
Long-Read Whole-Genome Sequencing as a Tool for Variant Detection in Inherited Retinal Dystrophies

 , , , , , ,
, , , , , ,  , ,
, ,  , add
Show full author list
, add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
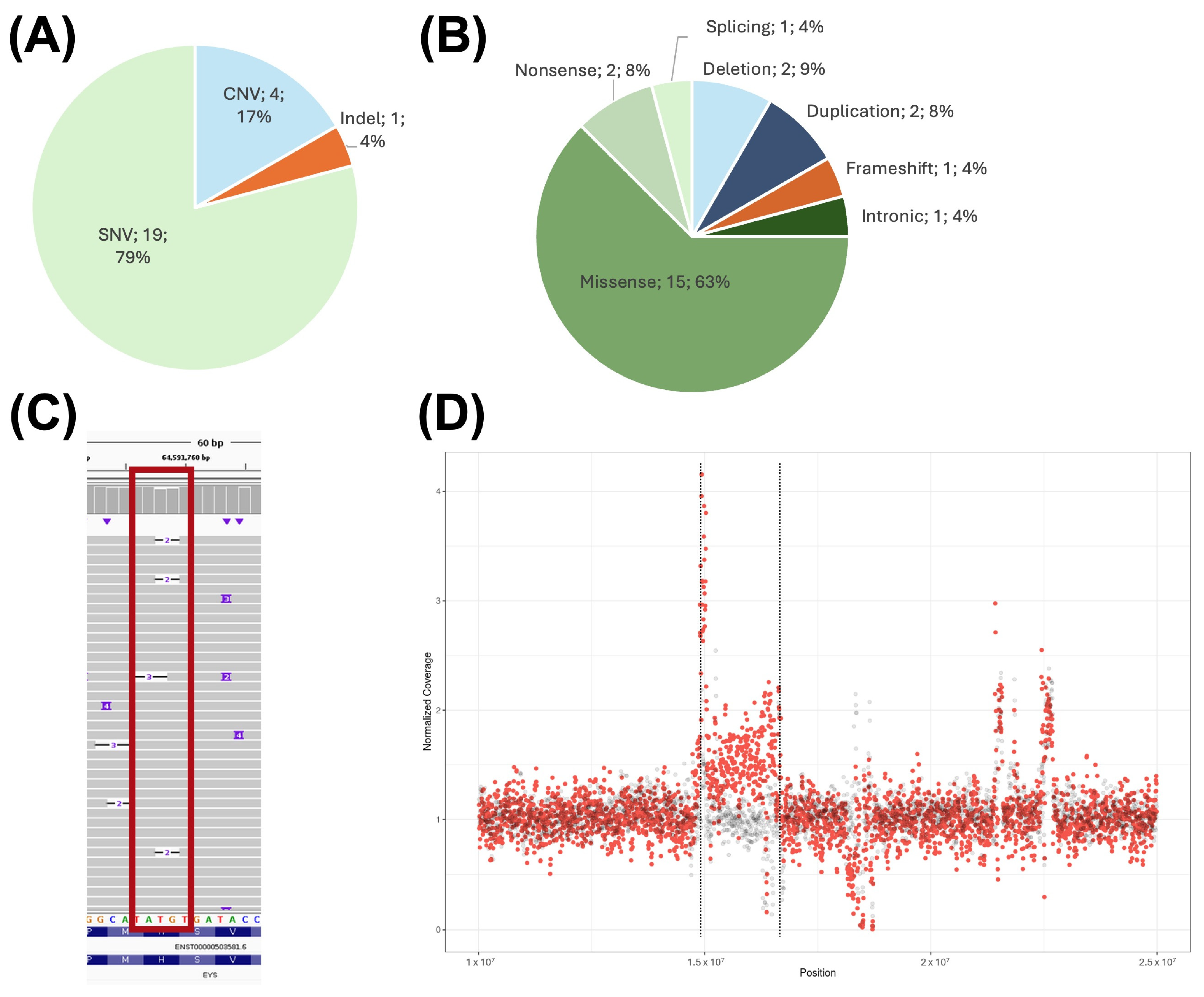
2.1. Confirmation of Previously Detected Variants in the Studied Cohort
2.2. Limitations of the Technique
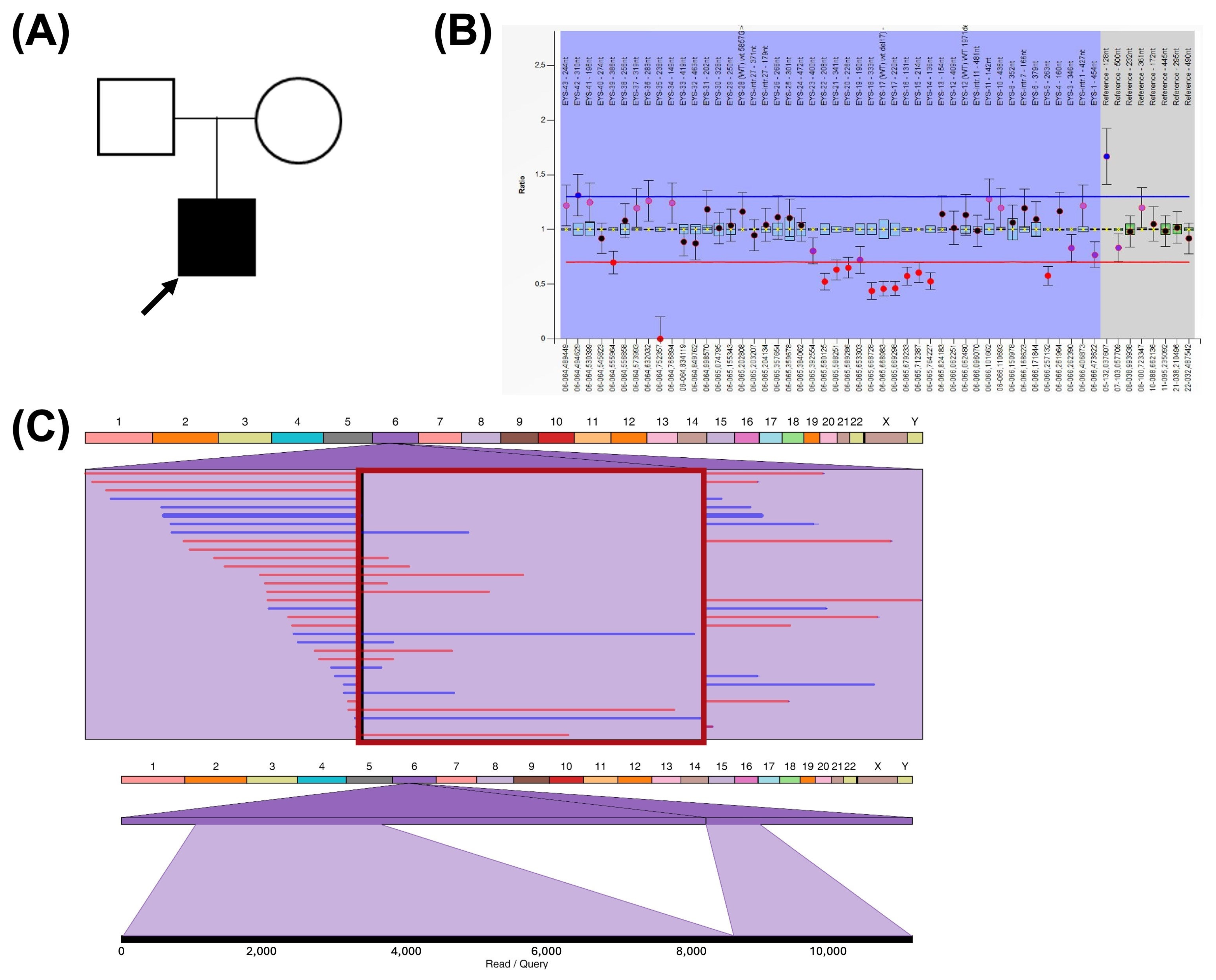
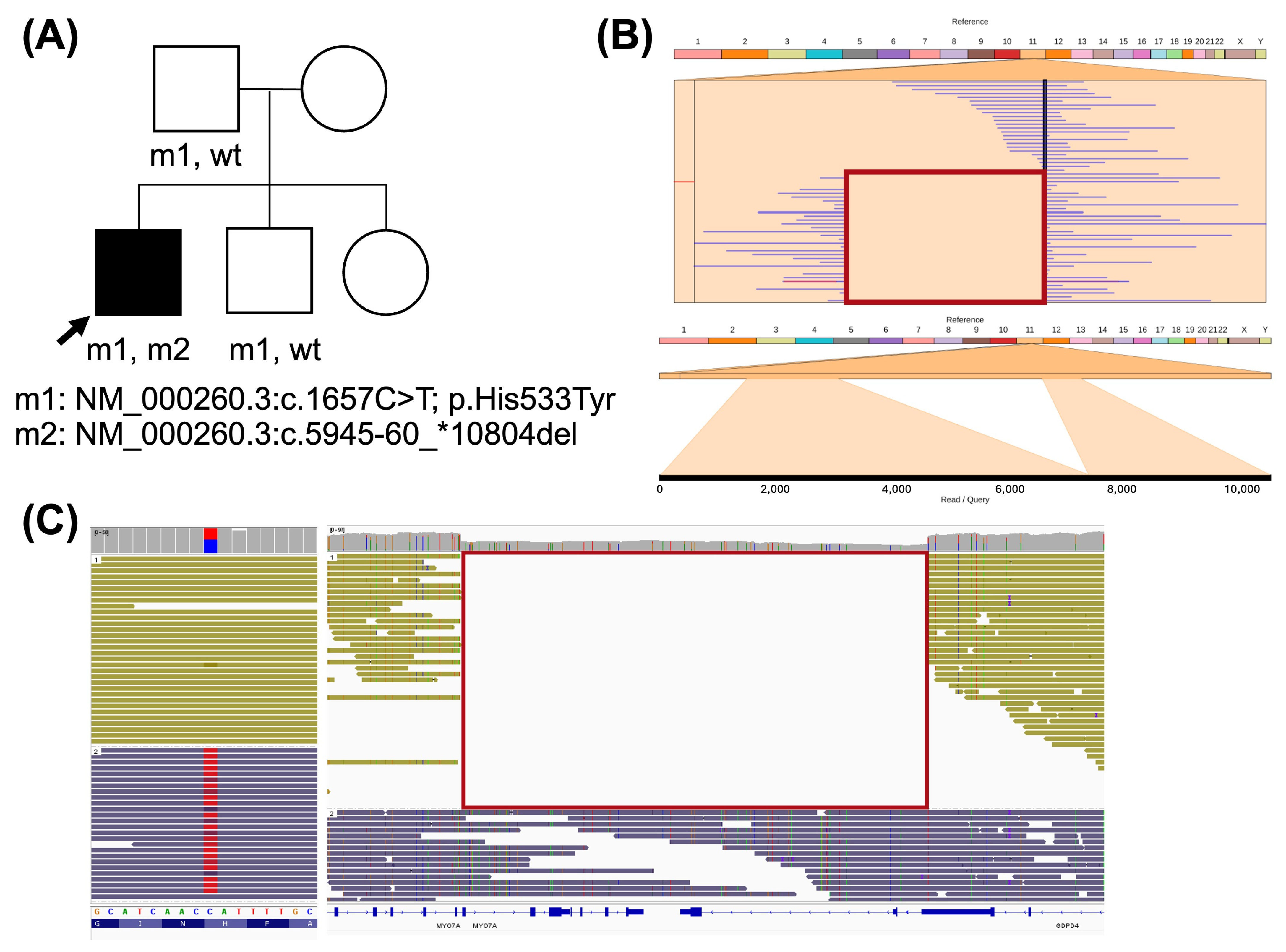
2.3. Genotype Characterization in Recessive IRDs
2.4. Repeat Mobile Element Variation
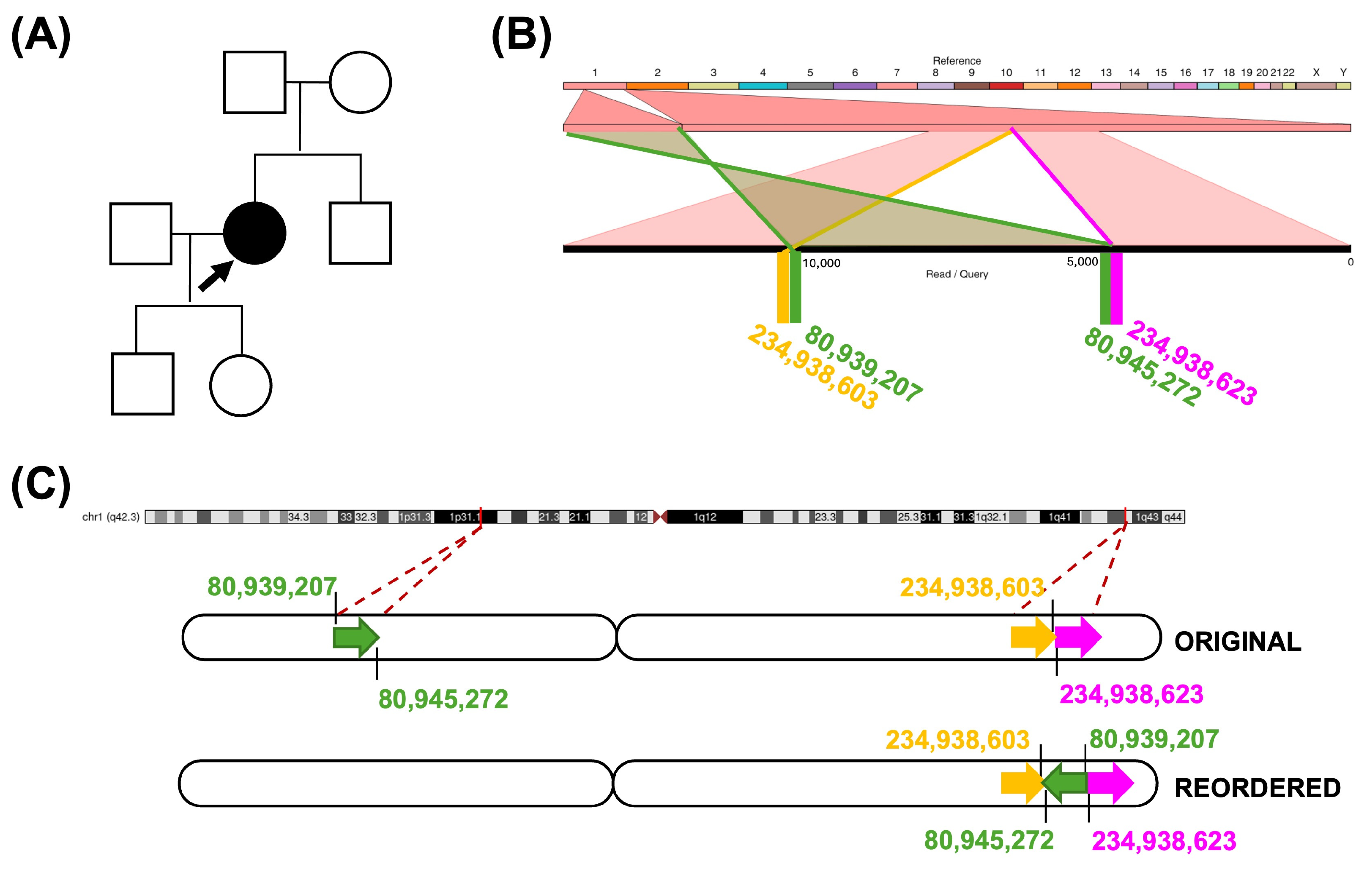
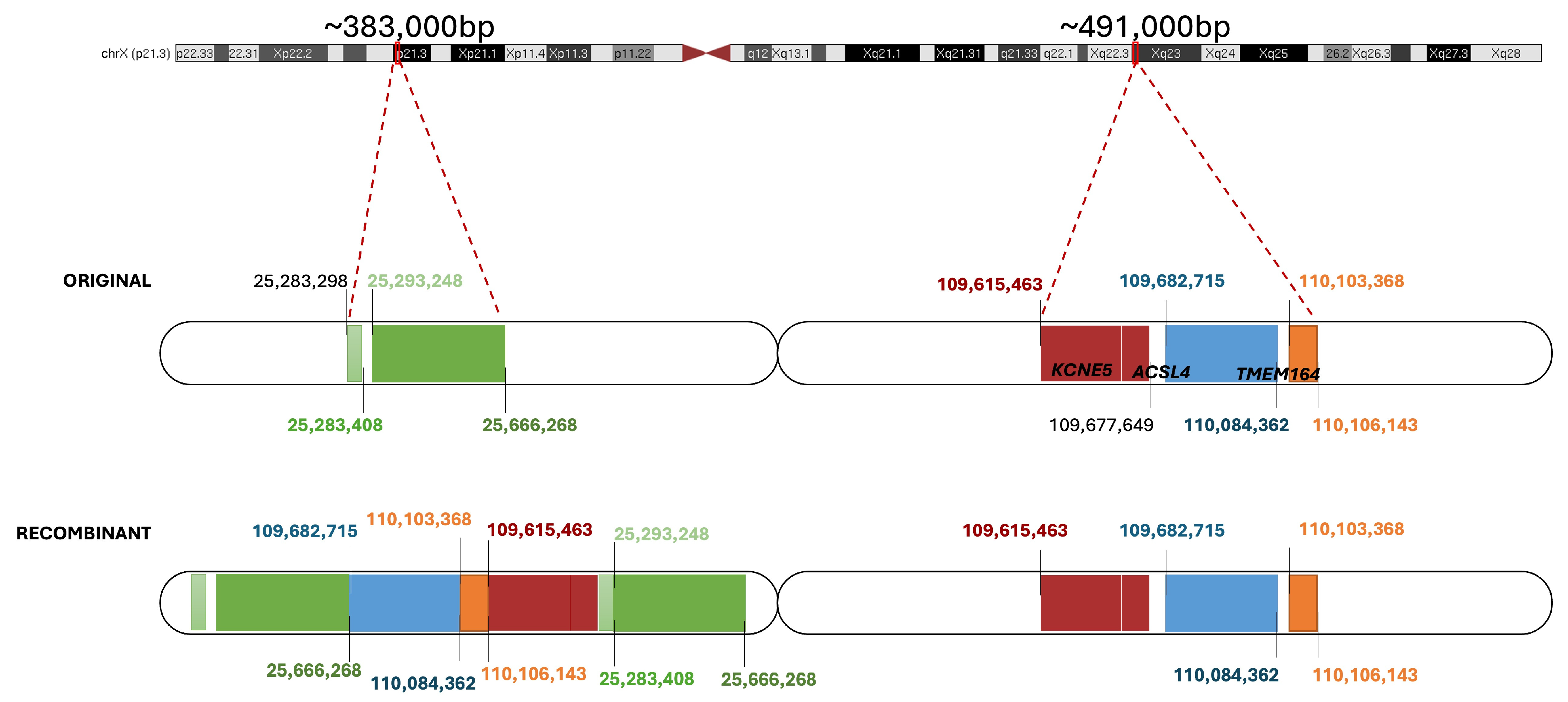
2.5. Non-Causative Complex Structural Rearrangement
3. Discussion
4. Materials and Methods
4.1. Subjects and Previous Studies
4.2. Clinical Evaluation
4.3. Library Preparation and Genomic Sequencing
4.4. Data Analyses
4.5. Variant Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aCGH | Array comparative genome hybridization |
| ACMG | American College of Medical Genetics and Genomics |
| CES | Clinical Exome Solution |
| CNV | Copy number variant |
| CSVS | Collaborative Spanish Variant Server |
| DNA | Deoxyribonucleic acid |
| FISH | Fluorescence in situ hybridization |
| FJD | Fundación Jiménez Díaz |
| GnomAD | Genome Aggregation Database |
| HGMD | Human Gene Mutation Database |
| HWM-DNA | High-molecular-weight DNA |
| IGV | Integrative Genomics Viewer |
| Indel | Insertion–deletion variant |
| IRD | Inherited retinal dystrophy |
| ISCN | International System for Human Cytogenomic Nomenclature |
| Kb | Kilobase |
| Kaviar | Known VARiants |
| L1HS | Long Interspersed Nuclear Element, subfamily H |
| LINE-1 | Long Interspersed Nuclear Element-1 |
| LOVD | Leiden Open Variation Database |
| LR-WGS | Long-read whole-genome sequencing |
| MLPA | Multiplex ligation-dependent probe amplification |
| NGS | Next-Generation Sequencing |
| ONT | Oxford Nanopore Technologies |
| Pb | Base pair |
| PEPPER | PEPPER-Margin-DeepVariant |
| RP | Retinitis pigmentosa |
| RPE | Retinal pigment epithelium |
| SD-OCT | Spectral Domain Optical Coherence Tomography |
| SNV | Single nucleotide variant |
| SV | Structural variant |
| VEP | Variant effect predictor |
| WES | Whole-exome sequencing |
| WGS | Whole-genome sequencing |
References
- Arteche-López, A.; Ávila-Fernández, A.; Romero, R.; Riveiro-Álvarez, R.; López-Martínez, M.A.; Giménez-Pardo, A.; Vélez-Monsalve, C.; Gallego-Merlo, J.; García-Vara, I.; Almoguera, B.; et al. Sanger Sequencing Is No Longer Always Necessary Based on a Single-Center Validation of 1109 NGS Variants in 825 Clinical Exomes. Sci. Rep. 2021, 11, 5697. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic Landscape of 6089 Inherited Retinal Dystrophies Affected Cases in Spain and Their Therapeutic and Extended Epidemiological Implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef]
- Gonzàlez-Duarte, R.; De Castro-Miró, M.; Tuson, M.; Ramírez-Castañeda, V.; Gils, R.V.; Marfany, G. Scaling New Heights in the Genetic Diagnosis of Inherited Retinal Dystrophies. In Retinal Degenerative Diseases; Bowes Rickman, C., Grimm, C., Anderson, R.E., Ash, J.D., LaVail, M.M., Hollyfield, J.G., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Germany, 2019; Volume 1185, pp. 215–219. ISBN 978-3-030-27377-4. [Google Scholar]
- Reurink, J.; Weisschuh, N.; Garanto, A.; Dockery, A.; Van Den Born, L.I.; Fajardy, I.; Haer-Wigman, L.; Kohl, S.; Wissinger, B.; Farrar, G.J.; et al. Whole Genome Sequencing for USH2A-Associated Disease Reveals Several Pathogenic Deep-Intronic Variants That Are Amenable to Splice Correction. Hum. Genet. Genom. Adv. 2023, 4, 100181. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; De Leeuw, N.; Mann, K.; Schuring-Blom, H.; Morgan, S.; Giardino, D.; Rack, K.; Hastings, R. European Guidelines for Constitutional Cytogenomic Analysis. Eur. J. Hum. Genet. 2019, 27, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Warburton, P.E.; Sebra, R.P. Long-Read DNA Sequencing: Recent Advances and Remaining Challenges. Annu. Rev. Genomics Hum. Genet. 2023, 24, 109–132. [Google Scholar] [CrossRef]
- Bonetti, G.; Cozza, W.; Bernini, A.; Kaftalli, J.; Mareso, C.; Cristofoli, F.; Medori, M.C.; Colombo, L.; Martella, S.; Staurenghi, G.; et al. Towards a Long-Read Sequencing Approach for the Molecular Diagnosis of RPGRORF15 Genetic Variants. Int. J. Mol. Sci. 2023, 24, 16881. [Google Scholar] [CrossRef] [PubMed]
- Sanchis-Juan, A.; Stephens, J.; French, C.E.; Gleadall, N.; Mégy, K.; Penkett, C.; Shamardina, O.; Stirrups, K.; Delon, I.; Dewhurst, E.; et al. Complex Structural Variants in Mendelian Disorders: Identification and Breakpoint Resolution Using Short- and Long-Read Genome Sequencing. Genome Med. 2018, 10, 95. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Billingsley, K.J.; Mastoras, M.; Meredith, M.; Monlong, J.; Lorig-Roach, R.; Asri, M.; Alvarez Jerez, P.; Malik, L.; Dewan, R.; et al. Scalable Nanopore Sequencing of Human Genomes Provides a Comprehensive View of Haplotype-Resolved Variation and Methylation. Nat. Methods 2023, 20, 1483–1492. [Google Scholar] [CrossRef]
- Chrystal, P.W.; Lambacher, N.J.; Doucette, L.P.; Bellingham, J.; Schiff, E.R.; Noel, N.C.L.; Li, C.; Tsiropoulou, S.; Casey, G.A.; Zhai, Y.; et al. The Inner Junction Protein CFAP20 Functions in Motile and Non-Motile Cilia and Is Critical for Vision. Nat. Commun. 2022, 13, 6595. [Google Scholar] [CrossRef]
- González-del Pozo, M.; Fernández-Suárez, E.; Bravo-Gil, N.; Méndez-Vidal, C.; Martín-Sánchez, M.; Rodríguez-de La Rúa, E.; Ramos-Jiménez, M.; Morillo-Sánchez, M.J.; Borrego, S.; Antiñolo, G. A Comprehensive WGS-Based Pipeline for the Identification of New Candidate Genes in Inherited Retinal Dystrophies. Npj Genomic Med. 2022, 7, 17. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ascari, G.; Rendtorff, N.D.; De Bruyne, M.; De Zaeytijd, J.; Van Lint, M.; Bauwens, M.; Van Heetvelde, M.; Arno, G.; Jacob, J.; Creytens, D.; et al. Long-Read Sequencing to Unravel Complex Structural Variants of CEP78 Leading to Cone-Rod Dystrophy and Hearing Loss. Front. Cell Dev. Biol. 2021, 9, 664317. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.; Stenton, S.L.; Berger, S.I.; Canigiula, P.; McNulty, B.; Violich, I.; Gardner, J.; Hillaker, T.; O’Rourke, S.M.; O’Leary, M.C.; et al. Advancing Long-Read Nanopore Genome Assembly and Accurate Variant Calling for Rare Disease Detection. Am. J. Hum. Genet. 2025, 112, 428–449. [Google Scholar] [CrossRef]
- Sano, Y.; Koyanagi, Y.; Wong, J.H.; Murakami, Y.; Fujiwara, K.; Endo, M.; Aoi, T.; Hashimoto, K.; Nakazawa, T.; Wada, Y.; et al. Likely Pathogenic Structural Variants in Genetically Unsolved Patients with Retinitis Pigmentosa Revealed by Long-Read Sequencing. J. Med. Genet. 2022, 59, 1133–1138. [Google Scholar] [CrossRef]
- Nakamichi, K.; Van Gelder, R.N.; Chao, J.R.; Mustafi, D. Targeted Adaptive Long-Read Sequencing for Discovery of Complex Phased Variants in Inherited Retinal Disease Patients. Sci. Rep. 2023, 13, 8535. [Google Scholar] [CrossRef]
- Natsuga, K.; Furuta, Y.; Takashima, S.; Nohara, T.; Huang, H.; Shinkuma, S.; Nakamura, H.; Katsuda, Y.; Higashi, H.; Hsu, C.; et al. Cas9-guided Haplotyping of Three Truncation Variants in Autosomal Recessive Disease. Hum. Mutat. 2022, 43, 877–881. [Google Scholar] [CrossRef]
- Fernández-Suárez, E.; González-del Pozo, M.; Méndez-Vidal, C.; Martín-Sánchez, M.; Mena, M.; De La Morena-Barrio, B.; Corral, J.; Borrego, S.; Antiñolo, G. Long-Read Sequencing Improves the Genetic Diagnosis of Retinitis Pigmentosa by Identifying an Alu Retrotransposon Insertion in the EYS Gene. Mob. DNA 2024, 15, 9. [Google Scholar] [CrossRef]
- Kwon, H.-J.; Lee, B.-H.; Lee, J.-Y. Detecting Alu Element Insertion Variant in RP1 Gene Using Whole Genome Sequencing in Patients with Retinitis Pigmentosa. Genes 2024, 15, 1290. [Google Scholar] [CrossRef]
- Gustafson, J.A.; Gibson, S.B.; Damaraju, N.; Zalusky, M.P.G.; Hoekzema, K.; Twesigomwe, D.; Yang, L.; Snead, A.A.; Richmond, P.A.; De Coster, W.; et al. High-Coverage Nanopore Sequencing of Samples from the 1000 Genomes Project to Build a Comprehensive Catalog of Human Genetic Variation. Genome Res. 2024, 34, 2061–2073. [Google Scholar] [CrossRef]
- De Bruijn, S.E.; Panneman, D.M.; Weisschuh, N.; Cadena, E.L.; Boonen, E.G.M.; Holtes, L.K.; Astuti, G.D.N.; Cremers, F.P.M.; Leijsten, N.; Corominas, J.; et al. Identification of Novel 3D-Genome Altering and Complex Structural Variants Underlying Retinitis Pigmentosa Type 17 through a Multistep and High-Throughput Approach. Front. Genet. 2024, 15, 1469686. [Google Scholar] [CrossRef] [PubMed]
- Hollox, E.J.; Zuccherato, L.W.; Tucci, S. Genome Structural Variation in Human Evolution. Trends Genet. 2022, 38, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Lu, H.; Lu, Y.; Zhou, G. Population-Scale Genotyping of Structural Variation in the Era of Long-Read Sequencing. Comput. Struct. Biotechnol. J. 2022, 20, 2639–2647. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Li, X.; Lai, Q.; Han, L.; et al. Comparison of the Two Up-to-Date Sequencing Technologies for Genome Assembly: HiFi Reads of Pacific Biosciences Sequel II System and Ultralong Reads of Oxford Nanopore. GigaScience 2020, 9, giaa123. [Google Scholar] [CrossRef]
- Redaelli, S.; Maitz, S.; Crosti, F.; Sala, E.; Villa, N.; Spaccini, L.; Selicorni, A.; Rigoldi, M.; Conconi, D.; Dalprà, L.; et al. Refining the Phenotype of Recurrent Rearrangements of Chromosome 16. Int. J. Mol. Sci. 2019, 20, 1095. [Google Scholar] [CrossRef]
- Allach El Khattabi, L.; Heide, S.; Caberg, J.-H.; Andrieux, J.; Doco Fenzy, M.; Vincent-Delorme, C.; Callier, P.; Chantot-Bastaraud, S.; Afenjar, A.; Boute-Benejean, O.; et al. 16p13.11 Microduplication in 45 New Patients: Refined Clinical Significance and Genotype–Phenotype Correlations. J. Med. Genet. 2020, 57, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Thiyagarajan, L.; Dudding-Byth, T.; Pinese, M.; Kirk, E.P. A Systematic Review and Pooled Analysis of Penetrance Estimates of Copy-Number Variants Associated with Neurodevelopment. Genet. Med. 2025, 27, 101227. [Google Scholar] [CrossRef]
- Damián, A.; Núñez-Moreno, G.; Jubin, C.; Tamayo, A.; De Alba, M.R.; Villaverde, C.; Fund, C.; Delépine, M.; Leduc, A.; Deleuze, J.F.; et al. Long-Read Genome Sequencing Identifies Cryptic Structural Variants in Congenital Aniridia Cases. Hum. Genomics 2023, 17, 45. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Kolmogorov, M.; Eizenga, J.M.; Miga, K.H.; et al. Haplotype-Aware Variant Calling with PEPPER-Margin-DeepVariant Enables High Accuracy in Nanopore Long-Reads. Nat. Methods 2021, 18, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, S.; Su, J.; Leung, A.W.-S.; Lam, T.-W.; Luo, R. Symphonizing Pileup and Full-Alignment for Deep Learning-Based Long-Read Variant Calling. Nat. Comput. Sci. 2022, 2, 797–803. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, Y.; Jiang, Y.; Li, J.; Gao, Y.; Cui, Z.; Liu, Y.; Liu, B.; Wang, Y. Long-Read-Based Human Genomic Structural Variation Detection with cuteSV. Genome Biol. 2020, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.; Vingron, M. SVIM: Structural Variant Identification Using Mapped Long Reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; Von Haeseler, A.; Schatz, M.C. Accurate Detection of Complex Structural Variations Using Single-Molecule Sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.; Paulin, L.F.; Grochowski, C.M.; Horner, D.W.; Mahmoud, M.; Behera, S.; Kalef-Ezra, E.; Gandhi, M.; Hong, K.; Pehlivan, D.; et al. Detection of Mosaic and Population-Level Structural Variants with Sniffles2. Nat. Biotechnol. 2024. [Google Scholar] [CrossRef]
- Romero, R.; De La Fuente, L.; Del Pozo-Valero, M.; Riveiro-Álvarez, R.; Trujillo-Tiebas, M.J.; Martín-Mérida, I.; Ávila-Fernández, A.; Iancu, I.-F.; Perea-Romero, I.; Núñez-Moreno, G.; et al. An Evaluation of Pipelines for DNA Variant Detection Can Guide a Reanalysis Protocol to Increase the Diagnostic Ratio of Genetic Diseases. Npj Genomic Med. 2022, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An Integrated Tool for Structural Variations Annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Harrison, S.M.; Biesecker, L.G.; Rehm, H.L. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 2019, 103, e93. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Peña-Chilet, M.; Roldán, G.; Perez-Florido, J.; Ortuño, F.M.; Carmona, R.; Aquino, V.; Lopez-Lopez, D.; Loucera, C.; Fernandez-Rueda, J.L.; Gallego, A.; et al. CSVS, a Crowdsourcing Database of the Spanish Population Genetic Variability. Nucleic Acids Res. 2021, 49, D1130–D1137. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Nattestad, M.; Aboukhalil, R.; Chin, C.-S.; Schatz, M.C. Ribbon: Intuitive Visualization for Complex Genomic Variation. Bioinformatics 2021, 37, 413–415. [Google Scholar] [CrossRef]
- ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020); McGowan-Jordan, J., Hastings, R.J., Moore, S., Eds.; S. Karger AG: Basel, Switzerland, 2020; ISBN 978-3-318-06706-4. Available online: https://karger.com/books/book/358/ISCN-2020An-International-System-for-Human (accessed on 14 April 2025).
- Patterson, M.; Marschall, T.; Pisanti, N.; Van Iersel, L.; Stougie, L.; Klau, G.W.; Schönhuth, A. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol. 2015, 22, 498–509. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodilla, C.; Núñez-Moreno, G.; Benitez, Y.; Rodríguez de Alba, M.; Blanco-Kelly, F.; López-Alcojor, A.; Fernández-Caballero, L.; Perea-Romero, I.; Del Pozo-Valero, M.; García-García, G.; et al. Long-Read Whole-Genome Sequencing as a Tool for Variant Detection in Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2025, 26, 3825. https://doi.org/10.3390/ijms26083825
Rodilla C, Núñez-Moreno G, Benitez Y, Rodríguez de Alba M, Blanco-Kelly F, López-Alcojor A, Fernández-Caballero L, Perea-Romero I, Del Pozo-Valero M, García-García G, et al. Long-Read Whole-Genome Sequencing as a Tool for Variant Detection in Inherited Retinal Dystrophies. International Journal of Molecular Sciences. 2025; 26(8):3825. https://doi.org/10.3390/ijms26083825
Chicago/Turabian StyleRodilla, Cristina, Gonzalo Núñez-Moreno, Yolanda Benitez, Marta Rodríguez de Alba, Fiona Blanco-Kelly, Aroa López-Alcojor, Lidia Fernández-Caballero, Irene Perea-Romero, Marta Del Pozo-Valero, Gema García-García, and et al. 2025. "Long-Read Whole-Genome Sequencing as a Tool for Variant Detection in Inherited Retinal Dystrophies" International Journal of Molecular Sciences 26, no. 8: 3825. https://doi.org/10.3390/ijms26083825
APA StyleRodilla, C., Núñez-Moreno, G., Benitez, Y., Rodríguez de Alba, M., Blanco-Kelly, F., López-Alcojor, A., Fernández-Caballero, L., Perea-Romero, I., Del Pozo-Valero, M., García-García, G., Balanzá, M., Villaverde, C., Zurita, O., Jubin, C., Fund, C., Delepine, M., Leduc, A., Deleuze, J.-F., Millán, J. M., ... Ayuso, C. (2025). Long-Read Whole-Genome Sequencing as a Tool for Variant Detection in Inherited Retinal Dystrophies. International Journal of Molecular Sciences, 26(8), 3825. https://doi.org/10.3390/ijms26083825