Future Perspectives. Biotechniques 2012, 53, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Kutluay, S.B. CLIP-Related Methodologies and Their Application to Retrovirology. Retrovirology 2018, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Y.; Zheng, L. Disease Ionomics: Understanding the Role of Ions in Complex Disease. Int. J. Mol. Sci. 2020, 21, 8646. [Google Scholar] [CrossRef] [PubMed]
- Punshon, T.; Ricachenevsky, F.K.; Hindt, M.N.; Socha, A.L.; Zuber, H. Methodological Approaches for Using Synchrotron X-Ray Fluorescence (SXRF) Imaging as a Tool in Ionomics: Examples from Arabidopsis Thaliana. Metallomics 2013, 5, 1133–1145. [Google Scholar] [CrossRef]
- Baxter, I.; Hosmani, P.S.; Rus, A.; Lahner, B.; Borevitz, J.O.; Muthukumar, B.; Mickelbart, M.V.; Schreiber, L.; Franke, R.B.; Salt, D.E. Root Suberin Forms an Extracellular Barrier That Affects Water Relations and Mineral Nutrition in Arabidopsis. PLoS Genet. 2009, 5, e1000492. [Google Scholar] [CrossRef]
- Gong, J.M.; Waner, D.A.; Horie, T.; Shi, L.L.; Horie, R.; Abid, K.B.; Schroeder, J.I. Microarray-Based Rapid Cloning of an Ion Accumulation Deletion Mutant in Arabidopsis Thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 15404–15409. [Google Scholar] [CrossRef]
- Regueira-Iglesias, A.; Balsa-Castro, C.; Blanco-Pintos, T.; Tomás, I. Critical Review of 16S RRNA Gene Sequencing Workflow in Microbiome Studies: From Primer Selection to Advanced Data Analysis. Mol. Oral. Microbiol. 2023, 38, 347–399. [Google Scholar] [CrossRef]
- Spotin, A.; Dalir, F.; Hazratian, T.; Shekarchi, A.A.; Mahami-Oskouei, M.; Farmani, M.; Dolatkhah, A.; Ahmadpour, E. Global Haplotype Distribution of Babesia Ovis Inferred by 18S RRNA Sequences; a Phylogeographical Systematic Review. Microb. Pathog. 2023, 181, 106179. [Google Scholar] [CrossRef]
- Gao, B.; Chi, L.; Zhu, Y.; Shi, X.; Tu, P.; Li, B.; Yin, J.; Gao, N.; Shen, W.; Schnabl, B. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules 2021, 11, 530. [Google Scholar] [CrossRef]
- Joseph, T.A.; Pe’er, I. An Introduction to Whole-Metagenome Shotgun Sequencing Studies. Methods Mol. Biol. 2021, 2243, 107–122. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-Cell Chromatin Accessibility Reveals Principles of Regulatory Variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex Single-Cell Profiling of Chromatin Accessibility by Combinatorial Cellular Indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-Cell Genome-Wide Bisulfite Sequencing for Assessing Epigenetic Heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-Cell ChIP-Seq Reveals Cell Subpopulations Defined by Chromatin State. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-Cell Hi-C Reveals Cell-to-Cell Variability in Chromosome Structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef]
- Frei, A.P.; Bava, F.A.; Zunder, E.R.; Hsieh, E.W.Y.; Chen, S.Y.; Nolan, G.P.; Gherardini, P.F. Highly Multiplexed Simultaneous Detection of RNAs and Proteins in Single Cells. Nat. Methods 2016, 13, 269–275. [Google Scholar] [CrossRef]
- Fessenden, M. Metabolomics: Small Molecules, Single Cells. Nature 2016, 540, 153–155. [Google Scholar] [CrossRef]
- Kulkarni, S.R.; Balachandran, S.M.; Ulaganathan, K.; Balakrishnan, D.; Praveen, M.; Prasad, A.S.H.; Fiyaz, R.A.; Senguttuvel, P.; Sinha, P.; Kale, R.R.; et al. Molecular Mapping of QTLs for Yield Related Traits in Recombinant Inbred Line (RIL) Population Derived from the Popular Rice Hybrid KRH-2 and Their Validation through SNP Genotyping. Sci. Rep. 2020, 10, 13695. [Google Scholar] [CrossRef]
- Zaw, H.; Raghavan, C.; Pocsedio, A.; Swamy, B.P.M.; Jubay, M.L.; Singh, R.K.; Bonifacio, J.; Mauleon, R.; Hernandez, J.E.; Mendioro, M.S.; et al. Exploring Genetic Architecture of Grain Yield and Quality Traits in a 16-Way Indica by Japonica Rice MAGIC Global Population. Sci. Rep. 2019, 9, 19605. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhou, X.; Wang, C.; Liu, A.; Sun, Z.; Li, S.; Shi, X.; Yang, S.; Guan, Y.; Cheng, J.; et al. Quantitative Trait Loci Detection for Three Tiller-Related Traits and the Effects on Wheat (Triticum aestivum L.) Yields. Theor. Appl. Genet. 2024, 137, 87. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, M.; Lin, K.; Xie, Y.; Guo, J.; Ye, L.; Zhuang, Y.; Teng, W.; Ran, X.; Tong, Y.; et al. The Bread Wheat Epigenomic Map Reveals Distinct Chromatin Architectural and Evolutionary Features of Functional Genetic Elements. Genome Biol. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, Z.; Luo, B.; Zhong, H.; Ma, P.; Zhang, H.; Hu, H.; Wang, Y.; Zhang, H.; Liu, D.; et al. Genetic Architecture of Maize Yield Traits Dissected by QTL Mapping and GWAS in Maize. Crop J. 2022, 10, 436–446. [Google Scholar] [CrossRef]
- Wang, D.; Sun, W.; Yuan, Z.; Sun, Q.; Fan, K.; Zhang, C.; Yu, S. Identification of a Novel QTL and Candidate Gene Associated with Grain Size Using Chromosome Segment Substitution Lines in Rice. Sci. Rep. 2021, 11, 189. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, Y.; Li, Z.; Zhu, J.; Liu, M.; Zhang, Y.; Dong, Z. Enhancer Transcription Detected in the Nascent Transcriptomic Landscape of Bread Wheat. Genome Biol. 2022, 23, 109. [Google Scholar] [CrossRef]
- Wei, J.; Fang, Y.; Jiang, H.; Wu, X.; Zuo, J.; Xia, X.; Li, J.; Stich, B.; Cao, H.; Liu, Y. Combining QTL Mapping and Gene Co-Expression Network Analysis for Prediction of Candidate Genes and Molecular Network Related to Yield in Wheat. BMC Plant Biol. 2022, 22, 288. [Google Scholar] [CrossRef]
- Chen, W.; Chen, L.; Zhang, X.; Yang, N.; Guo, J.; Wang, M.; Ji, S.; Zhao, X.; Yin, P.; Cai, L.; et al. Convergent Selection of a WD40 Protein That Enhances Grain Yield in Maize and Rice. Science 2022, 375, eabg7985. [Google Scholar] [CrossRef]
- Wang, S.; Xiao, Y.; Zhou, Z.W.; Yuan, J.; Guo, H.; Yang, Z.; Yang, J.; Sun, P.; Sun, L.; Deng, Y.; et al. High-Quality Reference Genome Sequences of Two Coconut Cultivars Provide Insights into Evolution of Monocot Chromosomes and Differentiation of Fiber Content and Plant Height. Genome Biol. 2021, 22, 304. [Google Scholar] [CrossRef]
- Wen, T.; Liu, C.; Wang, T.; Wang, M.; Tang, F.; He, L. Genomic Mapping and Identification of Candidate Genes Encoding Nulliplex-Branch Trait in Sea-Island Cotton (Gossypium barbadense L.) by Multi-Omics Analysis. Mol. Breed. 2021, 41, 34. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.L.; Schaefer, R.; Dang, P.; Jiang, T.; Chen, C. GWAS and Coexpression Network Reveal Ionomic Variation in Cultivated Peanut. J. Agric. Food Chem. 2019, 67, 12026–12036. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, L.; Chen, Y.; Wang, X.; Huai, D.; Kang, Y.; Jiang, H.; Liu, K.; Lei, Y.; Liao, B. Detection of a Major QTL and Development of KASP Markers for Seed Weight by Combining QTL-Seq, QTL-Mapping and RNA-Seq in Peanut. Theor. Appl. Genet. 2022, 135, 1779–1795. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, H.; Lu, S.; Yu, L.; Zhang, G.; Zhang, Y.; Yang, Q.Y.; Zhou, Y.; Wang, X.; Ma, W.; et al. Genome- and Transcriptome-Wide Association Studies Provide Insights into the Genetic Basis of Natural Variation of Seed Oil Content in Brassica Napus. Mol. Plant 2021, 14, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.; Yu, Z.; Zhang, Y.; Choe, K.; Uh, S.; Kim, K.; Jong, C.; Cha, J.; Kim, M.; Kim, Y.; et al. Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile. Int. J. Mol. Sci. 2024, 25, 5614. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Meng, X.; Zhao, X.; Ding, X.; Li, Y.; Cao, G.; Chu, Z.; Su, X.; Liu, Y.; Chen, X.; et al. Integrated Functional Omics Analysis of Flavonoid-Related Metabolism in AtMYB12 Transcript Factor Overexpressed Tomato. J. Agric. Food Chem. 2020, 68, 6776–6787. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Xiang, L.; Lai, J.; Li, C.; Zhong, Y.; Ye, W.; Yang, J.; Yang, J.; Wang, S. SlERF.H6 Mediates the Orchestration of Ethylene and Gibberellin Signaling That Suppresses Bitter-SGA Biosynthesis in Tomato. New Phytol. 2023, 239, 1353–1367. [Google Scholar] [CrossRef]
- Guo, H.; Li, C.; Lai, J.; Tong, H.; Cao, Z.; Wang, C.; Zhao, W.; He, L.; Wang, S.; Yang, J.; et al. Comprehensive Analysis of Metabolome and Transcriptome Reveals the Regulatory Network of Coconut Nutrients. Metabolites 2023, 13, 683. [Google Scholar] [CrossRef]
- Wang, S.; Shen, S.; Wang, C.; Wang, X.; Yang, C.; Zhou, S.; Zhang, R.; Zhou, Q.; Yu, H.; Guo, H.; et al. A Metabolomics Study in Citrus Provides Insight into Bioactive Phenylpropanoid Metabolism. Hortic. Res. 2023, 11, uhad267. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Chen, L.H.; Fan, B.L.; Xu, Z.; Wang, Q.; Zhao, B.Y.; Gao, M.; Yuan, M.H.; Tahir ul Qamar, M.; Jiang, Y.; et al. Integrative Multiomics Profiling of Passion Fruit Reveals the Genetic Basis for Fruit Color and Aroma. Plant Physiol. 2024, 194, 2491–2510. [Google Scholar] [CrossRef]
- Tang, K.; Karamat, U.; Li, G.; Guo, J.; Jiang, S.; Fu, M.; Yang, X. Integrated Metabolome and Transcriptome Analyses Reveal the Role of BoGSTF12 in Anthocyanin Accumulation in Chinese Kale (Brassica oleracea var. alboglabra). BMC Plant Biol. 2024, 24, 335. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, L.; Zhang, B.; Huang, W.; Zhang, Z.; An, B. Integrated Analysis of the Metabolome and Transcriptome Provides Insights into Anthocyanin Biosynthesis of Cashew Apple. Food Res. Int. 2024, 175, 113711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hu, F.; Cai, X.; Cheng, J.; Zhang, Y.; Lin, H.; Hu, K.; Wu, Z. Integrative Analysis of the Metabolome and Transcriptome of a Cultivated Pepper and Its Wild Progenitor Chiltepin (Capsicum annuum L. var. glabriusculum) Revealed the Loss of Pungency During Capsicum Domestication. Front. Plant Sci. 2022, 12, 783496. [Google Scholar] [CrossRef] [PubMed]
- Salami, M.; Heidari, B.; Alizadeh, B.; Batley, J.; Wang, J.; Tan, X.L.; Dadkhodaie, A.; Richards, C. Dissection of Quantitative Trait Nucleotides and Candidate Genes Associated with Agronomic and Yield-Related Traits under Drought Stress in Rapeseed Varieties: Integration of Genome-Wide Association Study and Transcriptomic Analysis. Front. Plant Sci. 2024, 15, 1342359. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.K.; Singh, V.; Anwar, K.; Pareek, A.; Jain, M. Integrated Transcriptome, Proteome and Metabolome Analyses Revealed Secondary Metabolites and Auxiliary Carbohydrate Metabolism Augmenting Drought Tolerance in Rice. Plant Physiol. Biochem. 2023, 201, 107849. [Google Scholar] [CrossRef]
- Cao, P.; Yang, J.; Xia, L.; Zhang, Z.; Wu, Z.; Hao, Y.; Liu, P.; Wang, C.; Li, C.; Yang, J.; et al. Two Gene Clusters and Their Positive Regulator SlMYB13 That Have Undergone Domestication-Associated Negative Selection Control Phenolamide Accumulation and Drought Tolerance in Tomato. Mol. Plant 2024, 17, 579–597. [Google Scholar] [CrossRef]
- Yu, P.; Li, C.; Li, M.; He, X.; Wang, D.; Li, H.; Marcon, C.; Li, Y.; Perez-Limón, S.; Chen, X.; et al. Seedling Root System Adaptation to Water Availability during Maize Domestication and Global Expansion. Nat. Genet. 2024, 56, 1245–1256. [Google Scholar] [CrossRef]
- Gui, S.; Wei, W.; Jiang, C.; Luo, J.; Chen, L.; Wu, S.; Li, W.; Wang, Y.; Li, S.; Yang, N.; et al. A Pan-Zea Genome Map for Enhancing Maize Improvement. Genome Biol. 2022, 23, 178. [Google Scholar] [CrossRef]
- Song, J.; Li, J.; Sun, J.; Hu, T.; Wu, A.; Liu, S.; Wang, W.; Ma, D.; Zhao, M. Genome-Wide Association Mapping for Cold Tolerance in a Core Collection of Rice (Oryza sativa L.) Landraces by Using High-Density Single Nucleotide Polymorphism Markers from Specific-Locus Amplified Fragment Sequencing. Front. Plant Sci. 2018, 9, 875. [Google Scholar] [CrossRef]
- Gu, S.; Zhuang, J.; Zhang, Z.; Chen, W.; Xu, H.; Zhao, M.; Ma, D. Multi-Omics Approach Reveals the Contribution of OsSEH1 to Rice Cold Tolerance. Front. Plant Sci. 2023, 13, 1110724. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, S.; Zhao, S.; Chen, D.; Tian, H.; Li, J.; Zhang, L.; Li, S.; Liu, L.; Shi, C.; et al. Global Crotonylatome and GWAS Revealed a TaSRT1-TaPGK Model Regulating Wheat Cold Tolerance through Mediating Pyruvate. Sci. Adv. 2024, 9, eadg1012. [Google Scholar] [CrossRef]
- Sun, Z.; Qi, F.; Liu, H.; Qin, L.; Xu, J.; Shi, L.; Zhang, Z.; Miao, L.; Huang, B.; Dong, W.; et al. QTL Mapping of Quality Traits in Peanut Using Whole-Genome Resequencing. Crop J. 2022, 10, 177–184. [Google Scholar] [CrossRef]
- Abdelrahman, M.; Nishiyama, R.; Tran, C.D.; Kusano, M.; Nakabayashi, R.; Okazaki, Y.; Matsuda, F.; Chávez Montes, R.A.; Mostofa, M.G.; Li, W.; et al. Defective Cytokinin Signaling Reprograms Lipid and Flavonoid Gene-to-Metabolite Networks to Mitigate High Salinity in Arabidopsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2105021118. [Google Scholar] [CrossRef]
- Hu, P.; Zheng, Q.; Luo, Q.; Teng, W.; Li, H.; Li, B.; Li, Z. Genome-Wide Association Study of Yield and Related Traits in Common Wheat under Salt-Stress Conditions. BMC Plant Biol. 2021, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.M.; Franzisky, B.L.; Messerer, M.; Du, B.; Lux, T.; White, P.J.; Carpentier, S.C.; Winkler, J.B.; Schnitzler, J.P.; El-Serehy, H.A.; et al. Integrative Multi-Omics Analyses of Date Palm (Phoenix dactylifera) Roots and Leaves Reveal How the Halophyte Land Plant Copes with Sea Water. Plant Genome 2024, 17, e20372. [Google Scholar] [CrossRef]
- Wu, W.; Dong, X.; Chen, G.; Lin, Z.; Chi, W.; Tang, W.; Yu, J.; Wang, S.; Jiang, X.; Liu, X.; et al. The Elite Haplotype OsGATA8-H Coordinates Nitrogen Uptake and Productive Tiller Formation in Rice. Nat. Genet. 2024, 56, 1516–1526. [Google Scholar] [CrossRef]
- Ichihashi, Y.; Date, Y.; Shino, A.; Shimizu, T.; Shibata, A.; Kumaishi, K.; Funahashi, F.; Wakayama, K.; Yamazaki, K.; Umezawa, A.; et al. Multi-Omics Analysis on an Agroecosystem Reveals the Significant Role of Organic Nitrogen to Increase Agricultural Crop Yield. Proc. Natl. Acad. Sci. USA 2020, 117, 14552–14560. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, J.; Wang, Y.; Wei, X.; Yin, Z.; Zhang, Y.; Pu, A.; Dong, Z.; Long, Y.; Wan, X. Mining Genic Resources Regulating Nitrogen-Use Efficiency Based on Integrative Biological Analyses and Their Breeding Applications in Maize and Other Crops. Plant J. 2024, 117, 1148–1164. [Google Scholar] [CrossRef]
- Zhao, E.; Dong, L.; Zhao, H.; Zhang, H.; Zhang, T.; Yuan, S.; Jiao, J.; Chen, K.; Sheng, J.; Yang, H.; et al. A Relationship Prediction Method for Magnaporthe Oryzae-Rice Multi-Omics Data Based on WGCNA and Graph Autoencoder. J Fungi 2023, 9, 1007. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, Y.; Li, Y.; Xing, M.; Lei, C.; Wang, S.; Nie, Y.; Wang, Y.; Zhao, M.; Han, Z.; et al. Haplotype-Resolved Gapless Genome and Chromosome Segment Substitution Lines Facilitate Gene Identification in Wild Rice. Nat. Commun. 2024, 15, 4573. [Google Scholar] [CrossRef]
- Liu, F.; Cai, S.; Ma, Z.; Yue, H.; Xing, L.; Wang, Y.; Feng, S.; Wang, L.; Dai, L.; Wan, H.; et al. RVE2, a New Regulatory Factor in Jasmonic Acid Pathway, Orchestrates Resistance to Verticillium Wilt. Plant Biotechnol. J. 2023, 21, 2507–2524. [Google Scholar] [CrossRef]
- He, Y.; Zhang, K.; Li, S.; Lu, X.; Zhao, H.; Guan, C.; Huang, X.; Shi, Y.; Kang, Z.; Fan, Y.; et al. Multiomics Analysis Reveals the Molecular Mechanisms Underlying Virulence in Rhizoctonia and Jasmonic Acid-Mediated Resistance in Tartary Buckwheat (Fagopyrum tataricum). Plant Cell 2023, 35, 2773–2798. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Yu, H.; Fu, Q.; Chen, H.; Ye, W.; Li, S.; Lou, Y. Comparative Transcriptome Analysis of Salivary Glands of Two Populations of Rice Brown Planthopper, Nilaparvata Lugens, That Differ in Virulence. PLoS ONE 2013, 8, e79612. [Google Scholar] [CrossRef]
- Ye, W.; Yu, H.; Jian, Y.; Zeng, J.; Ji, R.; Chen, H.; Lou, Y. A Salivary EF-Hand Calcium-Binding Protein of the Brown Planthopper Nilaparvata Lugens Functions as an Effector for Defense Responses in Rice. Sci. Rep. 2017, 7, 40498. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Ye, W.; Chen, H.; Zeng, J.; Li, H.; Yu, H.; Li, J.; Lou, Y. A Salivary Endo-β-1,4-Glucanase Acts as an Effector That Enables the Brown Planthopper to Feed on Rice. Plant Physiol. 2017, 173, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zha, W.; Yu, X.; Wu, Y.; Li, S.; Xu, H.; Li, P.; Li, C.; Liu, K.; Chen, J.; et al. Integrated Transcriptomics and Metabolomics Analysis Provide Insight into the Resistance Response of Rice against Brown Planthopper. Front. Plant Sci. 2023, 14, 1213257. [Google Scholar] [CrossRef] [PubMed]
- Gokulan, C.G.; Bangale, U.; Balija, V.; Ballichatla, S.; Potupureddi, G.; Rao, D.; Varma, P.; Magar, N.; Jallipalli, K.; Manthri, S.; et al. Multiomics-Assisted Characterization of Rice-Yellow Stem Borer Interaction Provides Genomic and Mechanistic Insights into Stem Borer Resistance in Rice. Theor. Appl. Genet. 2024, 137, 122. [Google Scholar] [CrossRef]
- Zhou, R.; Jiang, F.; Niu, L.; Song, X.; Yu, L.; Yang, Y.; Wu, Z. Increase Crop Resilience to Heat Stress Using Omic Strategies. Front. Plant Sci. 2022, 13, 891861. [Google Scholar] [CrossRef]
- Momeni, Z.; Hassanzadeh, E.; Saniee Abadeh, M.; Bellazzi, R. A Survey on Single and Multi Omics Data Mining Methods in Cancer Data Classification. J. Biomed. Inform. 2020, 107, 103466. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Method of the Year 2019: Single-Cell Multimodal Omics. Nat. Methods 2020, 17, 1. [CrossRef]
- Method of the Year 2020: Spatially Resolved Transcriptomics. Nat. Methods 2021, 18, 1. [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-Cell RNA Sequencing Technologies and Applications: A Brief Overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef] [PubMed]
- Maynard, K.R.; Jaffe, A.E.; Martinowich, K. Spatial Transcriptomics: Putting Genome-Wide Expression on the Map. Neuropsychopharmacology 2020, 45, 232–233. [Google Scholar] [CrossRef]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating Single-Cell and Spatial Transcriptomics to Elucidate Intercellular Tissue Dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef]
- Mo, Y.; Jiao, Y. Advances and Applications of Single-Cell Omics Technologies in Plant Research. Plant J. 2022, 110, 1551–1563. [Google Scholar] [CrossRef]
- Levitin, H.M.; Yuan, J.; Sims, P.A. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer 2018, 4, 264–268. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, J.; Zhou, Y.; Zhang, Y.; Qin, A.; Yu, X.; Zhao, Z.; Wu, R.; Guo, C.; Bawa, G.; et al. Identification of Novel Regulators Required for Early Development of Vein Pattern in the Cotyledons by Single-Cell RNA-Sequencing. Plant J. 2022, 110, 7–22. [Google Scholar] [CrossRef]
- Denyer, T.; Ma, X.; Klesen, S.; Scacchi, E.; Nieselt, K.; Timmermans, M.C.P. Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell RNA Sequencing. Dev. Cell 2019, 48, 840–852.e5. [Google Scholar] [CrossRef]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O’Malley, R.C.; Brady, S.M.; et al. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell Rep. 2019, 27, 2241–2247.e4. [Google Scholar] [CrossRef]
- Zhang, T.Q.; Xu, Z.G.; Shang, G.D.; Wang, J.W. A Single-Cell RNA Sequencing Profiles the Developmental Landscape of Arabidopsis Root. Mol. Plant 2019, 12, 648–660. [Google Scholar] [CrossRef]
- Song, Q.; Ando, A.; Jiang, N.; Ikeda, Y.; Chen, Z.J. Single-Cell RNA-Seq Analysis Reveals Ploidy-Dependent and Cell-Specific Transcriptome Changes in Arabidopsis Female Gametophytes. Genome Biol. 2020, 21, 178. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Du, Q.; Xu, M.; Du, F.; Jiao, Y. Single-Nucleus RNA-Seq Resolves Spatiotemporal Developmental Trajectories in the Tomato Shoot Apex. bioRxiv 2020, 2020.09.20.305029. [Google Scholar] [CrossRef]
- Nelms, B.; Walbot, V. Defining the Developmental Program Leading to Meiosis in Maize. Science 2019, 364, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Satterlee, J.W.; Strable, J.; Scanlon, M.J. Plant Stem-Cell Organization and Differentiation at Single-Cell Resolution. Proc. Natl. Acad. Sci. USA 2020, 117, 33689–33699. [Google Scholar] [CrossRef]
- Ma, X.; Denyer, T.; Timmermans, M.C.P. PscB: A Browser to Explore Plant Single Cell RNA-Sequencing Data Sets. Plant Physiol. 2020, 183, 464–467. [Google Scholar] [CrossRef]
- Huang, K.; Batish, M.; Teng, C.; Harkess, A.; Meyers, B.C.; Caplan, J.L. Quantitative Fluorescence In Situ Hybridization Detection of Plant MRNAs with Single-Molecule Resolution. Methods Mol. Biol. 2020, 2166, 23–33. [Google Scholar] [CrossRef]
- Zöllner, N.R.; Bezrutczyk, M.; Laureyns, R.; Nelissen, H.; Simon, R.; Frommer, W.B. An RNA in Situ Hybridization Protocol Optimized for Monocot Tissue. STAR Protoc. 2021, 2, 100398. [Google Scholar] [CrossRef]
- Yang, H.; Nukunya, K.; Ding, Q.; Thompson, B.E. Tissue-Specific Transcriptomics Reveal Functional Differences in Floral Development. Plant Physiol. 2022, 188, 1158–1173. [Google Scholar] [CrossRef]
- Giacomello, S.; Salmén, F.; Terebieniec, B.K.; Vickovic, S.; Navarro, J.F.; Alexeyenko, A.; Reimegård, J.; McKee, L.S.; Mannapperuma, C.; Bulone, V.; et al. Spatially Resolved Transcriptome Profiling in Model Plant Species. Nat. Plants 2017, 3, 17061. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Xia, K.; Sun, H.X.; Li, J.; Li, J.; Zhao, Y.; Chen, L.; Qin, C.; Chen, R.; Chen, Z.; Liu, G.; et al. The Single-Cell Stereo-Seq Reveals Region-Specific Cell Subtypes and Transcriptome Profiling in Arabidopsis Leaves. Dev. Cell 2022, 57, 1299–1310.e4. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal Transcriptomic Atlas of Mouse Organogenesis Using DNA Nanoball-Patterned Arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hu, Q.; Lv, T.; Wang, Y.; Lan, Q.; Xiang, R.; Tu, Z.; Wei, Y.; Han, K.; Shi, C.; et al. High-Resolution 3D Spatiotemporal Transcriptomic Maps of Developing Drosophila Embryos and Larvae. Dev. Cell 2022, 57, 1271–1283.e4. [Google Scholar] [CrossRef]
- Liu, C.; Li, R.; Li, Y.; Lin, X.; Zhao, K.; Liu, Q.; Wang, S.; Yang, X.; Shi, X.; Ma, Y.; et al. Spatiotemporal Mapping of Gene Expression Landscapes and Developmental Trajectories during Zebrafish Embryogenesis. Dev. Cell 2022, 57, 1284–1298.e5. [Google Scholar] [CrossRef]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch Effects in Single-Cell RNA-Sequencing Data Are Corrected by Matching Mutual Nearest Neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and Applications for Single-Cell and Spatial Multi-Omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Yu, X.; Liu, Z.; Sun, X. Single-Cell and Spatial Multi-Omics in the Plant Sciences: Technical Advances, Applications, and Perspectives. Plant Commun. 2023, 4, 100508. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Alexis, M.S.; Burge, C.B. Meta-Analysis of RNA-Seq Expression Data across Species, Tissues and Studies. Genome Biol. 2015, 16, 287. [Google Scholar] [CrossRef]
- Luo, C.; Fernie, A.R.; Yan, J. Single-Cell Genomics and Epigenomics: Technologies and Applications in Plants. Trends Plant Sci. 2020, 25, 1030–1040. [Google Scholar] [CrossRef]
- Tajik, M.; Baharfar, M.; Donald, W.A. Single-Cell Mass Spectrometry. Trends Biotechnol. 2022, 40, 1374–1392. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Multi-Omics | Method | Reference |
|---|---|---|
| Genomics | Sanger sequencing | [85] |
| Whole-genome sequencing (WGS) | [86] | |
| Whole-exome sequencing (WES) | [87] | |
| Replication sequencing (Repli-seq) | [88] | |
| PacBio Single-molecule real-time sequencing (SMRT) technology | [20] | |
| Nanopore DNA sequencing | [21] | |
| Epigenomics | Chromatin immunoprecipitation (ChIP-seq) | [89] |
| ChIP-exo | [90] | |
| Assay for transposase-accessible chromatin (ATAC-seq) | [53] | |
| Hi-C | [91] | |
| Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) | [92] | |
| Chromatin isolation by RNA purification sequencing (ChIRP-seq) | [93] | |
| Reduced representation bisulfite sequencing (RRBS-seq) | [94] | |
| Bisulfite sequencing (BS-seq) | [95] | |
| Methyl-CpG-Binding Domain Sequencing (MBD-seq) | [96] | |
| DNAse-seq | [97] | |
| Parallel Analysis of RNA Structure (PARS) | [98] | |
| Structure-seq | [99] | |
| Parallel analysis of RNA ends sequencing (PARE-seq) | [100] | |
| Massively parallel functional dissection sequencing (MPFD) | [101] | |
| Methylated RNA immunoprecipitation sequencing (MeRIP-seq) | [102] | |
| Single-molecule real-time sequencing (SMRT-seq) | [103] | |
| Transcriptomics | RNA sequencing (RNA-seq) | [104] |
| Isoform sequencing (Iso-seq) | [105] | |
| Targeted RNA sequencing | [106] | |
| Ribosome profiling (Ribo-seq) | [107] | |
| Global run-on sequencing (GRO-seq) | [108] | |
| Nascent-seq | [109] | |
| Native elongating transcript sequencing (NET-seq) | [110] | |
| PolyA-sequencing (PolyA-seq) | [111] | |
| Proteomics | Mass Spectrometry | [112] |
| Liquid Chromatography–Mass Spectrometry (LC-MS) | [113] | |
| Reverse Phase Protein Array (RPPA) | [114] | |
| Gel electrophoresis | [115] | |
| Isobaric tag for relative and absolute quantitation (iTRAQ) | [116] | |
| Stable isotope labeling by amino acids in cell culture (SILAC) | [117] | |
| Metabolomics | Mass Spectrometry | [118] |
| Nuclear Magnetic Resonance (NMR) | [68] | |
| Liquid Chromatography–Mass Spectrometry (LC-MS) | [70] | |
| Gas Chromatography–Mass Spectrometry (GC-MS) | [69] | |
| Interactomics | RNA on a massively parallel array (RNAMaP) | [119] |
| RNA immunoprecipitation sequencing (RIP-seq) | [120] | |
| ChIP-Seq | [89] | |
| Yeast Two-Hybrid (Y2H) | [121] | |
| Bimolecular fluorescence complementation (BiFC) | [122] | |
| Crosslinking-immunoprecipitation sequencing (CLIP) | [123] | |
| Ionomics | Inductively coupled plasma mass spectrometry (ICP-MS) | [124] |
| Synchrotron X-ray fluorescence (SXRF) | [125] | |
| Deletion mapping | [126] | |
| DNA microarray-based bulk segregant analysis (BSA) | [127] | |
| Microbiomics | 16S rRNA | [128] |
| 18S rRNA | [129] | |
| Internal transcribed spacer (ITS) | [130] | |
| Shotgun | [131] | |
| Metagenomics | [132] | |
| Single-cell | Single-cell RNA sequencing (scRNA-seq) | [133] |
| scATAC-seq | [134] | |
| scDNase-seq | [135] | |
| Single-cell genome-wide bisulfite sequencing | [136] | |
| Single-cell ChIP-seq | [137] | |
| Single-cell Hi-C | [138] | |
| proximity ligation assay for RNA (PLAYR) | [139] | |
| Mass Spectrometry for single-cell | [140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, B.-L.; Chen, L.-H.; Chen, L.-L.; Guo, H. Integrative Multi-Omics Approaches for Identifying and Characterizing Biological Elements in Crop Traits: Current Progress and Future Prospects. Int. J. Mol. Sci. 2025, 26, 1466. https://doi.org/10.3390/ijms26041466
Fan B-L, Chen L-H, Chen L-L, Guo H. Integrative Multi-Omics Approaches for Identifying and Characterizing Biological Elements in Crop Traits: Current Progress and Future Prospects. International Journal of Molecular Sciences. 2025; 26(4):1466. https://doi.org/10.3390/ijms26041466
Chicago/Turabian StyleFan, Bing-Liang, Lin-Hua Chen, Ling-Ling Chen, and Hao Guo. 2025. "Integrative Multi-Omics Approaches for Identifying and Characterizing Biological Elements in Crop Traits: Current Progress and Future Prospects" International Journal of Molecular Sciences 26, no. 4: 1466. https://doi.org/10.3390/ijms26041466
APA StyleFan, B.-L., Chen, L.-H., Chen, L.-L., & Guo, H. (2025). Integrative Multi-Omics Approaches for Identifying and Characterizing Biological Elements in Crop Traits: Current Progress and Future Prospects. International Journal of Molecular Sciences, 26(4), 1466. https://doi.org/10.3390/ijms26041466