
Novel Pathogenic Variant of the TRRAP Gene Detected in a Hungarian Family with Autosomal Dominant Non-Syndromic Hearing Loss

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
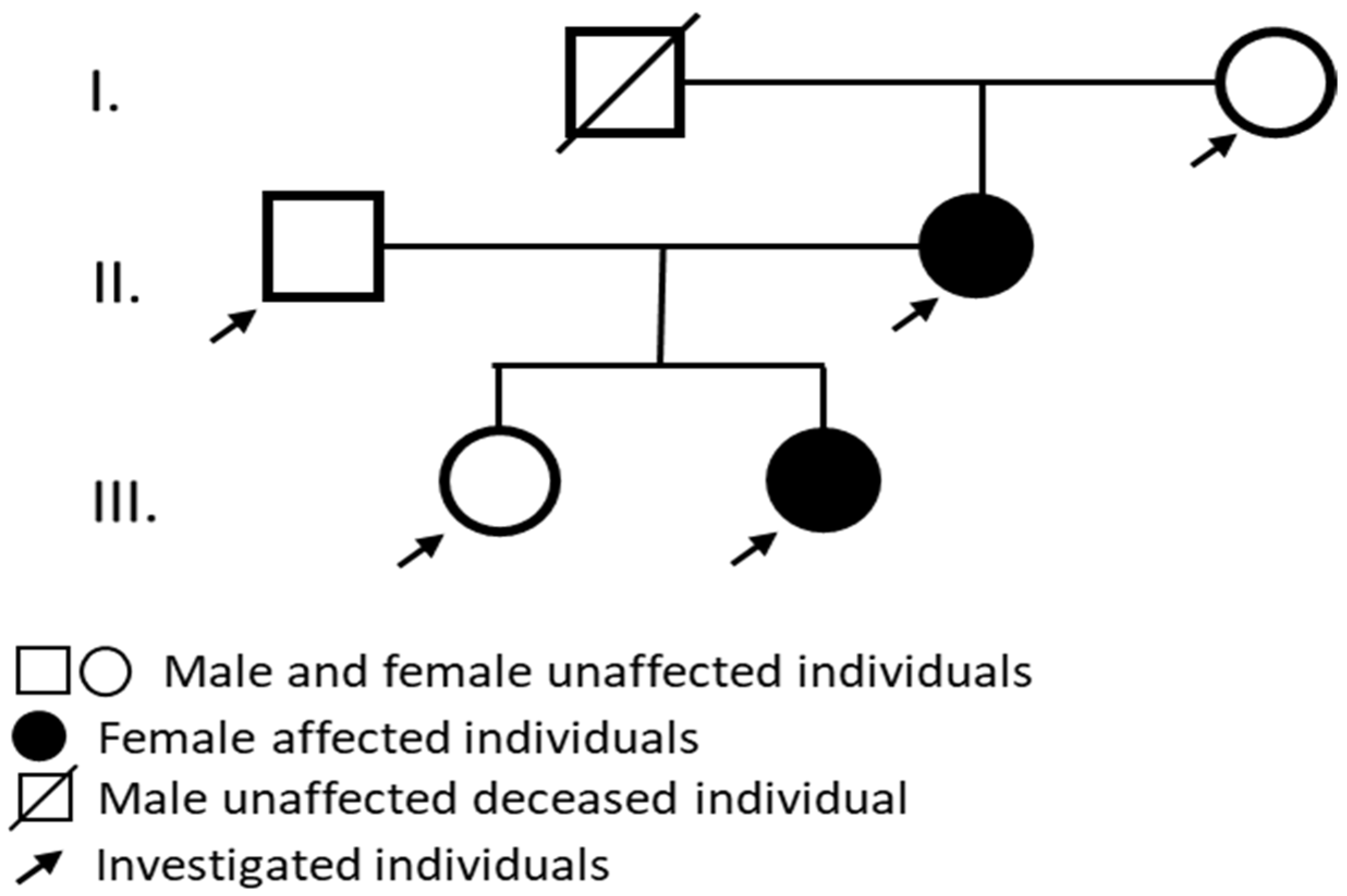
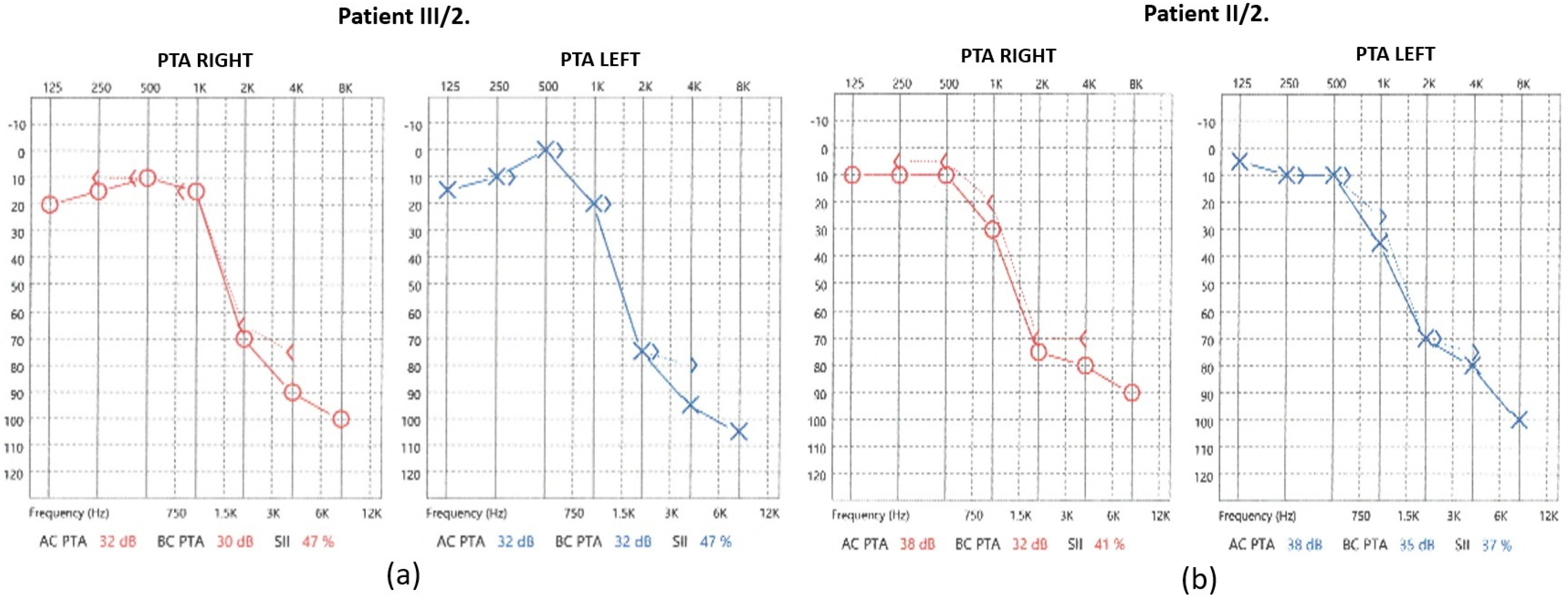
2.1. Audiological Investigations
2.2. Genetic Investigations
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Whole Exome Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Funamura, J.L. Evaluation and management of nonsyndromic congenital hearing loss. Curr. Opin. Otolaryngol. 2017, 25, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.S.; Emmett, S.D.; Robler, S.K.; Tucci, D.L. Global hearing loss prevention. Otolaryngol. Clin. N. Am. 2018, 51, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, L.L.; Tucci, D.L. Hearing loss in adults. N. Engl. J. Med. 2017, 377, 2465–2473. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, C.; Wang, M.M.; Xiao, Y.; Zhang, F.; Zhou, Y.; Li, J.; Zheng, Q.; Bai, X.; Wang, H. A novel nonsense mutation of POU4F3 gene causes autosomal dominant hearing loss. Neural Plast. 2016, 2016, 1512831, Erratum in Neural Plast. 2017, 2017, 9202847. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xia, W.; Hu, J.; Ma, J.; Huang, J.; Wang, X.; Jiang, N.; Zhang, J.; Ma, Z.; Ma, D. Novel TRRAP mutation causes autosomal dominant non-syndromic hearing loss. Clin. Genet. 2019, 96, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Templeton, G.W.; Moorhead, G.B.G. The phosphoinositide-3-OH-kinaserelated kinases of Arabidopsis thaliana. EMBO Rep. 2005, 6, 723–728. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murr, R.; Vaissiere, T.; Sawan, C.; Shukla, V.; Herceg, Z. Orchestration of chromatin-based processes: Mind the TRRAP. Oncogene 2007, 26, 5358–5372. [Google Scholar] [CrossRef] [PubMed]
- Loizou, J.I.; Murr, R.; Finkbeiner, M.G.; Sawan, C.; Wang, Z.Q.; Herceg, Z. Epigenetic information in chromatin: The code of entry for DNA repair. Cell Cycle 2006, 5, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Utley, R.T.; Workman, J.L.; Côté, J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003, 19, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Sharov, G.; Voltz, K.; Durand, A.; Kolesnikova, O.; Papai, G.; Myasnikov, A.G.; Dejaegere, A.; Ben Shem, A.; Schultz, P. Structure of the transcription activator target Tra1 within the chromatin modifying complex SAGA. Nat. Commun. 2017, 8, 1556. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Knutson, B.A.; Hahn, S. Domains of Tra1 important for activator recruitment and transcription coactivator functions of SAGA and NuA4 complexes. Mol. Cell Biol. 2011, 31, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ahmad, S.; Zhang, Z.; Côté, J.; Cai, G. Architecture of the Saccharomyces cerevisiae NuA4/TIP60 complex. Nat. Commun. 2018, 9, 1147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pál, M.; Nagy, D.; Neller, A.; Farkas, K.; Leprán-Török, D.; Nagy, N.; Füstös, D.; Nagy, R.; Németh, A.; Szilvássy, J.; et al. Genetic Etiology of Nonsyndromic Hearing Loss in Hungarian Patients. Int. J. Mol. Sci. 2023, 24, 7401. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, N.; Szalenko-Tőkés, Á.; Pál, M.; Bokor, B.A.; Nagy, R.; Jarabin, J.A.; Róvó, L.; Széll, M. Novel Pathogenic Variant of the TRRAP Gene Detected in a Hungarian Family with Autosomal Dominant Non-Syndromic Hearing Loss. Int. J. Mol. Sci. 2025, 26, 1583. https://doi.org/10.3390/ijms26041583
Nagy N, Szalenko-Tőkés Á, Pál M, Bokor BA, Nagy R, Jarabin JA, Róvó L, Széll M. Novel Pathogenic Variant of the TRRAP Gene Detected in a Hungarian Family with Autosomal Dominant Non-Syndromic Hearing Loss. International Journal of Molecular Sciences. 2025; 26(4):1583. https://doi.org/10.3390/ijms26041583
Chicago/Turabian StyleNagy, Nikoletta, Ágnes Szalenko-Tőkés, Margit Pál, Barbara Anna Bokor, Roland Nagy, János András Jarabin, László Róvó, and Márta Széll. 2025. "Novel Pathogenic Variant of the TRRAP Gene Detected in a Hungarian Family with Autosomal Dominant Non-Syndromic Hearing Loss" International Journal of Molecular Sciences 26, no. 4: 1583. https://doi.org/10.3390/ijms26041583
APA StyleNagy, N., Szalenko-Tőkés, Á., Pál, M., Bokor, B. A., Nagy, R., Jarabin, J. A., Róvó, L., & Széll, M. (2025). Novel Pathogenic Variant of the TRRAP Gene Detected in a Hungarian Family with Autosomal Dominant Non-Syndromic Hearing Loss. International Journal of Molecular Sciences, 26(4), 1583. https://doi.org/10.3390/ijms26041583