
Chondrosarcoma: New Molecular Insights, Challenges in Near-Patient Preclinical Modeling, and Therapeutic Approaches


Abstract
1. Introduction
2. CS Histological Classification and Clinical Aspects
3. CS Genetics
3.1. IDH1/IDH2 Mutations
3.2. TP53 Mutations
3.3. EPAS1 Gene Amplification and Hypoxia-Inducible Factor-2α (HIF-2α) Upregulation
3.4. MYC Amplification in CS
3.5. CS Multi-Omic Profiling
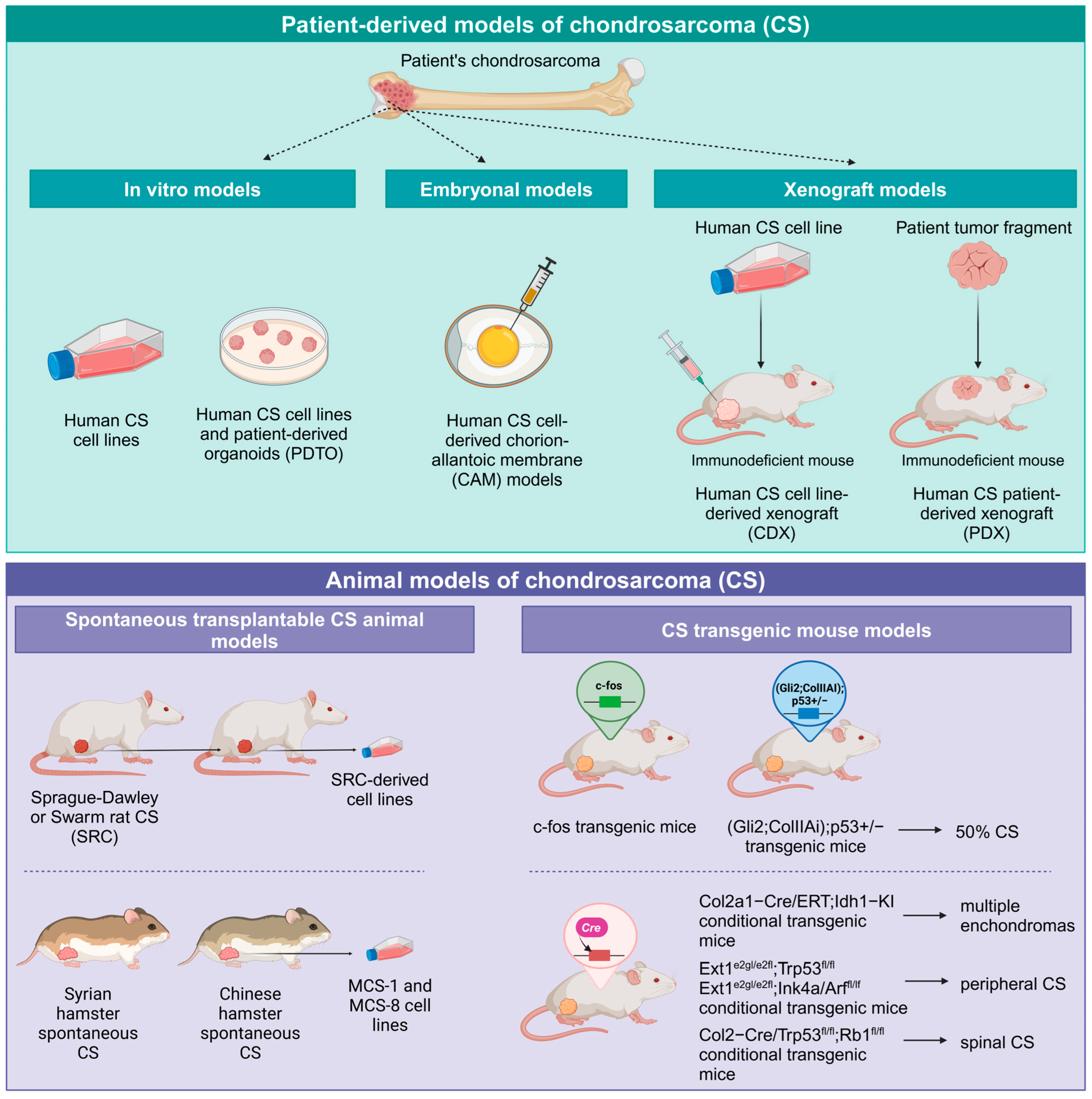
4. CS Preclinical Models and Near-Patient Models
4.1. CS Cell Lines
4.2. Organoids and 3D Models for CS
4.3. CS Chorion-Allantoic Membrane (CAM) Models
4.4. CS In Vivo Animal Models
4.5. Spontaneous Transplantable CS Animal Models
4.6. CS Transgenic Mouse Models
4.7. Human-Derived CS Xenograft Models in Mice
4.8. CS Cell Line-Derived Xenograft Models in Mice
4.9. CS Patient-Derived-Xenograft (PDX) Models
5. Innovative Therapies Currently Under Investigation in CS
5.1. Mutant IDH Targeted Therapies
5.2. HIF-2a and SIRT1 Targeted Therapies to Enhance CS Drug Sensitivity
5.3. Death Receptor 5 (DR5) Targeting in CS
5.4. Antiangiogenic Therapies
5.5. Chondroitin Sulfate Proteoglycan 4 Immunological Targeting
5.6. Immunotherapeutic Approaches in CS
5.7. Epigenetic Therapies
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Lou, S.; Yao, T. Trends in primary malignant bone cancer incidence and mortality in the United States, 2000–2017: A population-based study. J. Bone Oncol. 2024, 46, 100607. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, F.; Zhang, Y.; Yin, M.; Han, X.; Feng, J.; Wang, G. Twenty-year outcome of prevalence, incidence, mortality and survival rate in patients with malignant bone tumors. Int. J. Cancer 2024, 154, 226–240. [Google Scholar] [CrossRef]
- Van Praag (Veroniek), V.M.; Rueten-Budde, A.J.; Ho, V.; Dijkstra, P.D.S.; Fiocco, M.; Van De Sande, M.A.J.; Van Der Geest, I.C.; Bramer, J.A.; Schaap, G.R.; Jutte, P.C.; et al. Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg. Oncol. 2018, 27, 402–408. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lee, S.K. Classification of Chondrosarcoma: From Characteristic to Challenging Imaging Findings. Cancers 2023, 15, 1703. [Google Scholar] [CrossRef]
- Gerrand, C.; Amary, F.; Anwar, H.A.; Brennan, B.; Dileo, P.; Kalkat, M.S.; McCabe, M.G.; McCullough, A.L.; Parry, M.C.; Patel, A.; et al. UK guidelines for the management of bone sarcomas. Br. J. Cancer 2024, 132, 32–48. [Google Scholar] [CrossRef] [PubMed]
- De Andrea, C.E.; Zhu, J.; Jin, H.; Bovée, J.V.; Jones, K.B. Cell cycle deregulation and mosaic loss of Ext1 drive peripheral chondrosarcomagenesis in the mouse and reveal an intrinsic cilia deficiency. J. Pathol. 2015, 236, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, A.; Popovic, S.; Parasu, N.; Ghert, M. Chondrosarcoma: A Clinical Review. J. Clin. Med. 2023, 12, 2506. [Google Scholar] [CrossRef]
- El Beaino, M.; Roszik, J.; Livingston, J.A.; Wang, W.-L.; Lazar, A.J.; Amini, B.; Subbiah, V.; Lewis, V.; Conley, A.P. Mesenchymal Chondrosarcoma: A Review with Emphasis on its Fusion-Driven Biology. Curr. Oncol. Rep. 2018, 20, 37. [Google Scholar] [CrossRef]
- Klein, A.; Tauscher, F.; Birkenmaier, C.; Baur-Melnyk, A.; Knösel, T.; Jansson, V.; Dürr, H.R. Clear cell chondrosarcoma is an underestimated tumor: Report of 7 cases and meta-analysis of the literature. J. Bone Oncol. 2019, 19, 100267. [Google Scholar] [CrossRef]
- Flint, J.H.; Conley, A.P.; Rubin, M.L.; Feng, L.; Lin, P.P.; Moon, B.; Bird, J.; Satcher, R.L.; Lewis, V.O. Clear Cell Chondrosarcoma: Clinical Characteristics and Outcomes in 15 Patients. Sarcoma 2020, 2020, 2386191. [Google Scholar] [CrossRef]
- Brenca, M.; Stacchiotti, S.; Fassetta, K.; Sbaraglia, M.; Janjusevic, M.; Racanelli, D.; Polano, M.; Rossi, S.; Brich, S.; Dagrada, G.P.; et al. NR4A3 fusion proteins trigger an axon guidance switch that marks the difference between EWSR1 and TAF15 translocated extraskeletal myxoid chondrosarcomas. J. Pathol. 2019, 249, 90–101. [Google Scholar] [CrossRef]
- Drilon, A.D.; Popat, S.; Bhuchar, G.; D’Adamo, D.R.; Keohan, M.L.; Fisher, C.; Antonescu, C.R.; Singer, S.; Brennan, M.F.; Judson, I.; et al. Extraskeletal myxoid chondrosarcoma: A retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 2008, 113, 3364–3371. [Google Scholar] [CrossRef]
- Zając, A.E.; Kopeć, S.; Szostakowski, B.; Spałek, M.J.; Fiedorowicz, M.; Bylina, E.; Filipowicz, P.; Szumera-Ciećkiewicz, A.; Tysarowski, A.; Czarnecka, A.M.; et al. Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers 2021, 13, 2390. [Google Scholar] [CrossRef] [PubMed]
- Van Maldegem, A.; Conley, A.P.; Rutkowski, P.; Patel, S.R.; Lugowska, I.; Desar, I.M.E.; Bovée, J.V.M.G.; Gelderblom, H. Outcome of First-Line Systemic Treatment for Unresectable Conventional, Dedifferentiated, Mesenchymal, and Clear Cell Chondrosarcoma. Oncologist 2019, 24, 110–116. [Google Scholar] [CrossRef]
- Denu, R.A.; Yang, R.K.; Lazar, A.J.; Patel, S.S.; Lewis, V.O.; Roszik, J.; Livingston, J.A.; Wang, W.-L.; Shaw, K.R.; Ratan, R.; et al. Clinico-Genomic Profiling of Conventional and Dedifferentiated Chondrosarcomas Reveals TP53 Mutation to Be Associated with Worse Outcomes. Clin. Cancer Res. 2023, 29, 4844–4852. [Google Scholar] [CrossRef]
- Hompland, I.; Ferrari, S.; Bielack, S.; Palmerini, E.; Hall, K.S.; Picci, P.; Hecker-Nolting, S.; Donati, D.M.; Blattmann, C.; Bjerkehagen, B.; et al. Outcome in dedifferentiated chondrosarcoma for patients treated with multimodal therapy: Results from the EUROpean Bone Over 40 Sarcoma Study. Eur. J. Cancer 2021, 151, 150–158. [Google Scholar] [CrossRef]
- Stevenson, J.D.; Laitinen, M.K.; Parry, M.C.; Sumathi, V.; Grimer, R.J.; Jeys, L.M. The role of surgical margins in chondrosarcoma. Eur. J. Surg. Oncol. 2018, 44, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Amer, K.M.; Munn, M.; Congiusta, D.; Abraham, J.A.; Basu Mallick, A. Survival and Prognosis of Chondrosarcoma Subtypes: SEER Database Analysis. J. Orthop. Res. 2020, 38, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Matsumine, A.; Yamada, S.; Tsukushi, S.; Kawanami, K.; Ohno, T.; Katagiri, H.; Sugiura, H.; Yamada, K.; Yamada, Y.; et al. Oncological outcome after lung metastasis in patients presenting with localized chondrosarcoma at extremities: Tokai Musculoskeletal Oncology Consortium study. OncoTargets Ther. 2016, 9, 4747–4751. [Google Scholar] [CrossRef]
- Kim, S.; Ott, H.C.; Wright, C.D.; Wain, J.C.; Morse, C.; Gaissert, H.A.; Donahue, D.M.; Mathisen, D.J.; Lanuti, M. Pulmonary Resection of Metastatic Sarcoma: Prognostic Factors Associated with Improved Outcomes. Ann. Thorac. Surg. 2011, 92, 1780–1787. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Osanai, T.; Ogose, A.; Tamura, G.; Chano, T.; Kaneko, Y.; Ishikawa, A.; Orui, H.; Wada, T.; Ikeda, T.; et al. Methylation status of EXT1 and EXT2 promoters and two mutations of EXT2 in chondrosarcoma. Cancer Genet. Cytogenet. 2005, 158, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, S.; Liu, Y.; Yang, S. Mice with Trp53 and Rb1 deficiency in chondrocytes spontaneously develop chondrosarcoma via overactivation of YAP signaling. Cell Death Dis. 2022, 13, 570. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.G.; Nafa, K.; Agaram, N.; Zehir, A.; Benayed, R.; Sadowska, J.; Borsu, L.; Kelly, C.; Tap, W.D.; Fabbri, N.; et al. Genomic Profiling Identifies Association of IDH1/IDH2 Mutation with Longer Relapse-Free and Metastasis-Free Survival in High-Grade Chondrosarcoma. Clin. Cancer Res. 2020, 26, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Behjati, S.; Cooke, S.L.; Van Loo, P.; Wedge, D.C.; Pillay, N.; Marshall, J.; O’Meara, S.; Davies, H.; Nik-Zainal, S.; et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 2013, 45, 923–926. [Google Scholar] [CrossRef]
- Qi, W.; Rosikiewicz, W.; Yin, Z.; Xu, B.; Jiang, H.; Wan, S.; Fan, Y.; Wu, G.; Wang, L. Genomic profiling identifies genes and pathways dysregulated by HEY1–NCOA2 fusion and shines a light on mesenchymal chondrosarcoma tumorigenesis. J. Pathol. 2022, 257, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Dermawan, J.K.T.; Nafa, K.; Mohanty, A.; Xu, Y.; Rijo, I.; Casanova, J.; Villafania, L.; Benhamida, J.; Kelly, C.M.; Tap, W.D.; et al. Distinct IDH1/2-associated Methylation Profile and Enrichment of TP53 and TERT Mutations Distinguish Dedifferentiated Chondrosarcoma from Conventional Chondrosarcoma. Cancer Res. Commun. 2023, 3, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Carosi, F.; Broseghini, E.; Fabbri, L.; Corradi, G.; Gili, R.; Forte, V.; Roncarati, R.; Filippini, D.M.; Ferracin, M. Targeting Isocitrate Dehydrogenase (IDH) in Solid Tumors: Current Evidence and Future Perspectives. Cancers 2024, 16, 2752. [Google Scholar] [CrossRef]
- Ivanov, S.; Nano, O.; Hana, C.; Bonano-Rios, A.; Hussein, A. Molecular Targeting of the Isocitrate Dehydrogenase Pathway and the Implications for Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 7337. [Google Scholar] [CrossRef]
- Venneker, S.; Bovée, J.V.M.G. IDH Mutations in Chondrosarcoma: Case Closed or Not? Cancers 2023, 15, 3603. [Google Scholar] [CrossRef]
- Hirata, M.; Sasaki, M.; Cairns, R.A.; Inoue, S.; Puviindran, V.; Li, W.Y.; Snow, B.E.; Jones, L.D.; Wei, Q.; Sato, S.; et al. Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 2829–2834. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Trovarelli, G.; Sbaraglia, M.; Angelini, A.; Bellan, E.; Pala, E.; Belluzzi, E.; Pozzuoli, A.; Borga, C.; Dei Tos, A.P.; Ruggieri, P. Are IDH1 R132 Mutations Associated with Poor Prognosis in Patients with Chondrosarcoma of the Bone? Clin. Orthop. 2024, 482, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Cleven, A.H.G.; Suijker, J.; Agrogiannis, G.; Briaire-de Bruijn, I.H.; Frizzell, N.; Hoekstra, A.S.; Wijers-Koster, P.M.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. IDH1 or -2 mutations do not predict outcome and do not cause loss of 5-hydroxymethylcytosine or altered histone modifications in central chondrosarcomas. Clin. Sarcoma Res. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, E.; Wilding, C.; Engelman, B.; Huang, P.; Jones, R.L. Is the IDH Mutation a Good Target for Chondrosarcoma Treatment? Curr. Mol. Biol. Rep. 2020, 6, 1–9. [Google Scholar] [CrossRef]
- Kim, H.; Cho, Y.; Kim, H.-S.; Kang, D.; Cheon, D.; Kim, Y.-J.; Chang, M.J.; Lee, K.M.; Chang, C.B.; Kang, S.-B.; et al. A system-level approach identifies HIF-2α as a critical regulator of chondrosarcoma progression. Nat. Commun. 2020, 11, 5023. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, J.; Xu, J.; Xie, C.; Gao, F.; Li, Z. The expression of SIRT1 regulates the metastaticplasticity of chondrosarcoma cells by inducing epithelial-mesenchymal transition. Sci. Rep. 2017, 7, 41203. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Kim, H.; Min, J.; Yeon, H.J.; Hemberg, M.; Scimeca, L.; Wu, M.-R.; Kang, H.G.; Kim, Y.-J.; Kim, J.-H. Decoupling NAD+ metabolic dependency in chondrosarcoma by targeting the SIRT1-HIF-2α axis. Cell Rep. Med. 2024, 5, 101342. [Google Scholar] [CrossRef]
- Morrison, C.; Radmacher, M.; Mohammed, N.; Suster, D.; Auer, H.; Jones, S.; Riggenbach, J.; Kelbick, N.; Bos, G.; Mayerson, J. MYC Amplification and Polysomy 8 in Chondrosarcoma: Array Comparative Genomic Hybridization, Fluorescent In Situ Hybridization, and Association With Outcome. J. Clin. Oncol. 2005, 23, 9369–9376. [Google Scholar] [CrossRef]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef]
- Zhu, J.-X.; Xiao, J.-R. SF2523 inhibits human chondrosarcoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2019, 511, 559–565. [Google Scholar] [CrossRef]
- Nicolle, R.; Ayadi, M.; Gomez-Brouchet, A.; Armenoult, L.; Banneau, G.; Elarouci, N.; Tallegas, M.; Decouvelaere, A.-V.; Aubert, S.; Rédini, F.; et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat. Commun. 2019, 10, 4622. [Google Scholar] [CrossRef]
- Su, Z.; Ho, J.W.K.; Yau, R.C.H.; Lam, Y.L.; Shek, T.W.H.; Yeung, M.C.F.; Chen, H.; Oreffo, R.O.C.; Cheah, K.S.E.; Cheung, K.S.C. A single-cell atlas of conventional central chondrosarcoma reveals the role of endoplasmic reticulum stress in malignant transformation. Commun. Biol. 2024, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, M.; Tajima, K.; Pan, H.O.; Enomoto, M.; Kinoshita, A.; Suzuki, F.; Takano, Y.; Mori, Y. Establishment of a clonal human chondrosarcoma cell line with cartilage phenotypes. Cancer Res. 1989, 49, 3996–4002. [Google Scholar] [PubMed]
- Takigawa, M.; Pan, H.; Kinoshita, A.; Tajima, K.; Takano, Y. Establishment from a human chondrosarcoma of a new immortal cell line with high tumorigenicity in vivo, which is able to form proteoglycan-rich cartilage-like nodules and to respond to insulin in vitro. Int. J. Cancer 1991, 48, 717–725. [Google Scholar] [CrossRef]
- Kunisada, T.; Miyazaki, M.; Mihara, K.; Gao, C.; Kawai, A.; Inoue, H.; Namba, M. A new human chondrosarcoma cell line (OUMS-27) that maintains chondrocytic differentiation. Int. J. Cancer 1998, 77, 854–859. [Google Scholar] [CrossRef]
- Li, B.; Li, G.; Yan, X.; Zhu, D.; Lin, P.P.; Wang, Z.; Qu, H.; He, X.; Fu, Y.; Zhu, X.; et al. Fresh Tissue Multi-omics Profiling Reveals Immune Classification and Suggests Immunotherapy Candidates for Conventional Chondrosarcoma. Clin. Cancer Res. 2021, 27, 6543–6558. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, X.; He, L.; Gao, H.; Li, W.; Feng, H.; Zhao, Q.; Zhang, W.; Li, C.; Zhang, B.; et al. Establishment and characterization of a canine chondrosarcoma cell line: Mango. Anim. Dis. 2023, 3, 29. [Google Scholar] [CrossRef]
- Gil-Benso, R.; Lopez-Gines, C.; López-Guerrero, J.A.; Carda, C.; Callaghan, R.C.; Navarro, S.; Ferrer, J.; Pellín, A.; Llombart-Bosch, A. Establishment and Characterization of a Continuous Human Chondrosarcoma Cell Line, ch-2879: Comparative Histologic and Genetic Studies with Its Tumor of Origin. Lab. Investig. 2003, 83, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterwijk, J.G.; De Jong, D.; Van Ruler, M.A.; Hogendoorn, P.C.; Dijkstra, P.S.; Van Rijswijk, C.S.; Machado, I.; Llombart-Bosch, A.; Szuhai, K.; Bovée, J.V. Three new chondrosarcoma cell lines: One grade III conventional central chondrosarcoma and two dedifferentiated chondrosarcomas of bone. BMC Cancer 2012, 12, 375. [Google Scholar] [CrossRef]
- Nakagawa, M.; Nakatani, F.; Matsunaga, H.; Seki, T.; Endo, M.; Ogawara, Y.; Machida, Y.; Katsumoto, T.; Yamagata, K.; Hattori, A.; et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene 2019, 38, 6835–6849. [Google Scholar] [CrossRef]
- Clark, J.C.; Akiyama, T.; Dass, C.R.; Choong, P.F. New clinically relevant, orthotopic mouse models of human chondrosarcoma with spontaneous metastasis. Cancer Cell Int. 2010, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Kudawara, I.; Araki, N.; Myoui, A.; Kato, Y.; Uchida, A.; Yoshikawa, H. New cell lines with chondrocytic phenotypes from human chondrosarcoma. Virchows Arch. 2004, 444, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Ogose, A.; Hotta, T.; Kawashima, H.; Gu, W.; Umezu, H.; Toyama, T.; Endo, N. Establishment of novel human dedifferentiated chondrosarcoma cell line with osteoblastic differentiation. Virchows Arch. Int. J. Pathol. 2007, 451, 691–699. [Google Scholar] [CrossRef]
- Calabuig-Fariñas, S.; Benso, R.G.; Szuhai, K.; Machado, I.; López-Guerrero, J.A.; De Jong, D.; Peydró, A.; Miguel, T.S.; Navarro, L.; Pellín, A.; et al. Characterization of a New Human Cell Line (CH-3573) Derived from a Grade II Chondrosarcoma with Matrix Production. Pathol. Oncol. Res. 2012, 18, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, L.; Schaefer, K.; Gajewski, M.; Huckenbeck, W.; Baldus, S.; Rogel, U.; Mackintosh, C.; De Alava, E.; Myklebost, O.; Kresse, S.H.; et al. Molecular characterization of commonly used cell lines for bone tumor research: A trans-European EuroBoNet effort. Genes Chromosomes Cancer 2010, 49, 40–51. [Google Scholar] [CrossRef]
- Rey, V.; Menendez, S.T.; Estupiñan, O.; Rodriguez, A.; Santos, L.; Tornin, J.; Martinez-Cruzado, L.; Castillo, D.; Ordoñez, G.R.; Costilla, S.; et al. New Chondrosarcoma Cell Lines with Preserved Stem Cell Properties to Study the Genomic Drift During In Vitro/In Vivo Growth. J. Clin. Med. 2019, 8, 455. [Google Scholar] [CrossRef]
- Emori, M.; Nakahashi, N.; Takasawa, A.; Murata, K.; Murahashi, Y.; Shimizu, J.; Tsukahara, T.; Sugita, S.; Takada, K.; Hasegawa, T.; et al. Establishment and characterization of a novel dedifferentiated chondrosarcoma cell line, SMU-DDCS, harboring an IDH1 mutation. Hum. Cell 2023, 36, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Morioka, H.; Weissbach, L.; Vogel, T.; Nielsen, G.P.; Faircloth, G.T.; Shao, L.; Hornicek, F.J. Antiangiogenesis treatment combined with chemotherapy produces chondrosarcoma necrosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1211–1217. [Google Scholar]
- Monderer, D.; Luseau, A.; Bellec, A.; David, E.; Ponsolle, S.; Saiagh, S.; Bercegeay, S.; Piloquet, P.; Denis, M.G.; Lodé, L.; et al. New chondrosarcoma cell lines and mouse models to study the link between chondrogenesis and chemoresistance. Lab. Investig. J. Tech. Methods Pathol. 2013, 93, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Farges, M.; Mazeau, C.; Gioanni, J.; Ettore, F.; DeNovion, H.; Schneider, M. Establishment and characterization of a new cell line derived from a human chondrosarcoma. Oncol. Rep. 1997, 4, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, T.; Krueger, S.; Sel, S.; Werner, K.; Ropke, M.; Roessner, A. Differential expression of VEGF-A and angiopoietins in cartilage tumors and regulation by interleukin-1β. Cancer 2006, 106, 2028–2038. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garcia, J.; Jubelin, C.; Loussouarn, A.; Goumard, M.; Griscom, L.; Renodon-Cornière, A.; Heymann, M.-F.; Heymann, D. In vitro three-dimensional cell cultures for bone sarcomas. J. Bone Oncol. 2021, 30, 100379. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, V.A.; Mironov, V.A.; Van Kampen, K.A.; Karalkin, P.A.; Koudan, E.V.; Pereira, F.D.; Petrov, S.V.; Nezhurina, E.K.; Petrov, O.F.; Myasnikov, M.I.; et al. Scaffold-free and label-free biofabrication technology using levitational assembly in a high magnetic field. Biofabrication 2020, 12, 045022. [Google Scholar] [CrossRef]
- Palubeckaitė, I.; Venneker, S.; Van Den Akker, B.E.W.M.; Briaire-de Bruijn, I.H.; Boveé, J.V.M.G. Does PARP Inhibition Sensitize Chondrosarcoma Cell Lines to Chemotherapy or Radiotherapy? Results from a Three-dimensional Spheroid Cell Model. Clin. Orthop. 2023, 481, 608–619. [Google Scholar] [CrossRef]
- Minopoli, M.; Sarno, S.; Di Carluccio, G.; Azzaro, R.; Costantini, S.; Fazioli, F.; Gallo, M.; Apice, G.; Cannella, L.; Rea, D.; et al. Inhibiting Monocyte Recruitment to Prevent the Pro-Tumoral Activity of Tumor-Associated Macrophages in Chondrosarcoma. Cells 2020, 9, 1062. [Google Scholar] [CrossRef]
- Al Shihabi, A.; Tebon, P.J.; Nguyen, H.T.L.; Chantharasamee, J.; Sartini, S.; Davarifar, A.; Jensen, A.Y.; Diaz-Infante, M.; Cox, H.; Gonzalez, A.E.; et al. The landscape of drug sensitivity and resistance in sarcoma. Cell Stem Cell 2024, 31, 1524–1542.e4. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The chick embryo chorioallantoic membrane as a model for tumor biology. Exp. Cell Res. 2014, 328, 314–324. [Google Scholar] [CrossRef]
- Guder, W.K.; Hartmann, W.; Buhles, C.; Burdack, M.; Busch, M.; Dünker, N.; Hardes, J.; Dirksen, U.; Bauer, S.; Streitbürger, A. 5-ALA-mediated fluorescence of musculoskeletal tumors in a chick chorio-allantoic membrane model: Preclinical in vivo qualification analysis as a fluorescence-guided surgery agent in Orthopedic Oncology. J. Orthop. Surg. 2022, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoff, M.; Grunewald, S.; Schaefer, C.; Zöllner, S.K.; Plaumann, P.; Busch, M.; Dünker, N.; Ketzer, J.; Kersting, J.; Bauer, S.; et al. Evaluation of the Effect of Photodynamic Therapy on CAM-Grown Sarcomas. Bioengineering 2023, 10, 464. [Google Scholar] [CrossRef] [PubMed]
- Giusti, V.; Miserocchi, G.; Sbanchi, G.; Pannella, M.; Hattinger, C.M.; Cesari, M.; Fantoni, L.; Guerrieri, A.N.; Bellotti, C.; De Vita, A.; et al. Xenografting Human Musculoskeletal Sarcomas in Mice, Chick Embryo, and Zebrafish: How to Boost Translational Research. Biomedicines 2024, 12, 1921. [Google Scholar] [CrossRef]
- Clark, J.C.M.; Dass, C.R.; Choong, P.F.M. Development of chondrosarcoma animal models for assessment of adjuvant therapy. ANZ J. Surg. 2009, 79, 327–336. [Google Scholar] [CrossRef]
- Grimaud, E.; Damiens, C.; Rousselle, A.V.; Passuti, N.; Heymann, D.; Gouin, F. Bone remodelling and tumour grade modifications induced by interactions between bone and swarm rat chondrosarcoma. Histol. Histopathol. 2002, 17, 1103–1111. [Google Scholar] [CrossRef]
- Simard, F.A.; Richert, I.; Vandermoeten, A.; Decouvelaere, A.-V.; Michot, J.-P.; Caux, C.; Blay, J.-Y.; Dutour, A. Description of the immune microenvironment of chondrosarcoma and contribution to progression. OncoImmunology 2017, 6, e1265716. [Google Scholar] [CrossRef]
- Rüther, U.; Komitowski, D.; Schubert, F.R.; Wagner, E.F. c-fos expression induces bone tumors in transgenic mice. Oncogene 1989, 4, 861–865. [Google Scholar]
- Rüther, U. Induction of Bone Tumors in Transgenic Mice by C-FOS Depends on the Presence of a Retroviral Long Terminal Repeat. Cancer Genet. Cytogenet. 1998, 105, 123–127. [Google Scholar] [CrossRef]
- Ho, L.; Stojanovski, A.; Whetstone, H.; Wei, Q.X.; Mau, E.; Wunder, J.S.; Alman, B. Gli2 and p53 Cooperate to Regulate IGFBP-3- Mediated Chondrocyte Apoptosis in the Progression from Benign to Malignant Cartilage Tumors. Cancer Cell 2009, 16, 126–136. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells 2019, 8, 889. [Google Scholar] [CrossRef] [PubMed]
- Pathmanapan, S.; Ilkayeva, O.; Martin, J.T.; Loe, A.K.H.; Zhang, H.; Zhang, G.-F.; Newgard, C.B.; Wunder, J.S.; Alman, B.A. Mutant IDH and non-mutant chondrosarcomas display distinct cellular metabolomes. Cancer Metab. 2021, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Cao, S.; Mashl, R.J.; Mo, C.-K.; Zaccaria, S.; Wendl, M.C.; Davies, S.R.; Bailey, M.H.; Primeau, T.M.; Hoog, J.; et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment. Nat. Commun. 2021, 12, 5086. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Chawla, S.P.; Conley, A.P.; Wilky, B.A.; Tolcher, A.; Lakhani, N.J.; Berz, D.; Andrianov, V.; Crago, W.; Holcomb, M.; et al. Preclinical Characterization and Phase I Trial Results of INBRX-109, a Third-Generation, Recombinant, Humanized, Death Receptor 5 Agonist Antibody, in Chondrosarcoma. Clin. Cancer Res. 2023, 29, 2988–3003. [Google Scholar] [CrossRef] [PubMed]
- Safaric Tepes, P.; Segovia, D.; Jevtic, S.; Ramirez, D.; Lyons, S.K.; Sordella, R. Patient-derived xenografts and in vitro model show rationale for imatinib mesylate repurposing in HEY1-NCoA2-driven mesenchymal chondrosarcoma. Lab. Investig. 2022, 102, 1038–1049. [Google Scholar] [CrossRef]
- Cornillie, J.; Wozniak, A.; Li, H.; Wang, Y.; Boeckx, B.; Gebreyohannes, Y.K.; Wellens, J.; Vanleeuw, U.; Hompes, D.; Stas, M.; et al. Establishment and Characterization of Histologically and Molecularly Stable Soft-tissue Sarcoma Xenograft Models for Biological Studies and Preclinical Drug Testing. Mol. Cancer Ther. 2019, 18, 1168–1178. [Google Scholar] [CrossRef]
- Giordano, G.; Merlini, A.; Ferrero, G.; Mesiano, G.; Fiorino, E.; Brusco, S.; Centomo, M.L.; Leuci, V.; D’Ambrosio, L.; Aglietta, M.; et al. EphA2 Expression in Bone Sarcomas: Bioinformatic Analyses and Preclinical Characterization in Patient-Derived Models of Osteosarcoma, Ewing’s Sarcoma and Chondrosarcoma. Cells 2021, 10, 2893. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Hue, J.J.; Hajihassani, O.; Graor, H.J.; Katayama, E.S.; Loftus, A.W.; Bajor, D.; Rothermel, L.D.; Vaziri-Gohar, A.; Winter, J.M. Clinical development of IDH1 inhibitors for cancer therapy. Cancer Treat. Rev. 2022, 103, 102334. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.-L.; Sheth, C.M.; Charlab, R.; et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef]
- Casak, S.J.; Pradhan, S.; Fashoyin-Aje, L.A.; Ren, Y.; Shen, Y.-L.; Xu, Y.; Chow, E.C.Y.; Xiong, Y.; Zirklelbach, J.F.; Liu, J.; et al. FDA Approval Summary: Ivosidenib for the Treatment of Patients with Advanced Unresectable or Metastatic, Chemotherapy Refractory Cholangiocarcinoma with an IDH1 Mutation. Clin. Cancer Res. 2022, 28, 2733–2737. [Google Scholar] [CrossRef]
- FDA approves first IDH-targeted glioma drug. Nat. Biotechnol. 2024, 42, 1325. [CrossRef]
- Tap, W.D.; Villalobos, V.M.; Cote, G.M.; Burris, H.; Janku, F.; Mir, O.; Beeram, M.; Wagner, A.J.; Jiang, L.; Wu, B.; et al. Phase I Study of the Mutant IDH1 Inhibitor Ivosidenib: Safety and Clinical Activity in Patients with Advanced Chondrosarcoma. J. Clin. Oncol. 2020, 38, 1693–1701. [Google Scholar] [CrossRef]
- Griffith, T.; Stokes, B.; Kucaba, T.; Earel, J., Jr.; VanOosten, R.; Brincks, E.; Norian, L. TRAIL Gene Therapy: From Preclinical Development to Clinical Application. Curr. Gene Ther. 2009, 9, 9–19. [Google Scholar] [CrossRef]
- Subbiah, V.; Brown, R.E.; Buryanek, J.; Trent, J.; Ashkenazi, A.; Herbst, R.; Kurzrock, R. Targeting the Apoptotic Pathway in Chondrosarcoma Using Recombinant Human Apo2L/TRAIL (Dulanermin), a Dual Proapoptotic Receptor (DR4/DR5) Agonist. Mol. Cancer Ther. 2012, 11, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.T.; Kothambawala, T.; Wang, L.; Matthew, T.J.; Calhoun, S.E.; Saini, A.K.; Kotturi, M.F.; Hernandez, G.; Humke, E.W.; Peterson, M.S.; et al. Multimeric Anti-DR5 IgM Agonist Antibody IGM-8444 Is a Potent Inducer of Cancer Cell Apoptosis and Synergizes with Chemotherapy and BCL-2 Inhibitor ABT-199. Mol. Cancer Ther. 2021, 20, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Palmerini, E.; Lopez Pousa, A.; Grignani, G.; Redondo, A.; Hindi, N.; Provenzano, S.; Sebio, A.; Lopez Martin, J.A.; Valverde, C.; Martinez Trufero, J.; et al. Nivolumab and sunitinib in patients with advanced bone sarcomas: A multicenter, single-arm, phase 2 trial. Cancer 2025, 131, e35628. [Google Scholar] [CrossRef]
- Chow, W.; Frankel, P.; Ruel, C.; Araujo, D.M.; Milhem, M.; Okuno, S.; Hartner, L.; Undevia, S.; Staddon, A. Results of a prospective phase 2 study of pazopanib in patients with surgically unresectable or metastatic chondrosarcoma. Cancer 2020, 126, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Duffaud, F.; Italiano, A.; Bompas, E.; Rios, M.; Penel, N.; Mir, O.; Piperno-Neumann, S.; Chevreau, C.; Delcambre, C.; Bertucci, F.; et al. Efficacy and safety of regorafenib in patients with metastatic or locally advanced chondrosarcoma: Results of a non-comparative, randomised, double-blind, placebo controlled, multicentre phase II study. Eur. J. Cancer 2021, 150, 108–118. [Google Scholar] [CrossRef]
- Ingangi, V.; De Chiara, A.; Ferrara, G.; Gallo, M.; Catapano, A.; Fazioli, F.; Di Carluccio, G.; Peranzoni, E.; Marigo, I.; Carriero, M.V.; et al. Emerging Treatments Targeting the Tumor Microenvironment for Advanced Chondrosarcoma. Cells 2024, 13, 977. [Google Scholar] [CrossRef] [PubMed]
- Nota, S.P.F.T.; Osei-Hwedieh, D.O.; Drum, D.L.; Wang, X.; Sabbatino, F.; Ferrone, S.; Schwab, J.H. Chondroitin sulfate proteoglycan 4 expression in chondrosarcoma: A potential target for antibody-based immunotherapy. Front. Oncol. 2022, 12, 939166. [Google Scholar] [CrossRef]
- Tarone, L.; Giacobino, D.; Camerino, M.; Maniscalco, L.; Iussich, S.; Parisi, L.; Giovannini, G.; Dentini, A.; Bolli, E.; Quaglino, E.; et al. A chimeric human/dog-DNA vaccine against CSPG4 induces immunity with therapeutic potential in comparative preclinical models of osteosarcoma. Mol. Ther. 2023, 31, 2342–2359. [Google Scholar] [CrossRef] [PubMed]
- Kostine, M.; Cleven, A.H.; De Miranda, N.F.C.C.; Italiano, A.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef]
- Richert, I.; Gomez-Brouchet, A.; Bouvier, C.; Du Bouexic De Pinieux, G.; Karanian, M.; Blay, J.-Y.; Dutour, A. The immune landscape of chondrosarcoma—Potential for therapeutic targeting of CSFR1+ macrophages. J. Bone Oncol. 2020, 20, 100271. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Pollack, S.M.; Redman, M.W.; Baker, K.K.; Wagner, M.J.; Schroeder, B.A.; Loggers, E.T.; Trieselmann, K.; Copeland, V.C.; Zhang, S.; Black, G.; et al. Assessment of Doxorubicin and Pembrolizumab in Patients with Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 1778. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Ricciotti, R.W.; Mantilla, J.; Loggers, E.T.; Pollack, S.M.; Cranmer, L.D. Response to PD1 inhibition in conventional chondrosarcoma. J. Immunother. Cancer 2018, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Nowak, A.J.; Dressler, D.B.; Rock, A.; Mojica, K.; Woo, D.; Zuckerman, L.M.; Chow, W.; Agulnik, M. Role of immunotherapy in chondrosarcoma: A case report and review of the literature. Ther. Adv. Med. Oncol. 2023, 15, 17588359231199876. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Baharom, F.; Hermans, D.; Delamarre, L.; Seder, R.A. Vax-Innate: Improving therapeutic cancer vaccines by modulating T cells and the tumour microenvironment. Nat. Rev. Immunol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Lacuna, K.P.; Ingham, M.; Chen, L.; Das, B.; Lee, S.M.; Ge, L.; Druta, M.; Conley, A.P.; Keohan, M.L.; Agulnik, M.; et al. Correlative results from NCI CTEP/ETCTN 10330: A phase 2 study of belinostat with SGI-110 (guadecitabine) or ASTX727 (decitabine/cedazuridine) for advanced conventional chondrosarcoma (cCS). J. Clin. Oncol. 2024, 42, 11526. [Google Scholar] [CrossRef]
- Sheikh, T.N.; Chen, X.; Xu, X.; McGuire, J.T.; Ingham, M.; Lu, C.; Schwartz, G.K. Growth Inhibition and Induction of Innate Immune Signaling of Chondrosarcomas with Epigenetic Inhibitors. Mol. Cancer Ther. 2021, 20, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.J.; Hindi, N.; Palmerini, E.; Martínez-Trufero, J.; Lopez-Pousa, A.; Carrasco-Garcia, I.; Gonzalez-Billalabeitia, E.; Moura, D.S.; Ramos, R.; Tirabosco, R.; et al. ImmunoSarc II master trial (phase II of sunitinib and nivolumab): Results from the dedifferentiated chondrosarcoma (DDCS) cohort—A GEIS, ISG and UCL study. J. Clin. Oncol. 2024, 42, 11506. [Google Scholar] [CrossRef]

{kind=link}
| CS Type | Origin (% of CS Cases) | Location in Bone | Main Genetic Alterations | 5-Year Survival Rate |
|---|---|---|---|---|
| Conventional CS [3,4,5,14] | Bone, Primary (85%) | Central (great majority) | >50% IDH1/2 mutation | ACT: 93% Grade 1: 90% Grade 2: 80% Grade 3: 29% |
| Periosteal (Juxtacortical) (1%) | IDH1/2 mutation Altered hedgehog pathway | 68–93% | ||
| Bone, Secondary (<10%) | Central (deriving from enchondromas) | >50% IDH1/2 mutation | >90% | |
| Peripheral (deriving from osteochondromas) | EXT1/2 mutation | >90% | ||
| Dedifferentiated CS [7,14] | Bone (10–15%) | 80% IDH1/2 mutation | 10–25% | |
| Clear cell CS [7,9,10,14] | Bone (2–6%) | No specific driving mutations | 62–100% | |
| Mesenchymal CS [7,8,14] | Bone or soft tissues (2%) | HEY1::NCOA2 or IRF2BP2::CDX1 translocations | 37–50% | |
| Extraskeletal myxoid CS [11,12] | Soft tissues (3% of soft tissue sarcomas) | EWS::NR4A3, TAF15::NR4A3, or TCF12::NR4A3 translocations | 50% |
| CS Cell Line | Parental Tumor (Gender) | Main Genetic Features | Tumorigenicity | Ref. |
|---|---|---|---|---|
| JJ012 | Grade 2 conventional CS (male) | IDH1(R132G) heterozygous mutation | Yes, nude mice; subcutaneous (sc), and orthotopic tumors | [52] |
| L835 | Grade 3 central conventional CS (male) | IDH1(R132C) heterozygous mutation; homozygous deletion of the CDKN2A | No | [50] |
| SW1353 | Grade 2 conventional central CS (female) | IDH2(R172S) heterozygous mutation | Yes, SCID and nude mice; sc and orthotopic tumors; experimental metastasis after intrasplenic injection | [37] |
| OUMS27 | Grade 3 central conventional CS (male) | IDH1/2 wild-type, p53 mutated | Yes, nude mice | [46] |
| HCS-2/A and HCS-2/8 | Grade 1 Well-differentiated CS (male) | Not reported | HCS-2/A highly tumorigenic in nude mice; HCS-2/8 low tumorigenicity | [45] |
| HCS-TG | Grade 2 CS (male) | Not reported | Yes, nude mice | [53] |
| NDCS-1 | Dedifferentiated CS, metastasis (male) | IDH1/2 wild-type, p53 mutated | Yes, SCID mice | [54] |
| L2975 | Dedifferentiated CS, metastasis (male) | IDH2R172W heterozygous mutation; homozygous deletion of the CDKN2A | Yes, nude mice, slow-growing tumors | [50] |
| L3252 | Dedifferentiated CS, local recurrence (female) | IDH1/2 wild-type; homozygous deletion of CDKN2A | No | [50] |
| CH-3573 | Grade 2 conventional central CS (male) | IDH1/2 wild-type | Yes, nude mice | [55] |
| CH-2879 | Grade 3 conventional central CS | IDH1/2 wild-type | Yes, nude mice | [49,56] |
| CDS17 and T-CDS17 | Dedifferentiated CS (male) | IDH2R172G mutation | Yes, NOD/SCID mice, slow-growing tumors | [57] |
| SMU-DDCS | Dedifferentiated CS (female) | IDH1 mutation | Yes, nude mice | [58] |
| CHSA or CS1 | High grade CS (male) | IDH2 mutation | Yes, nude mice Highly tumorigenic | [59] |
| CH03 | Dedifferentiated CS (female) | IDH1/2 wild-type; TP53 deletion; p16ink4a deletion | No, nude mice | [60] |
| CH34 | Conventional CS grade 3 (male) | IDH1 mutation; TP53 wild-type; p16ink4a deletion; p14ARF deletion; MDM2 amplification | No, nude mice | [60] |
| CH56 | Conventional CS grade 3 (female) | IDH2 mutation; TP53 wild-type; p16ink4a deletion; p14ARF deletion; MDM2 amplification | Yes, nude mice | [60] |
| CAL78 | Dedifferentiated CS (male) | IDH1/2 wild-type | Yes, nude mice | [61] |
| C3842 | Secondary CS (male) | IDH1/2 wild-type | Not reported | [62] |
| Therapeutic Approach | Clinical Trial * | Drugs | Main Clinical Results | Ref. |
|---|---|---|---|---|
| Inhibition of mutant IDH1/2 | NCT04278781 Phase I/II trial IDH1 mutated advanced CS patients | Ivosidenib (IDH1 inhibitor) | Median PFS of 5.6 months. Best overall response: SD in 52% of the patients, PFS at 6 months was 0% for dedifferentiated CS and 54% for conventional CS | [90] |
| NCT06127407 CHONQUER study Phase III trial | Results awaited in 2028–2030 | [90] | ||
| DR5 targeting | NCT04553692 Phase I clinical trial | IGM-8444 (multimeric anti-DR5 IgM agonist antibody) | Estimated study completion by the end of 2027 | [93] |
| NCT03715933 Phase I trial | INBRX-109 (tetravalent single-domain agonistic antibody against DR5) | Disease control rate (SD and PR) was 87.1% with a median PFS of 7.6 months; durable clinical benefit in 40.7% (11 of 27) patients, including two PRs | [82] | |
| ChonDRAgon trial NCT04950075 Phase II trial | Estimated study completion by the end of 2026 | [82] | ||
| Antiangiogenic therapies | NCT01330966 Phase II trial | Pazopanib | Disease control rate (SD and PR) 43%, median PFS of 7.9 months, PFS rate at 6 months 55%, median overall survival 17.6 months | [95] |
| NCT02389244 Phase II trial | Regorafenib | Median PFS of 5 months, PFS rate at 6 months 43% | [96] | |
| NCT04055220 Efficacy and safety of Regorafenib as maintenance therapy | Recruiting, estimated study completion by the end of 2026 | [96] | ||
| Immunotherapy and combined therapies | SARC028 NCT 02301039 Phase II trial | Pembrolizumab anti-PD-1 antibody | 1PR and 1 SD out of 5 CS patients | [102] |
| IMMUNOSARC study NCT03277924 Phase II trial | Nivolumab and sunitinib (anti-PD-1 antibody + antiangiogenic drug) | In the dedifferentiated CS cohort median PFS of 5.6 months, PR 26.3%, and 52.6% SD | [94,110] | |
| Epigenetic therapies | NCT04340843 NCI 10330 | Belinostat with SGI-110 or ASTX727 (HDACinhibitor + DNMTinhibitor) | Failed to meet the endpoint of ORR | [108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landuzzi, L.; Ruzzi, F.; Lollini, P.-L.; Scotlandi, K. Chondrosarcoma: New Molecular Insights, Challenges in Near-Patient Preclinical Modeling, and Therapeutic Approaches. Int. J. Mol. Sci. 2025, 26, 1542. https://doi.org/10.3390/ijms26041542
Landuzzi L, Ruzzi F, Lollini P-L, Scotlandi K. Chondrosarcoma: New Molecular Insights, Challenges in Near-Patient Preclinical Modeling, and Therapeutic Approaches. International Journal of Molecular Sciences. 2025; 26(4):1542. https://doi.org/10.3390/ijms26041542
Chicago/Turabian StyleLanduzzi, Lorena, Francesca Ruzzi, Pier-Luigi Lollini, and Katia Scotlandi. 2025. "Chondrosarcoma: New Molecular Insights, Challenges in Near-Patient Preclinical Modeling, and Therapeutic Approaches" International Journal of Molecular Sciences 26, no. 4: 1542. https://doi.org/10.3390/ijms26041542
APA StyleLanduzzi, L., Ruzzi, F., Lollini, P.-L., & Scotlandi, K. (2025). Chondrosarcoma: New Molecular Insights, Challenges in Near-Patient Preclinical Modeling, and Therapeutic Approaches. International Journal of Molecular Sciences, 26(4), 1542. https://doi.org/10.3390/ijms26041542