
Comprehensive SHAP Values and Single-Cell Sequencing Technology Reveal Key Cell Clusters in Bovine Skeletal Muscle

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Construction of the Primitive Atlas for Bovine Skeletal Musculature
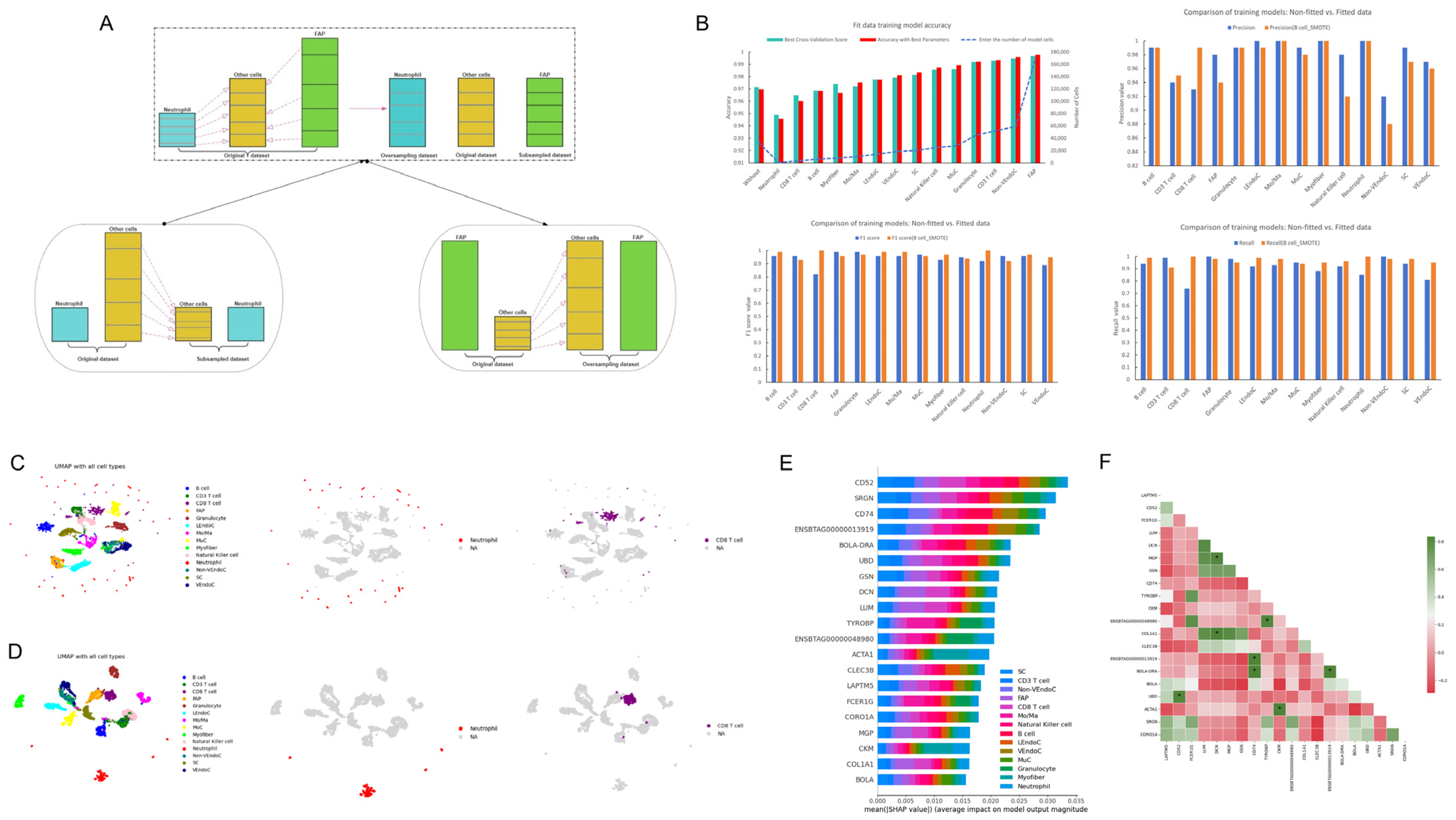
2.2. Multi-Model Optimization and the Identification of Key Genes in Skeletal Muscle
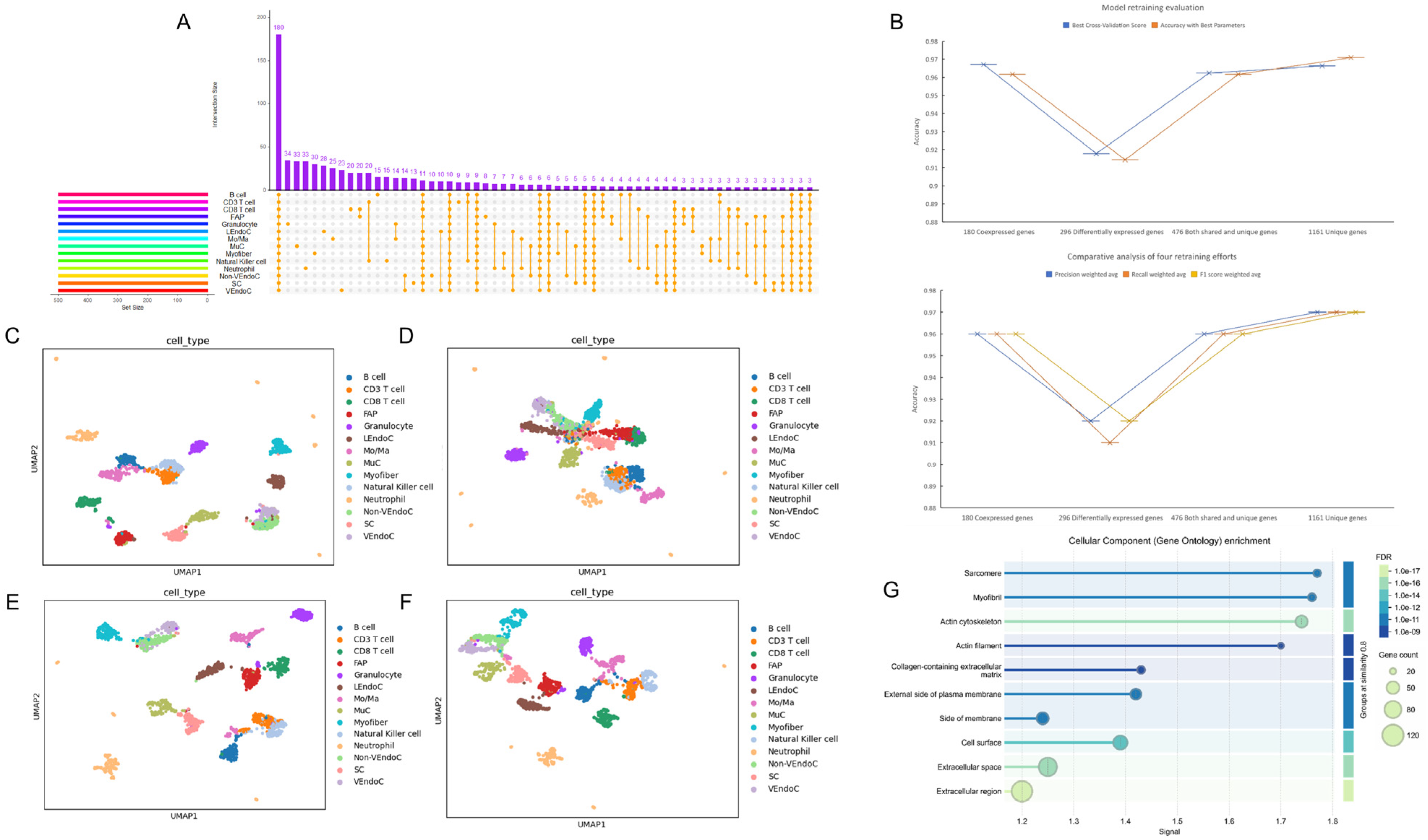
2.3. Reconstruction of Single-Cell Atlases and Model Optimization
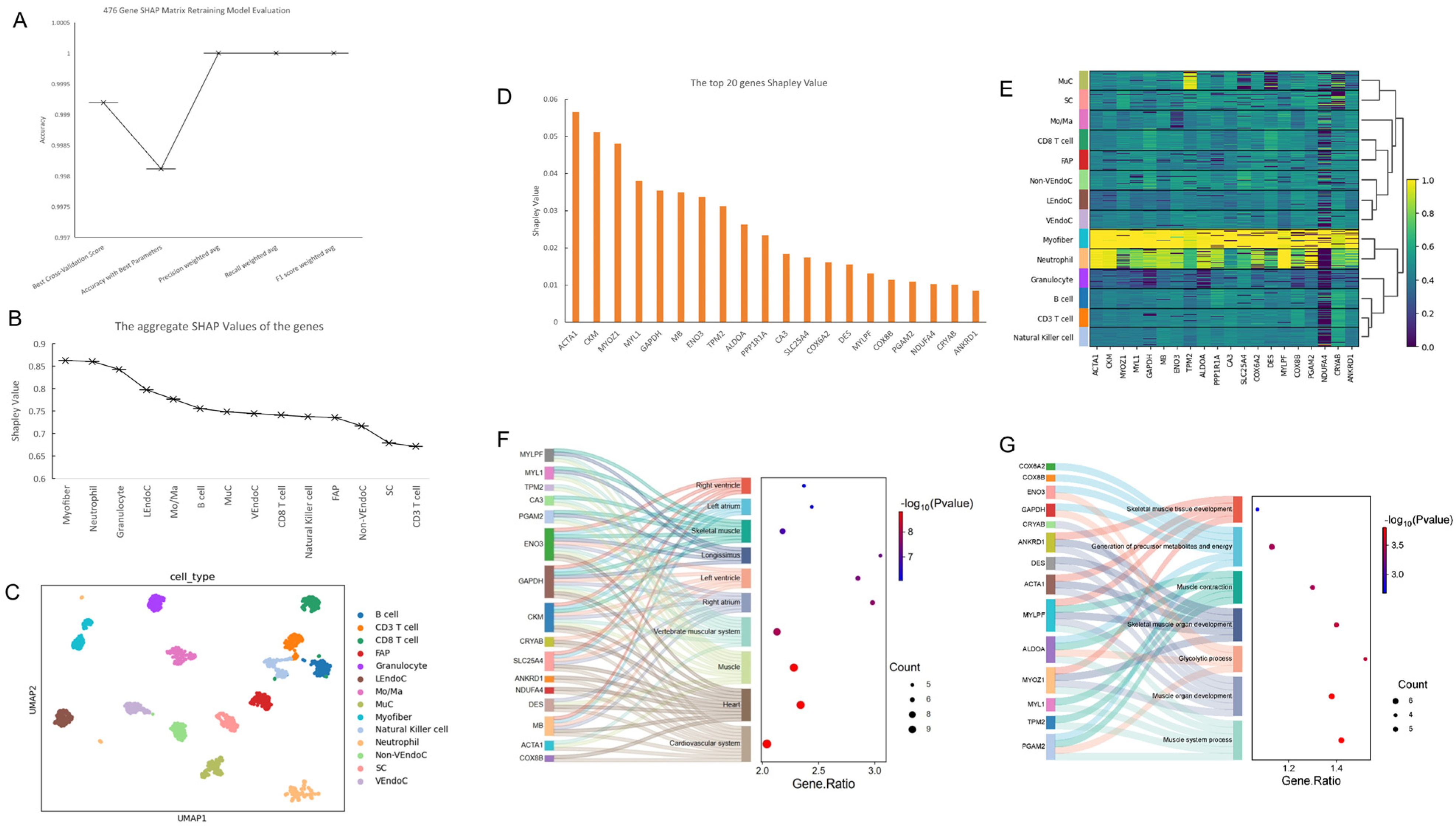
2.4. Construction of Single-Cell Atlas and Identification of Key Genes Based on SHAP Value Optimization
2.5. Comparative Analysis of Gene Expression and Functional Characteristics in Myofibers Relative to Other Cellular Types
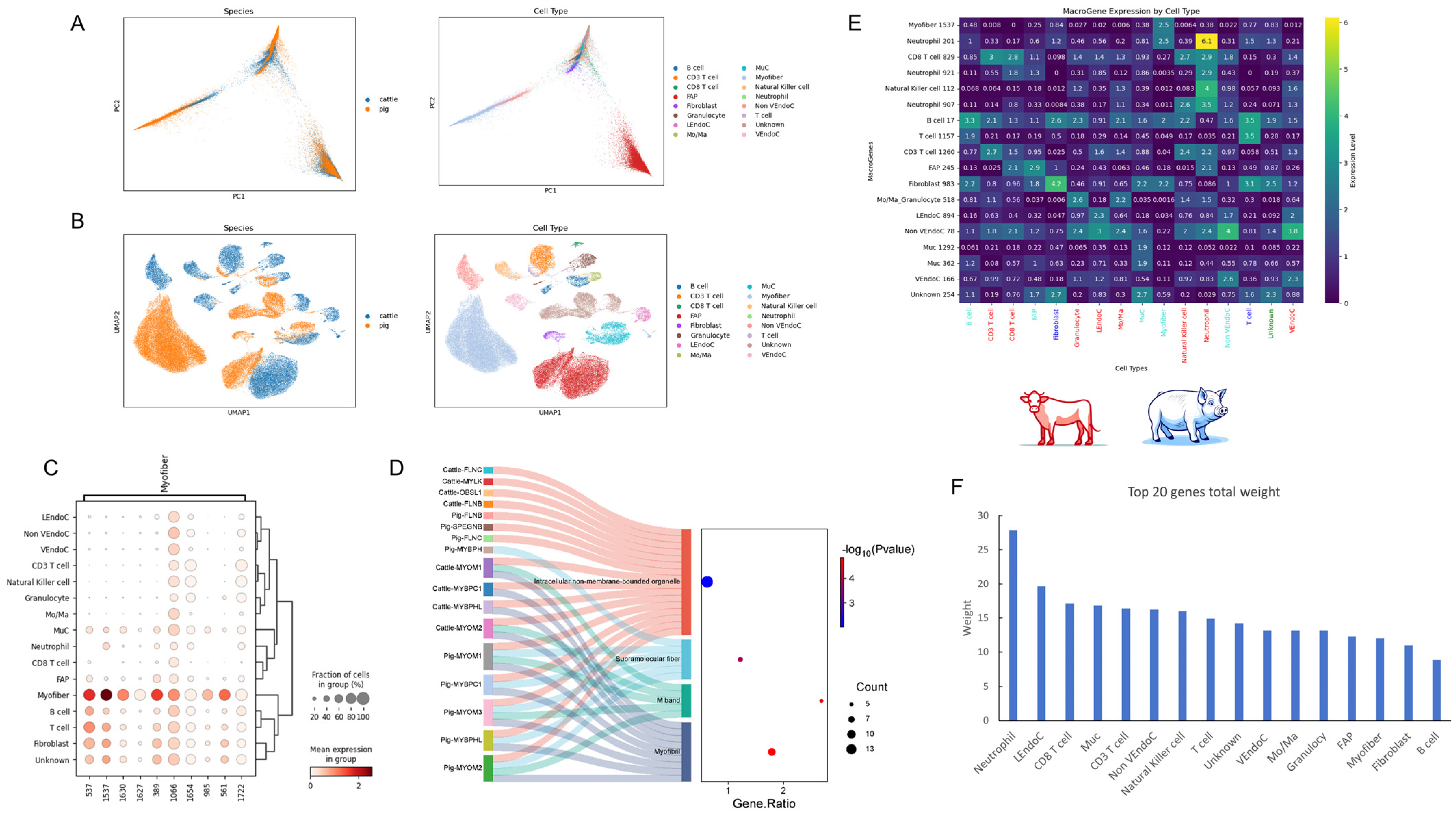
2.6. Cross-Species Single-Cell Analysis of Bovine and Porcine Skeletal Muscle
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Training of Bovine Skeletal Muscle Models
4.3. Analysis of SHAP Explainability in Skeletal Muscle Models
4.4. Model Training for 476 Co-Expressed and Specifically Expressed Genes
4.5. Cross-Species Comparison of Skeletal Muscle in Cattle and Pigs
4.6. Data Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Picard, B.; Gagaoua, M. Muscle Fiber Properties in Cattle and Their Relationships with Meat Qualities: An Overview. J. Agric. Food Chem. 2020, 22, 6021–6039. [Google Scholar] [CrossRef]
- Salim, A.P.A.A.; Ferrari, R.G.; Monteiro, M.L.G.; Mano, S.B. Effect of different feeding systems on color of longissimus muscle from Bos cattle: A systematic review and meta-analysis. Meat Sci. 2022, 192, 108871. [Google Scholar] [CrossRef]
- Duranti, G. Oxidative Stress and Skeletal Muscle Function. Int. J. Mol. Sci. 2023, 24, 10227. [Google Scholar] [CrossRef]
- Park, S.J.; Beak, S.H.; Jung, D.J.S.; Kim, S.Y.; Jeong, I.H.; Piao, M.Y.; Kang, H.J.; Fassah, D.M.; Na, S.W.; Yoo, S.P.; et al. Genetic, management, and nutritional factors affecting intramuscular fat deposition in beef cattle—A review. Asian-Australas. J. Anim. Sci. 2018, 7, 1043–1061. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Dong, Q.; Wu, B.; Yang, S.; Sun, J.; Cui, G.; Xu, W.; Zhao, M.; Zhang, Y.; Li, P.; et al. Global Landscape of m6A Methylation of Differently Expressed Genes in Muscle Tissue of Liaoyu White Cattle and Simmental Cattle. Front. Cell Dev. Biol. 2022, 10, 840513. [Google Scholar] [CrossRef] [PubMed]
- Bjercke, R.J.; Goll, D.E.; Robson, R.M.; Dutson, T.R. Relative roles of polysomes and cytoplasmic enzymes in regulating bovine skeletal muscle protein synthesis. J. Anim. Sci. 1984, 3, 684–696. [Google Scholar] [CrossRef]
- Kim, G.D.; Yang, H.S.; Jeong, J.Y. Comparison of Characteristics of Myosin Heavy Chain-based Fiber and Meat Quality among Four Bovine Skeletal Muscles. Korean J. Food Sci. Anim. Resour. 2016, 6, 819–828. [Google Scholar] [CrossRef] [PubMed]
- McKenna, C.; Keogh, K.; Porter, R.K.; Waters, S.M.; Cormican, P.; Kenny, D.A. An examination of skeletal muscle and hepatic tissue transcriptomes from beef cattle divergent for residual feed intake. Sci. Rep. 2021, 1, 8942. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Identification and Characterization of circRNA in Longissimus Dorsi of Different Breeds of Cattle. Front. Genet. 2020, 11, 565085. [Google Scholar] [CrossRef]
- Zhang, J.; Sheng, H.; Pan, C.; Wang, S.; Yang, M.; Hu, C.; Wei, D.; Wang, Y.; Ma, Y. Identification of key genes in bovine muscle development by co-expression analysis. PeerJ. 2023, 11, e15093. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Scuderi, F.; Provenzano, C.; Bartoccioni, E. Skeletal muscle cells: From local inflammatory response to active immunity. Gene Ther. 2011, 2, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, A.; Wojcik, S.; Pagano, T.B.; De Biase, D.; Russo, V.; Iovane, V.; Grieco, E.; Papparella, S.; Paciello, O. Age-Related Changes in Skeletal Muscle of Cattle. Vet. Pathol. 2016, 2, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.V.; Nguyen, O.C.; Malau-Aduli, A.E.O. Main regulatory factors of marbling level in beef cattle. Vet. Anim. Sci. 2021, 14, 100219. [Google Scholar] [CrossRef]
- Bhat, M.; Rabindranath, M.; Chara, B.S.; Simonetto, D.A. Artificial intelligence, machine learning, and deep learning in liver transplantation. J. Hepatol. 2023, 6, 1216–1233. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.W.; Torres Soto, J.; Glicksberg, B.S.; Shameer, K.; Miotto, R.; Ali, M.; Ashley, E.; Dudley, J.T. Artificial Intelligence in Cardiology. J. Am. Coll. Cardiol. 2018, 23, 2668–2679. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, D.; Liu, H.; Wen, M. CT-based radiomics analysis of different machine learning models for differentiating benign and malignant parotid tumors. Eur. Radiol. 2022, 10, 6953–6964. [Google Scholar] [CrossRef]
- Isles, P.D.F. A random forest approach to improve estimates of tributary nutrient loading. Water Res. 2024, 248, 120876. [Google Scholar] [CrossRef]
- Hu, J.; Szymczak, S. A review on longitudinal data analysis with random forest. Brief. Bioinform. 2023, 2, bbad002. [Google Scholar] [CrossRef]
- Pfeifer, B.; Sirocchi, C.; Bloice, M.D.; Kreuzthaler, M.; Urschler, M. Federated unsupervised random forest for privacy-preserving patient stratification. Bioinformatics 2024, 2, ii198–ii207. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Shao, L.; Lang, Y. A study on the factors influencing the utilization of public health services by China’s migrant population based on the Shapley value method. BMC Public Health 2023, 1, 2328. [Google Scholar] [CrossRef]
- Shickel, B.; Tighe, P.J.; Bihorac, A.; Rashidi, P. Deep EHR: A Survey of Recent Advances in Deep Learning Techniques for Electronic Health Record (EHR) Analysis. IEEE J. Biomed. Health Inform. 2018, 5, 1589–1604. [Google Scholar] [CrossRef]
- Lim, S.; Lee, S.; Piao, Y.; Choi, M.; Bang, D.; Gu, J.; Kim, S. On modeling and utilizing chemical compound information with deep learning technologies: A task-oriented approach. Comput. Struct. Biotechnol. J. 2022, 20, 4288–4304. [Google Scholar] [CrossRef]
- Mousavi, S.M.; Beroza, G.C. Deep-learning seismology. Science 2022, 6607, eabm4470. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef] [PubMed]
- Rosen, Y.; Brbić, M.; Roohani, Y.; Swanson, K.; Li, Z.; Leskovec, J. Toward universal cell embeddings: Integrating single-cell RNA-seq datasets across species with SATURN. Nat. Methods 2024, 8, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S. Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol. 2017, 5, 637–650. [Google Scholar] [CrossRef]
- Misra, P.; Jadhav, A.R.; Bapat, S.A. Single-cell sequencing: A cutting edge tool in molecular medical research. Med. J. Armed Forces India 2022, 78 (Suppl. S1), S7–S13. [Google Scholar] [CrossRef] [PubMed]
- Aliya, S.; Lee, H.; Alhammadi, M.; Umapathi, R.; Huh, Y.S. An Overview on Single-Cell Technology for Hepatocellular Carcinoma Diagnosis. Int. J. Mol. Sci. 2022, 3, 1402. [Google Scholar] [CrossRef]
- Raja, G.; Ramkumar, B.; Rajendiran, B.; Prathiba, S.B.; Arumugam, T.; Karuppanan, K.; Nkenyereye, L.; Dev, K. Synergistic Analysis of Lung Cancer’s Impact on Cardiovascular Disease Using ML-Based Techniques. IEEE J. Biomed. Health Inform. 2024. [Google Scholar] [CrossRef]
- Dablain, D.; Krawczyk, B.; Chawla, N.V. DeepSMOTE: Fusing Deep Learning and SMOTE for Imbalanced Data. IEEE Trans. Neural. Netw. Learn. Syst. 2023, 9, 6390–6404. [Google Scholar] [CrossRef]
- Choudhary, S.; Satija, R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 2022, 1, 27. [Google Scholar] [CrossRef]
- Vallejo, J.; Cochain, C.; Zernecke, A.; Ley, K. Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-Seq. Cardiovasc. Res. 2021, 13, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Yu, J.; Li, J.; Li, N.; Liu, J.; Xiu, L.; Zeng, J.; Wang, T.; Wu, L. Integration of scRNA-Seq and Bulk RNA-Seq to Analyse the Heterogeneity of Ovarian Cancer Immune Cells and Establish a Molecular Risk Model. Front. Oncol. 2021, 11, 711020. [Google Scholar] [CrossRef]
- Harvey, H.B.; Sotardi, S.T. The Pareto Principle. J. Am. Coll. Radiol. 2018, 6, 931. [Google Scholar] [CrossRef]
- Xiao, Q.; Niu, B.; Tan, Y.; Yang, Z.; Chen, X. Generative Upper-Level Policy Imitation Learning With Pareto-Improvement for Energy-Efficient Advanced Machining Systems. IEEE Trans. Neural Netw. Learn. Syst. 2024. [Google Scholar] [CrossRef]
- Suzuki, A.; Hayama, S.; Okada, M. Composition of myofiber types in the vocalis muscles involved in rapid closure of the glottis in Japanese macaques. Z. Morphol. Anthropol. 2002, 83, 161–169. [Google Scholar] [CrossRef]
- Wright, S.A.; Ramos, P.; Johnson, D.D.; Scheffler, J.M.; Elzo, M.A.; Mateescu, R.G.; Bass, A.L.; Carr, C.C.; Scheffler, T.L. Brahman genetics influence muscle fiber properties, protein degradation, and tenderness in an Angus-Brahman multibreed herd. Meat Sci. 2018, 135, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.L.; de Carvalho, E.B.; Ramos, E.M.; Pereira, L.J.; Zangeronimo, M.G. Relationship between beta-adrenergic agonists, calpain system activity and beef texture: A systematic review. J. Anim. Physiol. Anim. Nutr. 2021, 3, 442–451. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Li, Y.; Tang, M.; Deng, Q.; Mao, J.; Du, L. Estrogen Regulates Glucose Metabolism in Cattle Neutrophils Through Autophagy. Front. Vet. Sci. 2021, 8, 773514. [Google Scholar] [CrossRef]
- Morrell, E.L.; Odriozola, E.; Dorsch, M.A.; Fiorentino, M.A.; Rivera, M.E.; Poppenga, R.; Navarro, M.A.; Uzal, F.A.; Cantón, G. A review of cardiac blackleg in cattle, and report of 2 cases without skeletal muscle involvement in Argentina. J. Vet. Diagn. Investig. 2022, 6, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 2, 1223–1248. [Google Scholar] [CrossRef]
- Ilkovski, B.; Cooper, S.T.; Nowak, K.; Ryan, M.M.; Yang, N.; Schnell, C.; Durling, H.J.; Roddick, L.G.; Wilkinson, I.; Kornberg, A.J.; et al. Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene. Am. J. Hum. Genet. 2001, 6, 1333–1343. [Google Scholar] [CrossRef]
- Goebel, H.H.; Brockmann, K.; Bönnemann, C.G.; Warlo, I.A.; Hanefeld, F.; Labeit, S.; Durling, H.J.; Laing, N.G. Actin-related myopathy without any missense mutation in the ACTA1 gene. J. Child. Neurol. 2004, 2, 149–153. [Google Scholar] [CrossRef]
- Nowak, K.J.; Ravenscroft, G.; Jackaman, C.; Filipovska, A.; Davies, S.M.; Lim, E.M.; Squire, S.E.; Potter, A.C.; Baker, E.; Clément, S.; et al. Rescue of skeletal muscle alpha-actin-null mice by cardiac (fetal) alpha-actin. J. Cell Biol. 2009, 5, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for gene expression and epigenomics data sets: 23-year update. Nucleic Acids Res. 2024, 52, D138–D144. [Google Scholar] [CrossRef]
- Kurtaliaj, I.; Hoppe, E.D.; Huang, Y.; Ju, D.; Sandler, J.A.; Yoon, D.; Smith, L.J.; Betancur, S.T.; Effiong, L.; Gardner, T.; et al. Python tooth-inspired fixation device for enhanced rotator cuff repair. Sci. Adv. 2024, 26, 5270. [Google Scholar] [CrossRef]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Zisi, C.; Pappa-Louisi, A.; Nikitas, P. Separation optimization in HPLC analysis implemented in R programming language. J. Chromatogr. A 2020, 1617, 460823. [Google Scholar] [CrossRef]
- Rocheta, M.; Dionísio, F.M.; Fonseca, L.; Pires, A.M. Paternity analysis in Excel. Comput. Methods Programs Biomed. 2007, 3, 234–248. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Ma, F.; Li, P.; Guo, L.; Liu, Z.; Huo, C.; Shi, C.; Zhu, L.; Gu, M.; Na, R.; et al. Comprehensive SHAP Values and Single-Cell Sequencing Technology Reveal Key Cell Clusters in Bovine Skeletal Muscle. Int. J. Mol. Sci. 2025, 26, 2054. https://doi.org/10.3390/ijms26052054
Guo Y, Ma F, Li P, Guo L, Liu Z, Huo C, Shi C, Zhu L, Gu M, Na R, et al. Comprehensive SHAP Values and Single-Cell Sequencing Technology Reveal Key Cell Clusters in Bovine Skeletal Muscle. International Journal of Molecular Sciences. 2025; 26(5):2054. https://doi.org/10.3390/ijms26052054
Chicago/Turabian StyleGuo, Yaqiang, Fengying Ma, Peipei Li, Lili Guo, Zaixia Liu, Chenxi Huo, Caixia Shi, Lin Zhu, Mingjuan Gu, Risu Na, and et al. 2025. "Comprehensive SHAP Values and Single-Cell Sequencing Technology Reveal Key Cell Clusters in Bovine Skeletal Muscle" International Journal of Molecular Sciences 26, no. 5: 2054. https://doi.org/10.3390/ijms26052054
APA StyleGuo, Y., Ma, F., Li, P., Guo, L., Liu, Z., Huo, C., Shi, C., Zhu, L., Gu, M., Na, R., & Zhang, W. (2025). Comprehensive SHAP Values and Single-Cell Sequencing Technology Reveal Key Cell Clusters in Bovine Skeletal Muscle. International Journal of Molecular Sciences, 26(5), 2054. https://doi.org/10.3390/ijms26052054