
Identification and Molecular Mechanism of Novel α-Glucosidase Inhibitory Peptides from the Hydrolysate of Hemp Seed Proteins: Peptidomic Analysis, Molecular Docking, and Dynamics Simulation
 ,
,
Abstract
1. Introduction
2. Results and Discussion
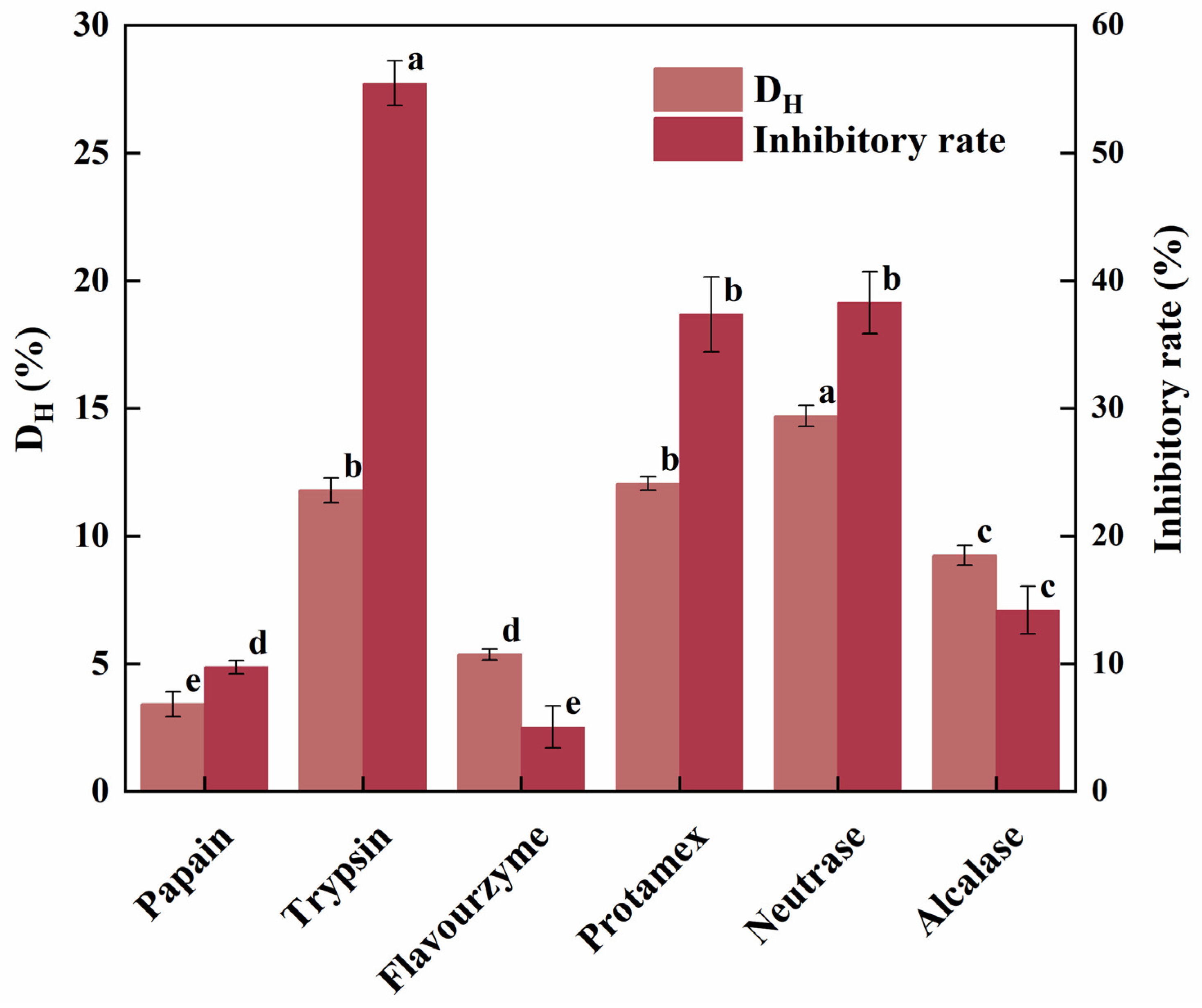
2.1. Protease Screening for HSP Hydrolysis
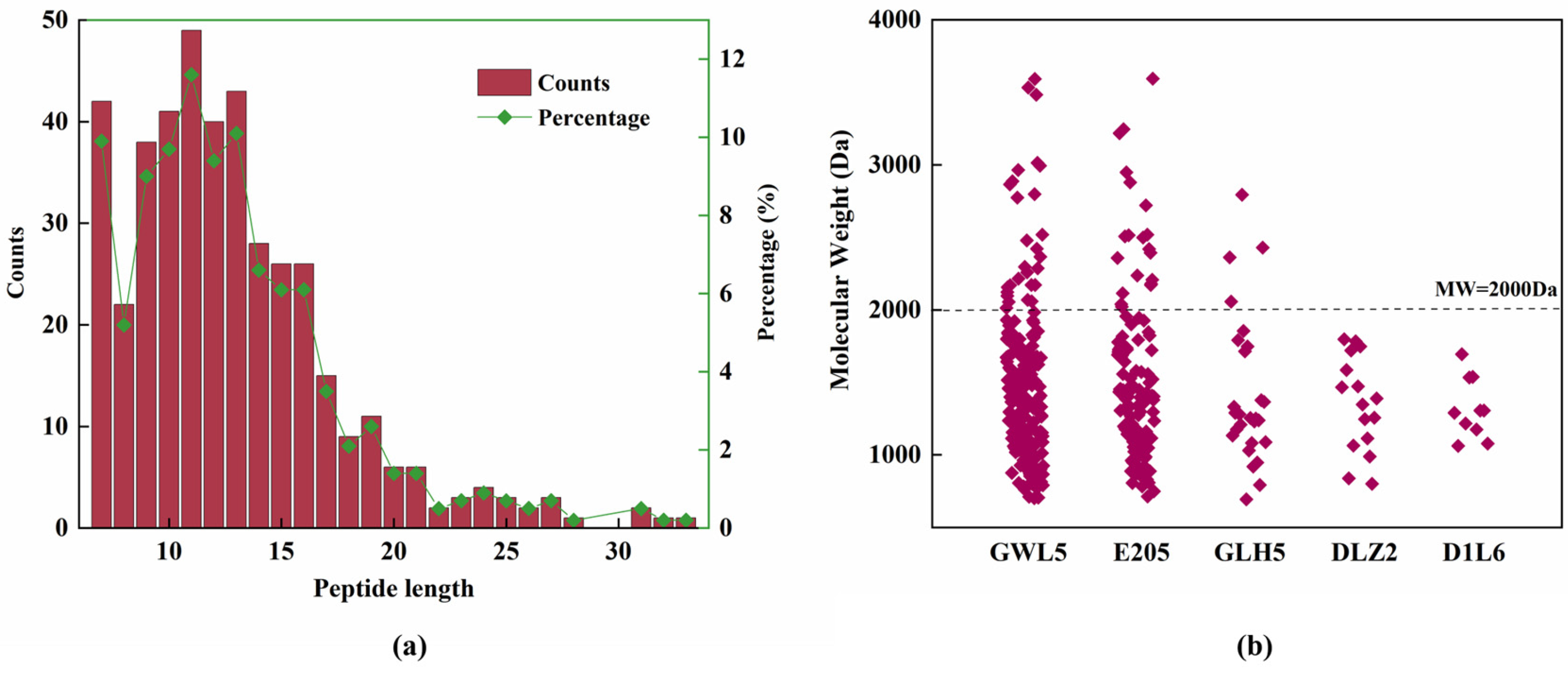
2.2. Peptidomics Analysis
2.3. Prediction of Potential Activity of the Peptides in Tryptic Hydrolysates of HSP
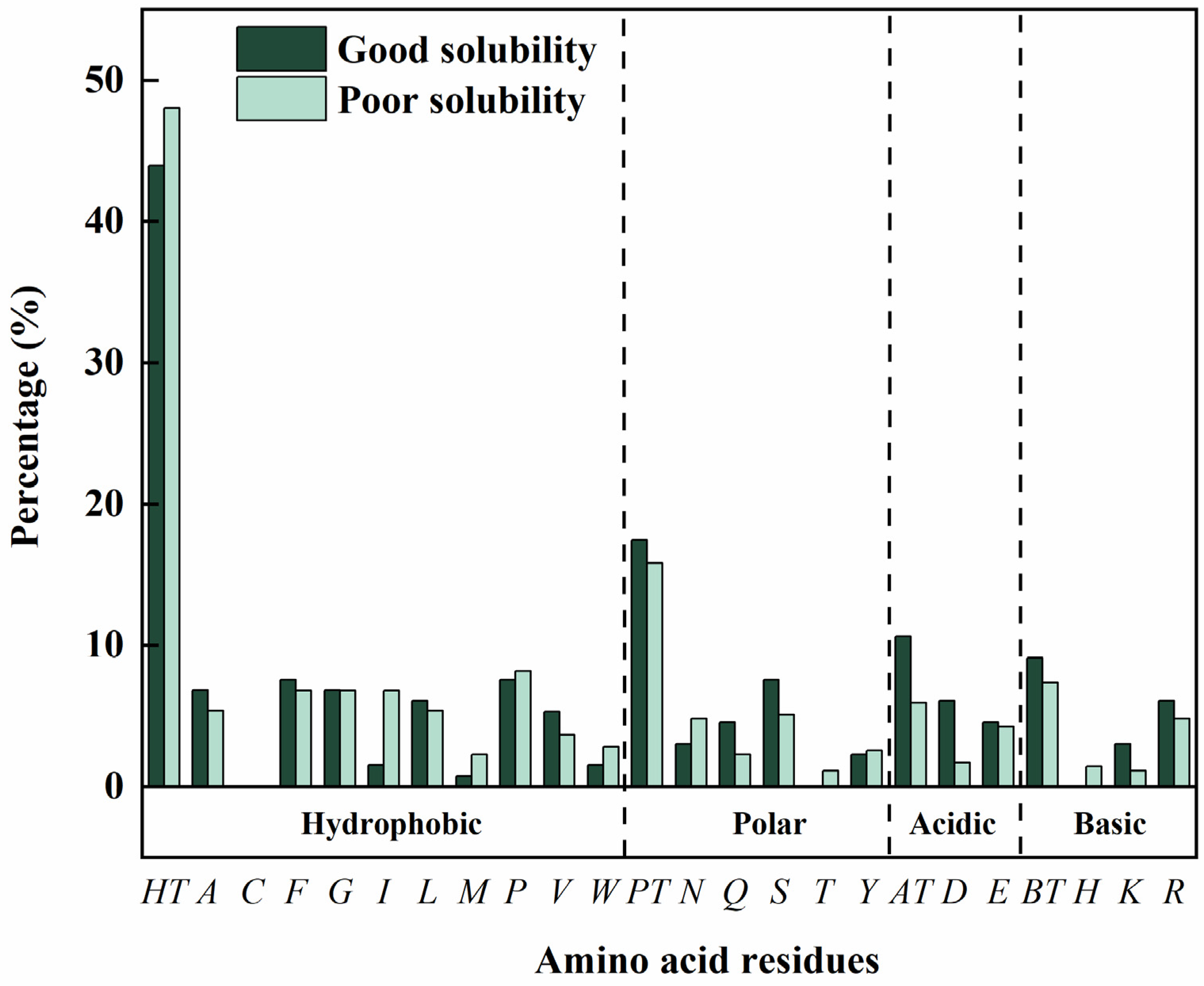
2.4. Analysis of Physicochemical Properties of the Peptides
2.5. Toxicity and ADMET Property Analysis of the Peptides
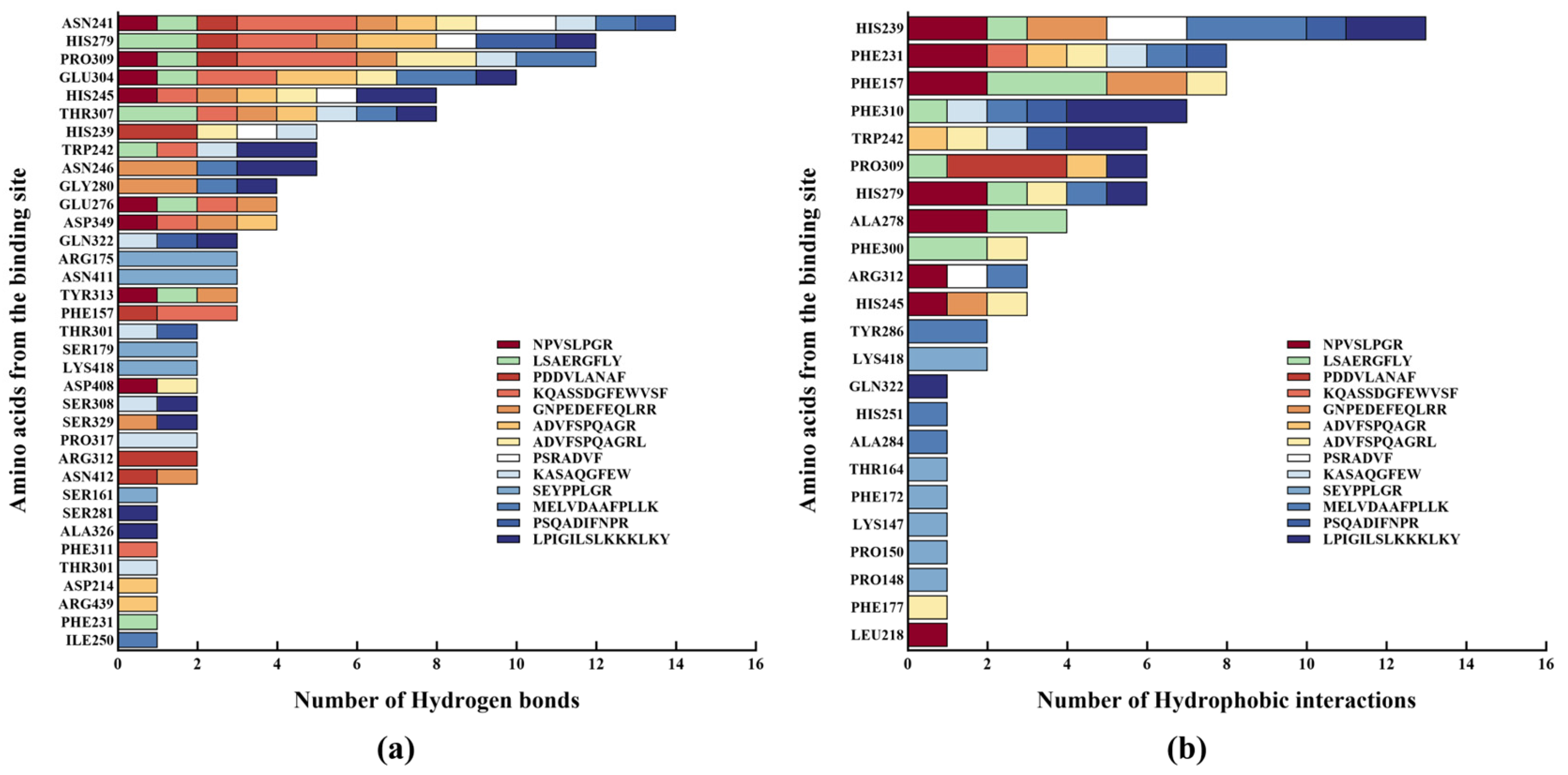
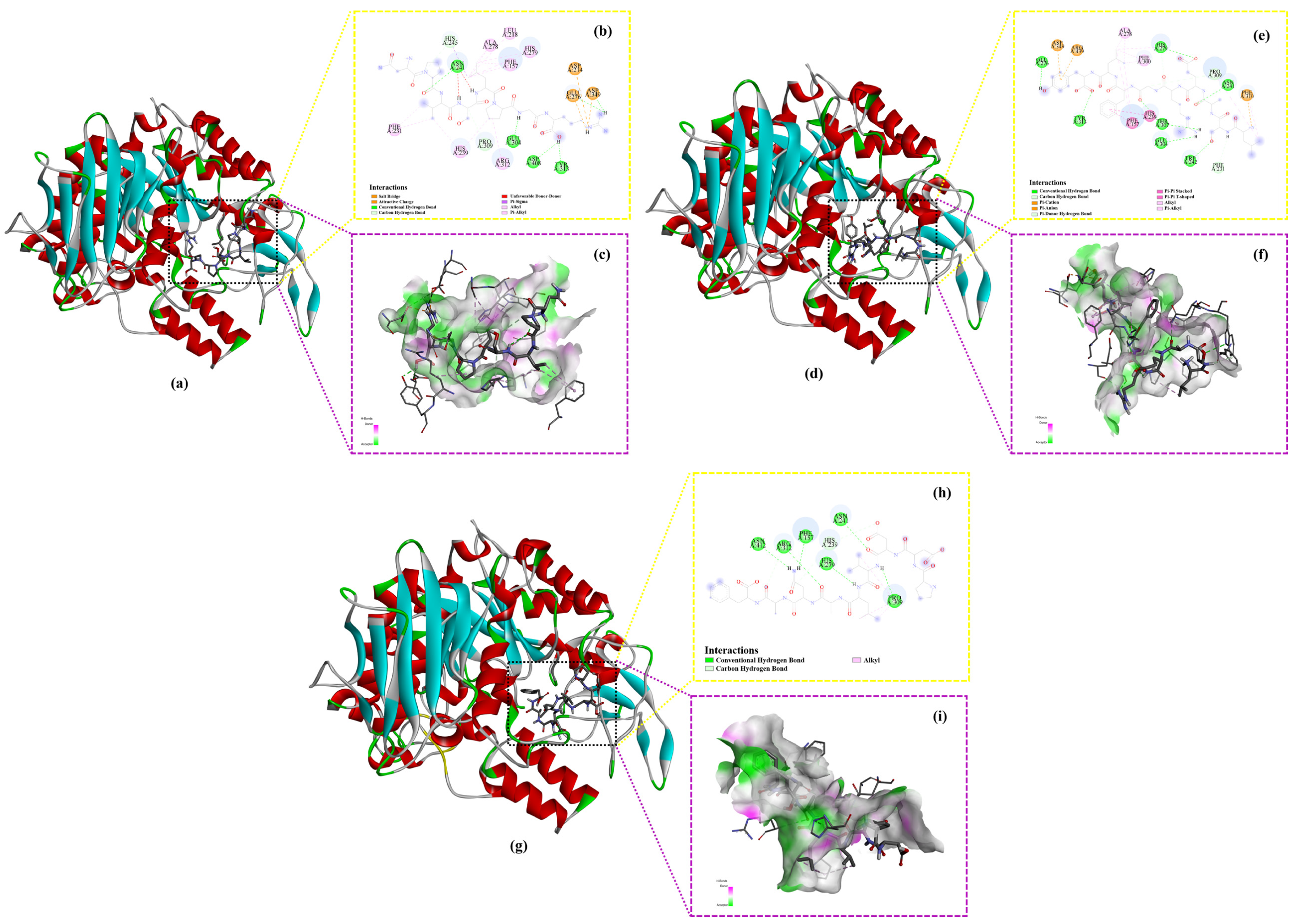
2.6. Molecular Docking
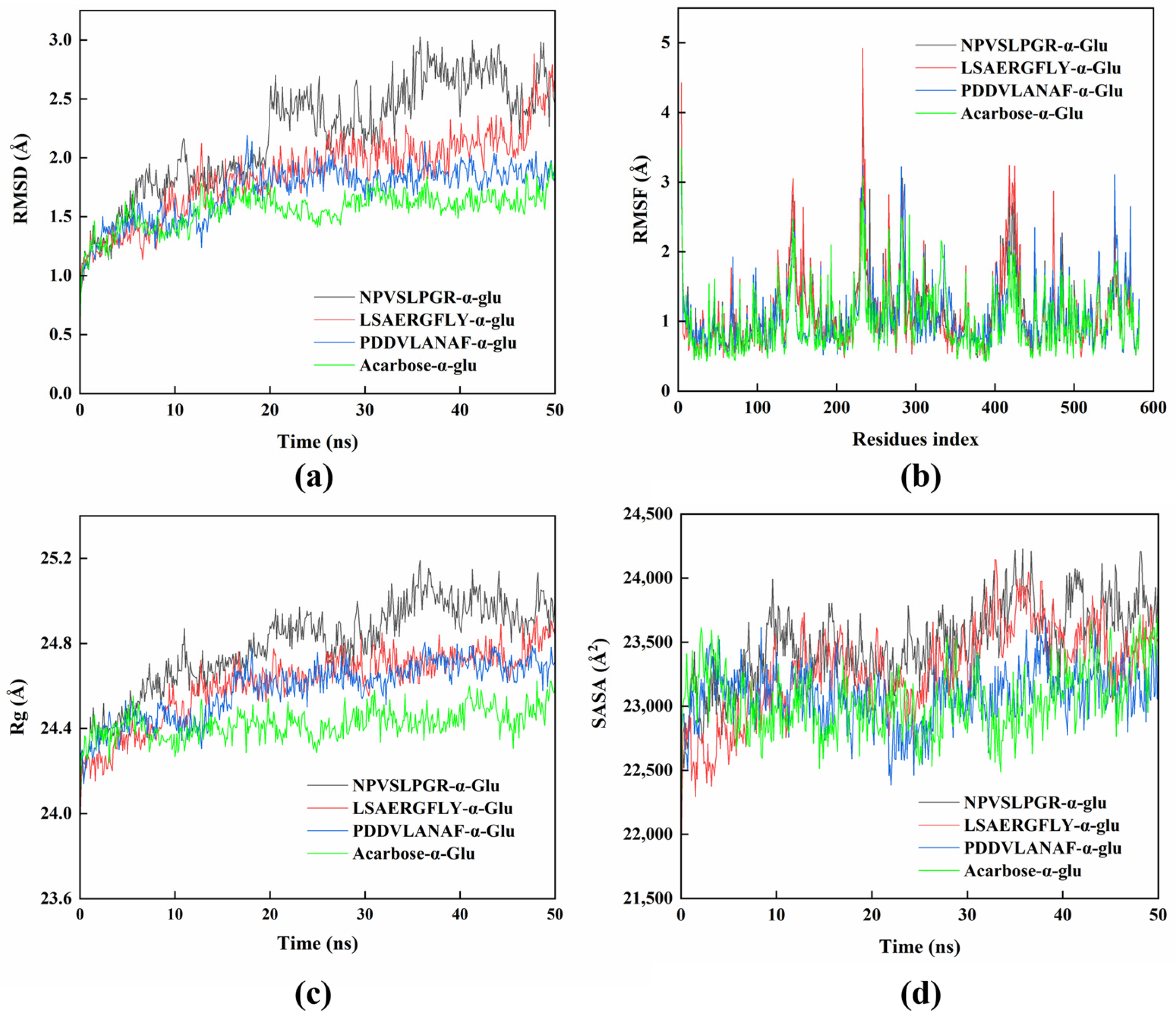
2.7. Molecular Dynamics
3. Materials and Methods
3.1. Materials and Reagents
3.2. Hydrolysis of HSP
3.3. Determination of Degree of Hydrolysis (DH)
3.4. Determination of α-Glucosidase Inhibitory Activity
3.5. Analysis of Peptide Profile
3.6. Evaluation of Potential Biological Activity of the Peptides
3.7. Analysis of Physicochemical Properties of the Peptides
3.8. Peptide Toxicity Analysis
3.9. ADMET Evaluation
3.10. Molecular Docking
3.11. Molecular Dynamics (MD) Simulation
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Xie, T.; Wu, Q.; Hu, Z.; Luo, Y.; Luo, F. α-Glucosidase Inhibitory Peptides: Sources, Preparations, Identifications, and Action Mechanisms. Nutrients 2023, 15, 4267. [Google Scholar] [CrossRef] [PubMed]
- Uuh Narvaez, J.J.; Segura Campos, M.R. Combination Therapy of Bioactive Compounds with Acarbose: A Proposal to Control Hyperglycemia in Type 2 Diabetes. J. Food Biochem. 2022, 46, e14268. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Standl, E.; Tong, N.; Shah, P.; Kalra, S.; Rathod, R. Therapeutic Potential of α-Glucosidase Inhibitors in Type 2 Diabetes Mellitus: An Evidence-Based Review. Expert Opin. Pharmacother. 2015, 16, 1959–1981. [Google Scholar] [CrossRef]
- Tsunoda, T.; Samadi, A.; Burade, S.; Mahmud, T. Complete Biosynthetic Pathway to the Antidiabetic Drug Acarbose. Nat. Commun. 2022, 13, 3455. [Google Scholar] [CrossRef]
- Dong, Y.; Sui, L.; Yang, F.; Ren, X.; Xing, Y.; Xiu, Z. Reducing the Intestinal Side Effects of Acarbose by Baicalein through the Regulation of Gut Microbiota: An in Vitro Study. Food Chem. 2022, 394, 133561. [Google Scholar] [CrossRef]
- Yang, X.; Wang, D.; Dai, Y.; Zhao, L.; Wang, W.; Ding, X. Identification and Molecular Binding Mechanism of Novel α-Glucosidase Inhibitory Peptides from Hot-Pressed Peanut Meal Protein Hydrolysates. Foods 2023, 12, 663. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, H.-N.; Zhang, M.; Mu, T.-H. Production and Characterization of α-Glucosidase Inhibitory Peptides from Sweet Potato Protein by Ultrasound-Assisted Enzymatic Hydrolysis and in Vitro Gastrointestinal Digestion. Eur. Food Res. Technol. 2024, 251, 257–267. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, Y.; Ma, C.; Bian, X.; Liu, X.; Wang, Y.; Chen, F.; Wang, B.; Zhang, G.; Zhang, N. Characterization of the Structure, Antioxidant Activity and Hypoglycemic Activity of Soy (Glycine max L.) Protein Hydrolysates. Food Res. Int. 2023, 173, 113473. [Google Scholar] [CrossRef]
- Wang, X.; Deng, Y.; Xie, P.; Liu, L.; Zhang, C.; Cheng, J.; Zhang, Y.; Liu, Y.; Huang, L.; Jiang, J. Novel Bioactive Peptides from Ginkgo Biloba Seed Protein and Evaluation of Their α-Glucosidase Inhibition Activity. Food Chem. 2023, 404, 134481. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Z.; Wang, H.; Zhang, F.; Guo, S.; Shen, Q. Comparison of the Generation of α-Glucosidase Inhibitory Peptides Derived from Prolamins of Raw and Cooked Foxtail Millet: In Vitro Activity, de Novo Sequencing, and in Silico Docking. Food Chem. 2023, 411, 135378. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Wu, Y.; Dai, Q.; Yan, X.; Liu, Y.; Sun, D.; Yu, Z.; Jiang, S.; Ma, Q.; Jiang, W. Extraction, Identification, and Molecular Mechanisms of α-Glucosidase Inhibitory Peptides from Defatted Antarctic Krill (Euphausia superba) Powder Hydrolysates. Int. J. Biol. Macromol. 2024, 266, 131126. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Qiu, L.; Song, H.; Xiao, T.; Luo, T.; Deng, Z.; Zheng, L. Inhibition Mechanism of Oligopeptides from Soft-Shelled Turtle Egg against α-Glucosidase and Their Gastrointestinal Digestive Properties. J. Food Biochem. 2022, 46, e14328. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-H.; Pan, R.-Y.; Ren, G.-Y.; Zhou, M.; Zhang, B.; Fan, J.-L.; Qiu, Z.-J. A Novel Antidiabetic Peptide GPAGAP from Andrias davidianus Collagen Hydrolysates: Screening, Action Mechanism Prediction and Improving Insulin Resistance in HepG2 Cells. Food Med. Homol. 2024, 1, 9420010. [Google Scholar] [CrossRef]
- Zhao, Q.; Wei, G.; Li, K.; Duan, S.; Ye, R.; Huang, A. Identification and Molecular Docking of Novel α-Glucosidase Inhibitory Peptides from Hydrolysates of Binglangjiang Buffalo Casein. LWT 2022, 156, 113062. [Google Scholar] [CrossRef]
- Callaway, J.C. Hempseed as a Nutritional Resource: An Overview. Euphytica 2004, 140, 65–72. [Google Scholar] [CrossRef]
- Rizzo, G.; Storz, M.A.; Calapai, G. The Role of Hemp (Cannabis sativa L.) as a Functional Food in Vegetarian Nutrition. Foods 2023, 12, 3505. [Google Scholar] [CrossRef]
- Huang, X.; Liao, J.; Shi, P.; Pei, X.; Wang, C. Virtual Screening and Directional Preparation of Xanthine Oxidase Inhibitory Peptides Derived from Hemp Seed Protein. Food Sci. Hum. Wellness 2024, 13, 3652–3660. [Google Scholar] [CrossRef]
- Zhang, Z.; Ge, J.; Wei, L.; Shi, J.; Ji, Y.; Xing, X.; Shi, Y.; Dong, Y. Isolation and Identification of Novel Hemp Seed Protein-Derived Pancreatic Lipase Inhibitory Peptides. Food Biosci. 2025, 63, 105834. [Google Scholar] [CrossRef]
- Montserrat-de la Paz, S.; Rivero-Pino, F.; Villanueva, A.; Toscano-Sanchez, R.; Martin, M.E.; Millan, F.; Millan-Linares, M.C. Nutritional Composition, Ultrastructural Characterization, and Peptidome Profile of Antioxidant Hemp Protein Hydrolysates. Food Biosci. 2023, 53, 102561. [Google Scholar] [CrossRef]
- Montserrat-de la Paz, S.; Villanueva-Lazo, A.; Millan, F.; Martin-Santiago, V.; Rivero-Pino, F.; Millan-Linares, M.C. Production and Identification of Immunomodulatory Peptides in Intestine Cells Obtained from Hemp Industrial By-Products. Food Res. Int. 2023, 174, 113616. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Li, W.; Wang, Y.; Xu, B.; Hu, X.; Li, X.-B.; Liu, J.-Y.; Zhang, C.; Zhang, C.-Y.; Xing, X.-H. Mining and Validation of Novel Hemp Seed-Derived DPP-IV-Inhibiting Peptides Using a Combination of Multi-Omics and Molecular Docking. J. Agric. Food Chem. 2023, 71, 9164–9174. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wu, S.; Jia, C.; Cui, C.; Sun-Waterhouse, D. Active Peptides with Hypoglycemic Effect Obtained from Hemp (Cannabis sativa L) Protein through Identification, Molecular Docking, and Virtual Screening. Food Chem. 2023, 429, 136912. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wu, S.; Jia, C.; Cui, C. Hydrolysates of Hemp (Cannabis sativa L.) Seed Meal: Characterization and Their Inhibitory Effect on α-Glucosidase Activity and Glucose Transport in Caco-2 Cells. Ind. Crops Prod. 2023, 205, 117559. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, F.; He, Z.; Fang, X.; Liu, X. Optimization and Molecular Mechanism of Novel α-Glucosidase Inhibitory Peptides Derived from Camellia Seed Cake through Enzymatic Hydrolysis. Foods 2023, 12, 393. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Bester, M.J.; Neitz, A.W.H.; Gaspar, A.R.M. Structural Properties of Bioactive Peptides with α-glucosidase Inhibitory Activity. Chem. Biol. Drug Des. 2018, 91, 370–379. [Google Scholar] [CrossRef]
- Di Stefano, E.; Oliviero, T.; Udenigwe, C.C. Functional Significance and Structure–Activity Relationship of Food-Derived α-Glucosidase Inhibitors. Curr. Opin. Food Sci. 2018, 20, 7–12. [Google Scholar] [CrossRef]
- Fan, Y.; Yu, Z.; Zhao, W.; Ding, L.; Zheng, F.; Li, J.; Liu, J. Identification and Molecular Mechanism of Angiotensin-Converting Enzyme Inhibitory Peptides from Larimichthys Crocea Titin. Food Sci. Hum. Wellness 2020, 9, 257–263. [Google Scholar] [CrossRef]
- Wang, Y.; Luan, J.; Tang, X.; Zhu, W.; Xu, Y.; Bu, Y.; Li, J.; Cui, F.; Li, X. Identification of Umami Peptides Based on Virtual Screening and Molecular Docking from Atlantic Cod (Gadus morhua). Food Funct. 2023, 14, 1510–1519. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, T.; Ning, Y.; Wang, D.; Li, F.; Fan, Y.; Yao, J.; Ren, G.; Zhang, B. Identification and Molecular Mechanism of Novel Tyrosinase Inhibitory Peptides from the Hydrolysate of “Fengdan” Peony (Paeonia ostii) Seed Meal Proteins: Peptidomics and in Silico Analysis. LWT 2023, 180, 114695. [Google Scholar] [CrossRef]
- Okella, H.; Okello, E.; Mtewa, A.G.; Ikiriza, H.; Kaggwa, B.; Aber, J.; Ndekezi, C.; Nkamwesiga, J.; Ajayi, C.O.; Mugeni, I.M.; et al. ADMET Profiling and Molecular Docking of Potential Antimicrobial Peptides Previously Isolated from African Catfish, Clarias gariepinus. Front. Mol. Biosci. 2022, 9, 1039286. [Google Scholar] [CrossRef] [PubMed]
- Nadavapalli, P.; Nadavapalli, P.; Bojja, K.S.; Gawli, K. Befunolol as a Potential Inhibitor of Glycogen Phosphorylase: An in Silico Approach. J. Appl. Biol. Biotechnol. 2024, 12, 127–132. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Y.; Liu, J.; Meng, Z.; Huang, A.; Xu, F.; Wang, X. Identification, Characterization and in Vitro Activity of Hypoglycemic Peptides in Whey Hydrolysates from Rubing Cheese by-Product. Food Res. Int. 2023, 164, 112382. [Google Scholar] [CrossRef]
- Yu, Z.; Fan, Y.; Zhao, W.; Ding, L.; Li, J.; Liu, J. Novel Angiotensin-Converting Enzyme Inhibitory Peptides Derived from Oncorhynchus Mykiss Nebulin: Virtual Screening and in Silico Molecular Docking Study. J. Food Sci. 2018, 83, 2375–2383. [Google Scholar] [CrossRef]
- Dang, K.; Lan, J.; Wang, Y.; Pan, D.; Du, L.; Suo, S.; Dang, Y.; Gao, X. Screening and Evaluation of Novel DPP-IV Inhibitory Peptides in Goat Milk Based on Molecular Docking and Molecular Dynamics Simulation. Food Chem. X 2025, 25, 102217. [Google Scholar] [CrossRef]
- Yu, Z.; Cao, Y.; Kan, R.; Ji, H.; Zhao, W.; Wu, S.; Liu, J.; Shiuan, D. Identification of Egg Protein-Derived Peptides as Xanthine Oxidase Inhibitors: Virtual Hydrolysis, Molecular Docking, and in Vitro Activity Evaluation. Food Sci. Hum. Wellness 2022, 11, 1591–1597. [Google Scholar] [CrossRef]
- Li, X.; You, Q. Sanguinarine Identified as a Natural Dual Inhibitor of AURKA and CDK2 through Network Pharmacology and Bioinformatics Approaches. Sci. Rep. 2024, 14, 29608. [Google Scholar] [CrossRef]
- Tang, H.; Huang, L.; Sun, C.; Zhao, D. Exploring the Structure-Activity Relationship and Interaction Mechanism of Flavonoids and α-Glucosidase Based on Experimental Analysis and Molecular Docking Studies. Food Funct. 2020, 11, 3332–3350. [Google Scholar] [CrossRef]
- Dai, T.; Li, T.; He, X.; Li, X.; Liu, C.; Chen, J.; McClements, D.J. Analysis of Inhibitory Interaction between Epigallocatechin Gallate and α-Glucosidase: A Spectroscopy and Molecular Simulation Study. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2020, 230, 118023. [Google Scholar] [CrossRef]
- Ur Rehman, N.; Rafiq, K.; Khan, A.; Ahsan Halim, S.; Ali, L.; Al-Saady, N.; Hilal Al-Balushi, A.; Al-Busaidi, H.K.; Al-Harrasi, A. α-Glucosidase Inhibition and Molecular Docking Studies of Natural Brominated Metabolites from Marine Macro Brown Alga Dictyopteris Hoytii. Mar. Drugs 2019, 17, 666. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Satish, A.M.; Shbeer, A.M.; Ageel, M.; Al-Ghorbani, M.; Ranganatha, L.; Parameswaran, S.; Ramu, R. Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches. Molecules 2022, 27, 3888. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Akhter, S.; Fatema, K.; Hossain, M.R.; Sultana, T.; Uzzaman, M. Selective Modification of Diclofenac to Reduce the Adverse Effects; A Computer-Aided Drug Design Approach. Inform. Med. Unlocked 2023, 36, 101159. [Google Scholar] [CrossRef]
- Wang, T.; Bo, N.; Sha, G.; Guan, Y.; Yang, D.; Shan, X.; Lv, Z.; Chen, Q.; Yang, G.; Gong, S.; et al. Identification and Molecular Mechanism of Novel Hypoglycemic Peptide in Ripened Pu-Erh Tea: Molecular Docking, Dynamic Simulation, and Cell Experiments. Food Res. Int. Ott. Ont 2024, 194, 114930. [Google Scholar] [CrossRef] [PubMed]
- Al-Masri, A.A.; Ameen, F.; Davella, R.; Mamidala, E. Antidiabetic Effect of Flavonoid from Rumex Vesicarius on Alloxan Induced Diabetes in Male Albino Wistar Rats and Its Validation through in Silico Molecular Docking and Dynamic Simulation Studies. Biotechnol. Genet. Eng. Rev. 2024, 40, 4479–4494. [Google Scholar] [CrossRef]
- Selvaraju, K.; Raguraman, V.; Yadav, H.N.; Hariprasad, P.; Malik, A. Spectral Characterization and Binding Dynamics of Bioactive Compounds from Chlorella Minutissima against α-Glucosidase: An in Vitro and in Silico Approach. Algal Res. 2023, 75, 103281. [Google Scholar] [CrossRef]
- Wang, X.; Deng, Y.; Zhang, Y.; Zhang, C.; Liu, L.; Liu, Y.; Jiang, J.; Xie, P.; Huang, L. Screening and Evaluation of Novel α-Glucosidase Inhibitory Peptides from Ginkgo Biloba Seed Cake Based on Molecular Docking Combined with Molecular Dynamics Simulation. J. Agric. Food Chem. 2023, 71, 10326–10337. [Google Scholar] [CrossRef]
- Pan, J.; Nawaz, M.; Liu, J.; Liu, H.; Lv, Z.; Yang, W.; Jiao, Z.; Zhang, Q. Exploring Synergistic Inhibitory Mechanisms of Flavonoid Mixtures on α-Glucosidase by Experimental Analysis and Molecular Dynamics Simulation. Food Chem. 2025, 464, 141560. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, Q.; Liu, H.-J.; Li, S.-Z.; Jiang, Z.-Q. Characterization of Actinidin from Chinese Kiwifruit Cultivars and Its Applications in Meat Tenderization and Production of Angiotensin I-Converting Enzyme (ACE) Inhibitory Peptides. LWT 2017, 78, 1–7. [Google Scholar] [CrossRef]
- Liu, L.; Chen, J.; Li, X. Novel Peptides with α-Glucosidase Inhibitory Activity from Changii radix Hydrolysates. Process Biochem. 2021, 111, 200–206. [Google Scholar] [CrossRef]
- Mooney, C.; Haslam, N.J.; Pollastri, G.; Shields, D.C. Towards the Improved Discovery and Design of Functional Peptides: Common Features of Diverse Classes Permit Generalized Prediction of Bioactivity. PLoS ONE 2012, 7, e45012. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence | PeptideRanker Score | No. | Sequence | PeptideRanker Score |
|---|---|---|---|---|---|
| 1 | NAPMMFY | 0.944 | 25 | NPVSLPGR | 0.613 |
| 2 | NLPILRF | 0.853 | 26 | DIIAIPAGM | 0.608 |
| 3 | RGLLLPSFLNAPM | 0.806 | 27 | AYEPVWAIGTGK | 0.605 |
| 4 | LHFPPHR | 0.806 | 28 | KQASSDGFEWVSF | 0.603 |
| 5 | GFEWVSF | 0.803 | 29 | EGDIIAIPAGM | 0.587 |
| 6 | PSQADIFNPR | 0.779 | 30 | KASAQGFEW | 0.583 |
| 7 | RGLLLPSFL | 0.772 | 31 | FHLAGNPHR | 0.579 |
| 8 | PPSGGRFTQIL | 0.767 | 32 | AMPDDVLANAF | 0.572 |
| 9 | AGLQFPVGR | 0.763 | 33 | PSRADVF | 0.570 |
| 10 | EGDIIAIPAGMAYW | 0.755 | 34 | PDDVLANAF | 0.558 |
| 11 | NLPILSFLR | 0.748 | 35 | LINPVSLPGRFEPF | 0.556 |
| 12 | SEYPPLGR | 0.738 | 36 | EGDIIAIPAGMAY | 0.547 |
| 13 | NLPILSFL | 0.732 | 37 | SAQGFEWIAVK | 0.546 |
| 14 | SYNLPILR | 0.694 | 38 | GNPEDEFEQLRR | 0.532 |
| 15 | ADVFSPQAGRL | 0.693 | 39 | GFEWIAVK | 0.525 |
| 16 | HAQGSGGTIWPF | 0.686 | 40 | ASAQGFEWIAVK | 0.522 |
| 17 | IAGNPHQEFPQSMM | 0.679 | 41 | ADVFSPQAGR | 0.522 |
| 18 | INPVSLPGRFEPF | 0.669 | 42 | HAQGSGGTIWPFGPETR | 0.518 |
| 19 | QASSDGFEWVSF | 0.668 | 43 | LINPVSLPGRFEPFY | 0.513 |
| 20 | PAGVAYW | 0.660 | 44 | LSAERGFLY | 0.509 |
| 21 | YNLPILR | 0.658 | 45 | LINPVSLPGR | 0.506 |
| 22 | LPIGILSLKKKLKY | 0.644 | 46 | MELVDAAFPLLK | 0.506 |
| 23 | NNYNLPILR | 0.622 | 47 | RIGFLEANPNAF | 0.502 |
| 24 | NLPILSF | 0.613 |
| No. | Sequence | pI | Net Charge at pH 7.0 | Water Solubility | No. | Sequence | pI | Net Charge at pH 7.0 | Water Solubility |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NAPMMFY | 3.24 | 0.0 | Poor | 25 | NPVSLPGR | 10.42 | 1.0 | Good |
| 2 | NLPILRF | 10.42 | 1.0 | Poor | 26 | DIIAIPAGM | 0.78 | −1.0 | Poor |
| 3 | RGLLLPSFLNAPM | 10.55 | 1.0 | Poor | 27 | AYEPVWAIGTGK | 6.84 | 0.0 | Poor |
| 4 | LHFPPHR | 10.84 | 1.2 | Poor | 28 | KQASSDGFEWVSF | 3.93 | −1.0 | Good |
| 5 | GFEWVSF | 0.99 | −1.0 | Poor | 29 | EGDIIAIPAGM | 0.71 | −2.0 | Poor |
| 6 | PSQADIFNPR | 7.08 | 0.0 | Good | 30 | KASAQGFEW | 6.65 | 0.0 | Good |
| 7 | RGLLLPSFL | 10.55 | 1.0 | Poor | 31 | FHLAGNPHR | 10.59 | 1.2 | Poor |
| 8 | PPSGGRFTQIL | 11.29 | 1.0 | Poor | 32 | AMPDDVLANAF | 0.61 | −2.0 | Poor |
| 9 | AGLQFPVGR | 10.90 | 1.0 | Poor | 33 | PSRADVF | 7.08 | 0.0 | Good |
| 10 | EGDIIAIPAGMAYW | 0.60 | −2.0 | Poor | 34 | PDDVLANAF | 0.61 | −2.0 | Good |
| 11 | NLPILSFLR | 10.42 | 1.0 | Poor | 35 | LINPVSLPGRFEPF | 6.86 | 0.0 | Poor |
| 12 | SEYPPLGR | 6.58 | 0.0 | Good | 36 | EGDIIAIPAGMAY | 0.67 | −2.0 | Poor |
| 13 | NLPILSFL | 3.21 | 0.0 | Poor | 37 | SAQGFEWIAVK | 6.59 | 0.0 | Poor |
| 14 | SYNLPILR | 9.57 | 1.0 | Poor | 38 | GNPEDEFEQLRR | 4.04 | −2.0 | Good |
| 15 | ADVFSPQAGRL | 6.71 | 0.0 | Good | 39 | GFEWIAVK | 6.85 | 0.0 | Poor |
| 16 | HAQGSGGTIWPF | 7.56 | 0.1 | Poor | 40 | ASAQGFEWIAVK | 6.91 | 0.0 | Poor |
| 17 | IAGNPHQEFPQSMM | 5.10 | −0.9 | Poor | 41 | ADVFSPQAGR | 6.71 | 0.0 | Good |
| 18 | INPVSLPGRFEPF | 6.87 | 0.0 | Poor | 42 | HAQGSGGTIWPFGPETR | 7.57 | 0.1 | Poor |
| 19 | QASSDGFEWVSF | 0.70 | −2.0 | Poor | 43 | LINPVSLPGRFEPFY | 6.81 | 0.0 | Poor |
| 20 | PAGVAYW | 3.78 | 0.0 | Poor | 44 | LSAERGFLY | 6.81 | 0.0 | Good |
| 21 | YNLPILR | 9.57 | 1.0 | Poor | 45 | LINPVSLPGR | 10.84 | 1.0 | Poor |
| 22 | LPIGILSLKKKLKY | 10.77 | 4.0 | Good | 46 | MELVDAAFPLLK | 3.93 | −1.0 | Good |
| 23 | NNYNLPILR | 9.41 | 1.0 | Poor | 47 | RIGFLEANPNAF | 6.58 | 0.0 | Poor |
| 24 | NLPILSF | 3.28 | 0.0 | Poor |
| No. | Sequence | Toxicity (SVM Score) | BBB 1 | HIA 2 | CYP450 2C9 Substrate 3 | CYP450 2C9 Inhibitor 4 |
|---|---|---|---|---|---|---|
| 1 | PSQADIFNPR | non-toxicity (1.190) | -(0.982) | +(0.743) | -(0.825) | -(0.878) |
| 2 | SEYPPLGR | non-toxicity (0.210) | -(0.983) | +(0.637) | -(0.813) | -(0.921) |
| 3 | ADVFSPQAGRL | non-toxicity (0.800) | -(0.986) | -(0.539) | -(0.801) | -(0.867) |
| 4 | LPIGILSLKKKLKY | non-toxicity (1.320) | -(0.993) | +(0.829) | -(0.866) | -(0.938) |
| 5 | NPVSLPGR | non-toxicity (0.890) | -(0.979) | -(0.622) | -(0.832) | -(0.892) |
| 6 | KQASSDGFEWVSF | non-toxicity (0.810) | -(0.936) | +(0.802) | -(0.838) | -(0.884) |
| 7 | KASAQGFEW | non-toxicity (1.030) | -(0.860) | +(0.765) | -(0.850) | -(0.908) |
| 8 | PSRADVF | non-toxicity (0.520) | -(0.920) | -(0.577) | -(0.787) | -(0.911) |
| 9 | PDDVLANAF | non-toxicity (0.940) | -(0.901) | +(0.771) | -(0.822) | -(0.928) |
| 10 | GNPEDEFEQLRR | non-toxicity (1.160) | -(0.992) | +(0.673) | -(0.770) | -(0.876) |
| 11 | ADVFSPQAGR | non-toxicity (0.710) | -(0.986) | -(0.663) | -(0.796) | -(0.892) |
| 12 | LSAERGFLY | non-toxicity (0.870) | -(0.953) | +(0.529) | -(0.738) | -(0.839) |
| 13 | MELVDAAFPLLK | non-toxicity (1.620) | -(0.990) | +(0.759) | -(0.827) | -(0.910) |
| Sequence | Binding Energy (kcal/mol) | Amino Acid Residues Involved in Hydrogen Bond (Number of Hydrogen Bonds) | Amino Acid Residues Involved in Electrostatic Interactions (Number of Electrostatic Interactions) | Amino Acid Residues Involved in Hydrophobic Interactions (Number of Hydrophobic Interactions) |
|---|---|---|---|---|
| NPVSLPGR | −8.7 | Asn241, His245, Glu276, Glu304, Pro309, Tyr313, Asp349, Asp408 (8) | Asp214, Glu276, Asp349 (3) | His245, Ala278, Leu218, His279, Phe157, Arg312, His239, Phe231 (13) |
| LSAERGFLY | −8.5 | Phe231, Asn241, Trp242, Glu276, His279, Glu304, Thr307, Pro309, Tyr313 (11) | Asp349, Arg439, Phe310 (3) | Ala278, His279, Phe300, Pro309, Phe310, His239, Phe157 (11) |
| PDDVLANAF | −8.4 | Phe157, His239, Asn241, His279, Pro309, Arg312, Asn412 (9) | Not involved (0) | Pro309 (3) |
| Acarbose | −8.1 | Ile217, Lys262, Asn263, His258, Ala289, Tyr292, Glu293, Ser295 (9) | Not involved (0) | Lys262 (1) |
| KQASSDGFEWVSF | −8.0 | Phe157, Asn241, Trp242, His245, His279, Glu276, Glu304, Thr307, Pro309, Phe311, Asp349 (18) | Asp349, Asp408, Glu276, Asp214 (5) | Phe231 (1) |
| GNPEDEFEQLRR | −8.0 | Asp214, Asn241, His245, Asn246, Glu276, His279, Gly280, Thr307, Pro309, Tyr313, Ser329, Asp349, Asn412, Arg439 (16) | Asp214, Asp349, Glu276, Asp 408 (4) | His245, His239, Phe157 (5) |
| ADVFSPQAGR | −7.7 | Asn241, His245, His279, Glu304, Thr307, Asp349 (8) | Glu276, Asp349 (2) | Phe231, Trp242 Pro309 (3) |
| ADVFSPQAGRL | −7.6 | His239, Asn241, His245, Glu304, Pro309, Asp408 (7) | Asp408 (1) | His279, Trp242, His245, Phe231, Phe177, Phe157, Phe300 (7) |
| PSRADVF | −7.6 | His239, Asn241, His245, His279 (5) | Not involved (0) | His239, Arg312 (3) |
| KASAQGFEW | −7.1 | His239, Asn241, Trp242, Thr301, Thr307, Ser308, Pro309, Pro317, Gln322 (10) | Not involved (0) | Phe231, Phe310, Trp242 (3) |
| SEYPPLGR | −7.0 | Ser161, Arg175, Ser179, Asn411, Lys418 (11) | Glu414 (1) | Pro148, Pro150, Lys147, Phe172, Lys418, Thr164 (7) |
| MELVDAAFPLLK | −6.9 | Asn241, Asn246, Ile250, Gly280, Glu304, Thr307, Pro309 (9) | Not involved (0) | Phe310, His239, His279, Arg312, Phe231, Ala284, Tyr286, His251 (11) |
| PSQADIFNPR | −6.0 | Asn241, His279, Gln322, Thr301 (5) | Glu325 (1) | Trp242, His 239, Phe231, Phe310 (4) |
| LPIGILSLKKKLKY | −6.0 | Trp242, His245, Asn246, His279, Gly280, Ser281, Glu304, Thr307, Ser308, Gln322, Ala326, Ser329 (14) | Glu304, Glu325 (4) | Gln322, Phe310, Pro309, His279, Trp242, His239 (10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mengyuan, Z.; Chen, C.; Feng, W.; Ning, Z.; Wanyu, Y.; Tianrong, Z.; Guoyan, R.; Zhijun, Q.; Bin, Z. Identification and Molecular Mechanism of Novel α-Glucosidase Inhibitory Peptides from the Hydrolysate of Hemp Seed Proteins: Peptidomic Analysis, Molecular Docking, and Dynamics Simulation. Int. J. Mol. Sci. 2025, 26, 2222. https://doi.org/10.3390/ijms26052222
Mengyuan Z, Chen C, Feng W, Ning Z, Wanyu Y, Tianrong Z, Guoyan R, Zhijun Q, Bin Z. Identification and Molecular Mechanism of Novel α-Glucosidase Inhibitory Peptides from the Hydrolysate of Hemp Seed Proteins: Peptidomic Analysis, Molecular Docking, and Dynamics Simulation. International Journal of Molecular Sciences. 2025; 26(5):2222. https://doi.org/10.3390/ijms26052222
Chicago/Turabian StyleMengyuan, Zhang, Chen Chen, Wei Feng, Zhao Ning, Yang Wanyu, Zhang Tianrong, Ren Guoyan, Qiu Zhijun, and Zhang Bin. 2025. "Identification and Molecular Mechanism of Novel α-Glucosidase Inhibitory Peptides from the Hydrolysate of Hemp Seed Proteins: Peptidomic Analysis, Molecular Docking, and Dynamics Simulation" International Journal of Molecular Sciences 26, no. 5: 2222. https://doi.org/10.3390/ijms26052222
APA StyleMengyuan, Z., Chen, C., Feng, W., Ning, Z., Wanyu, Y., Tianrong, Z., Guoyan, R., Zhijun, Q., & Bin, Z. (2025). Identification and Molecular Mechanism of Novel α-Glucosidase Inhibitory Peptides from the Hydrolysate of Hemp Seed Proteins: Peptidomic Analysis, Molecular Docking, and Dynamics Simulation. International Journal of Molecular Sciences, 26(5), 2222. https://doi.org/10.3390/ijms26052222