

InfoScan: A New Transcript Identification Tool Based on scRNA-Seq and Its Application in Glioblastoma

 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
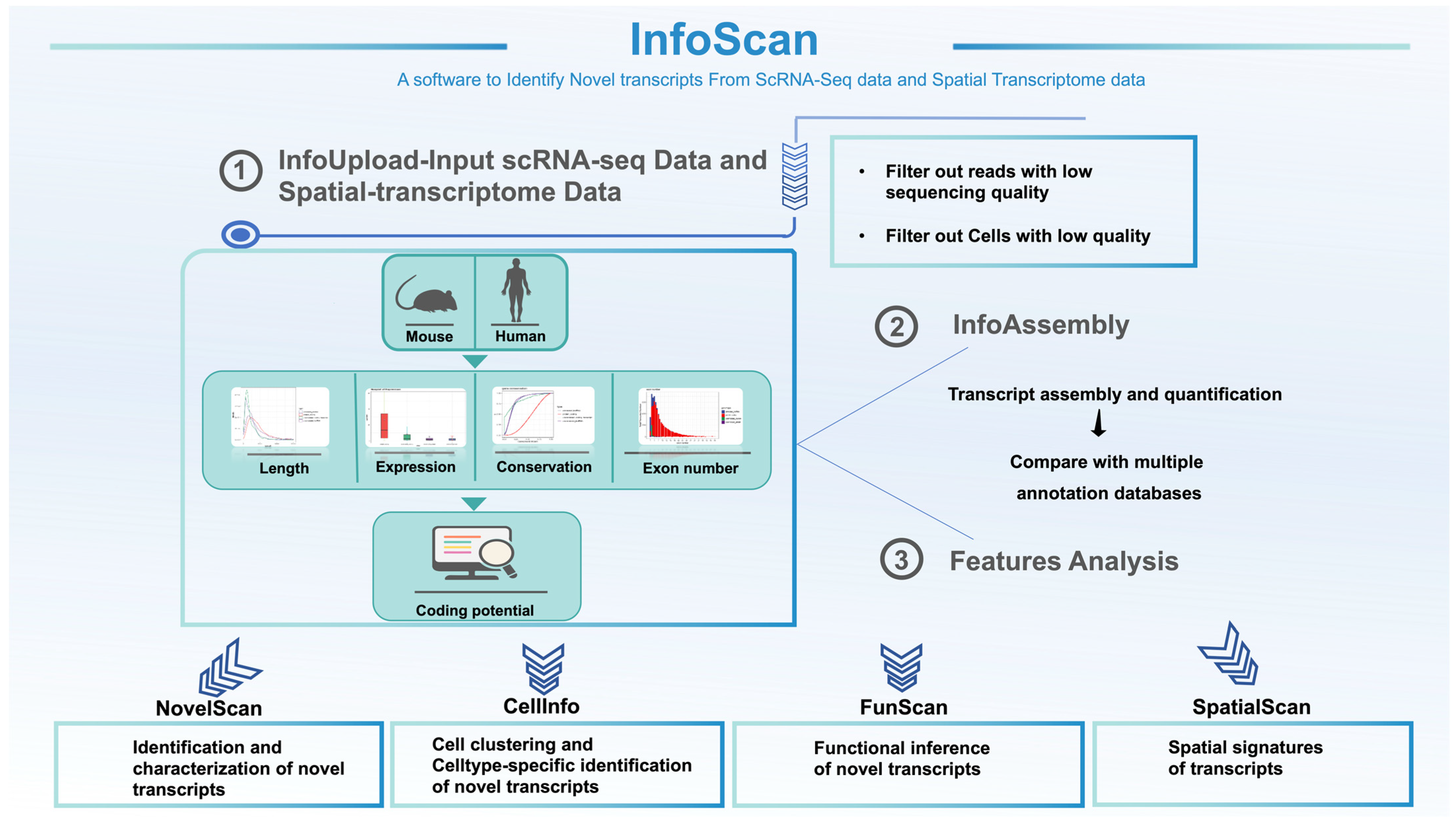
2.1. Establishment of the InfoScan Tool
2.2. InfoScan Reveals the Abundance of Unannotated lncRNAs in GBM
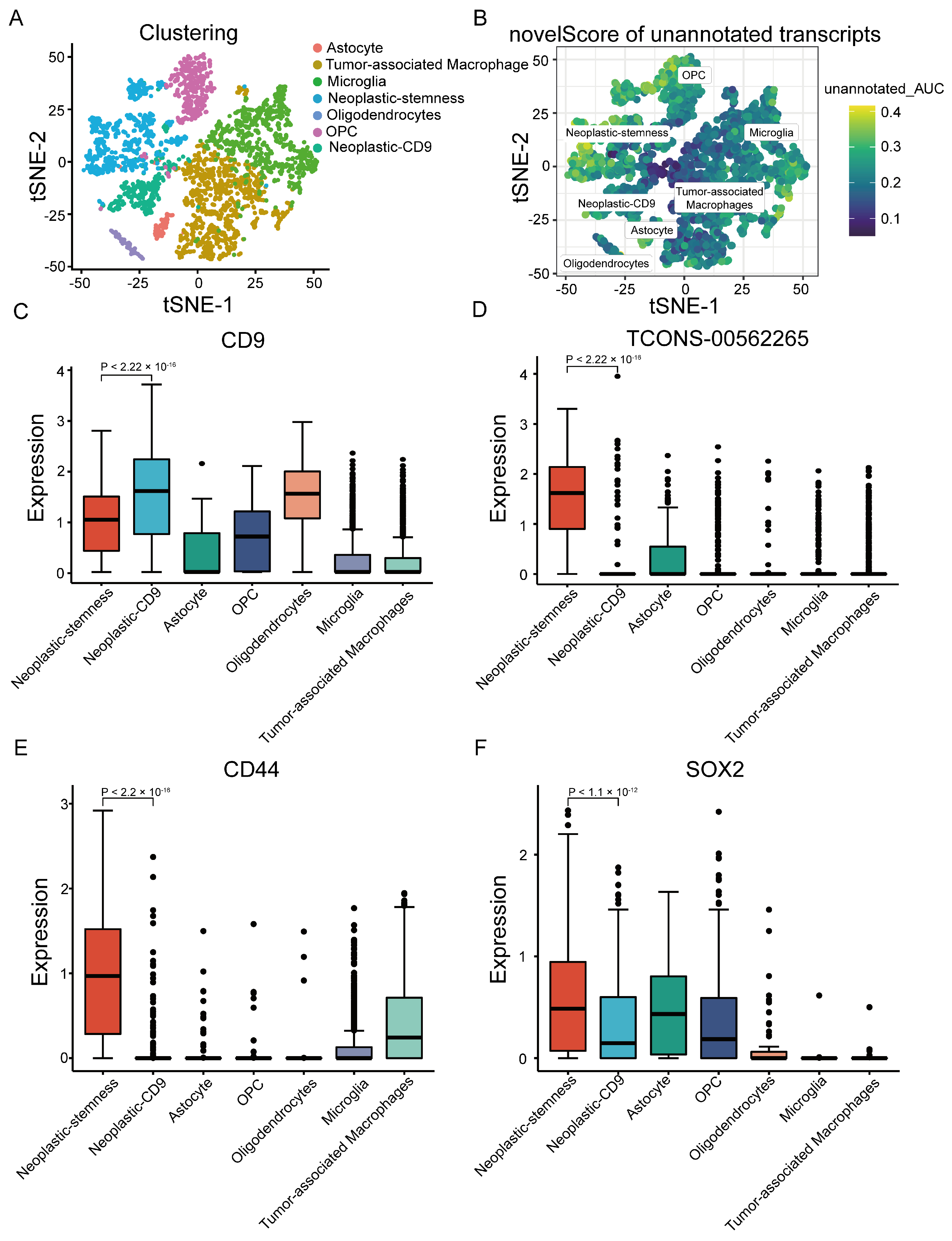
2.3. Unannotated lncRNAs Mark a Neoplastic-Stemness Subpopulation
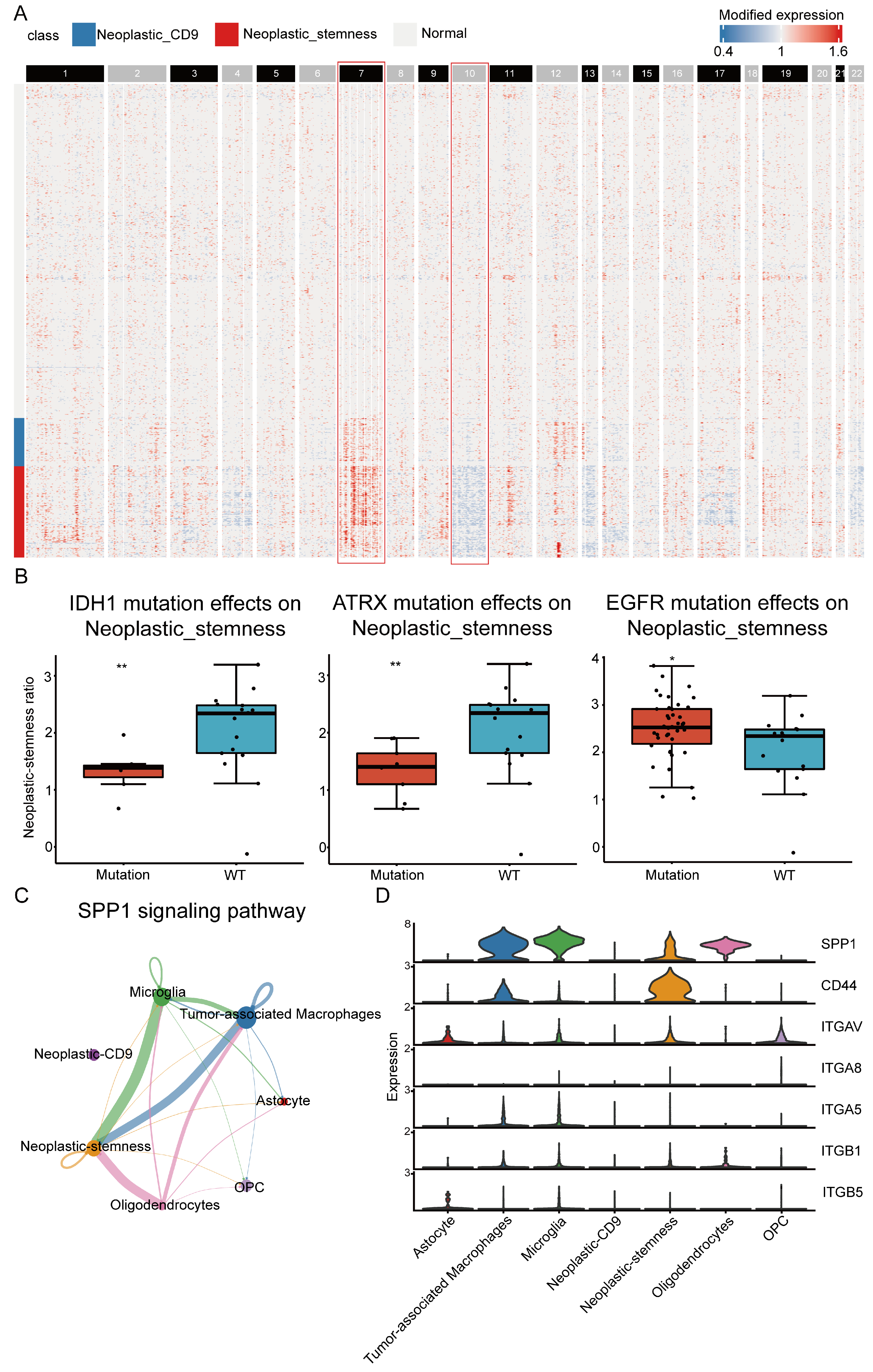
2.4. Validation of Neoplastic-Stemness Stem Cell Characteristics Through Integrated InfoScan, Enrichment Analysis, and Spatial Transcriptomic Profiling
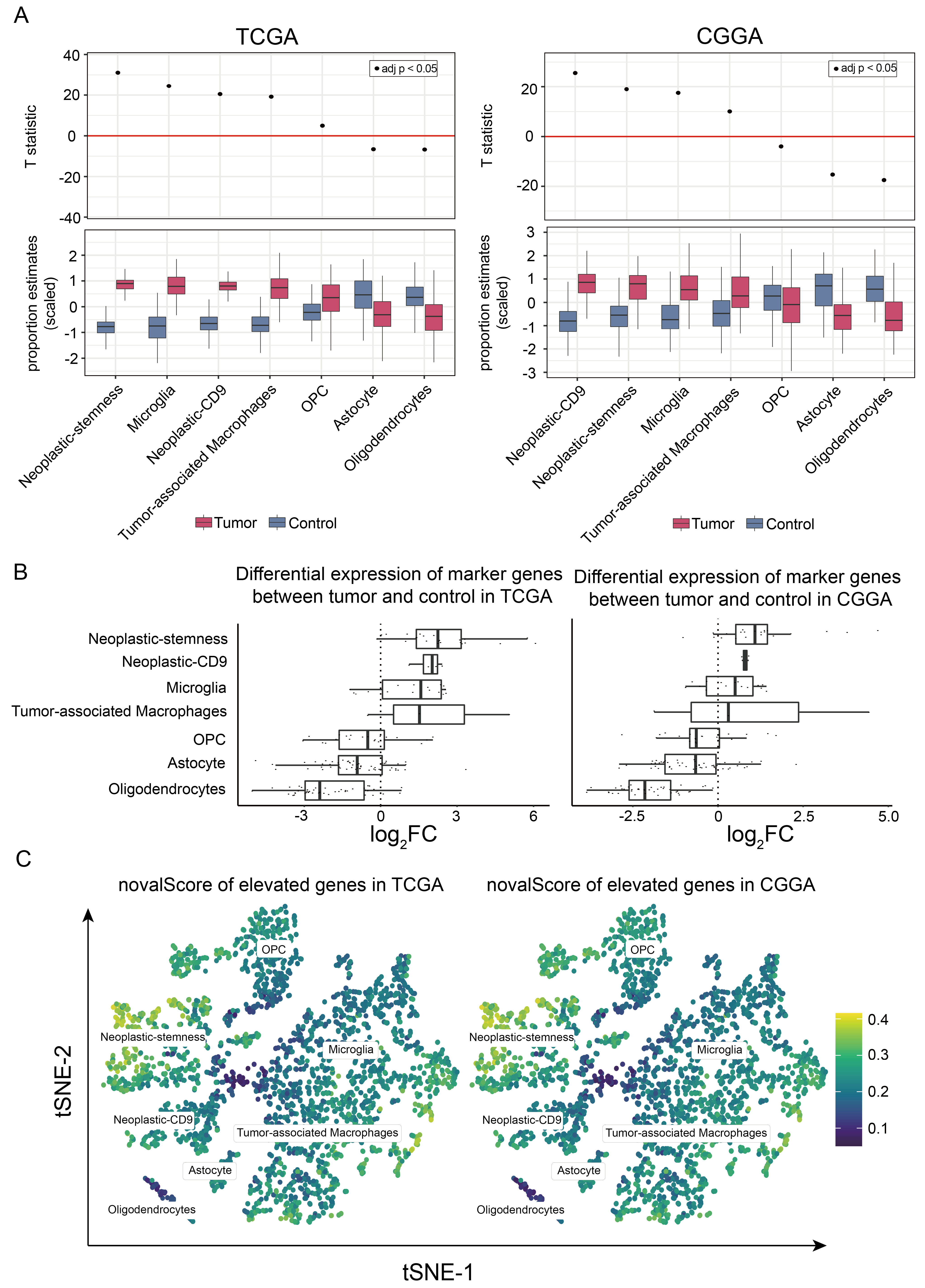
2.5. Neoplastic-Stemness Significantly Increased in Large Cohorts of GBM Patients
2.6. Increased Proportion of Neoplastic-Stemness Correlates with Reduced Survival in GBM Patients
2.7. Tumor Microenvironment Induce Neoplastic-Stemness via the SPP1–CD44 Axis
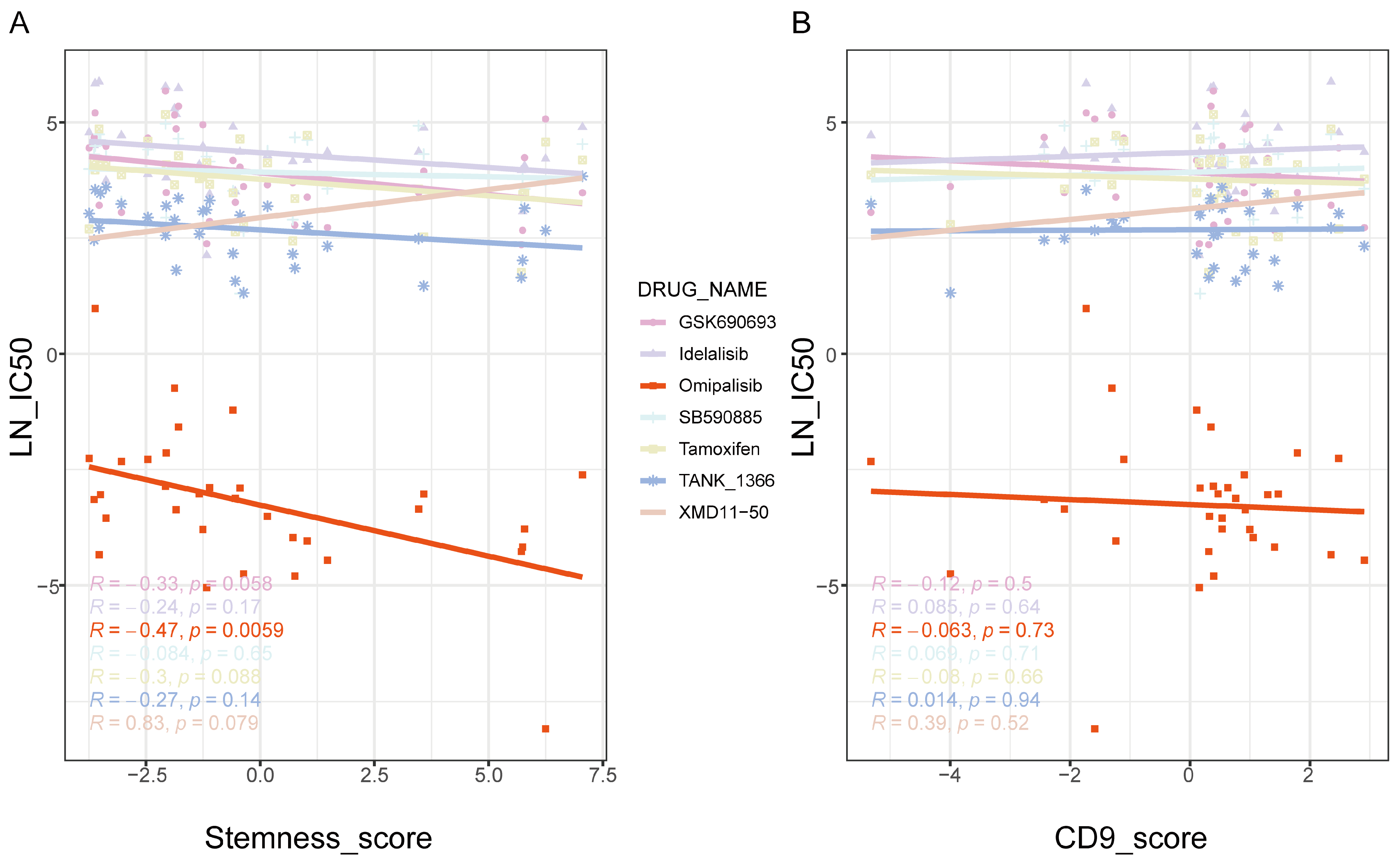
2.8. The Therapeutic Effects of Omipalisib on Neoplastic-Stemness Cells
2.9. Application of InfoScan in Breast Cancer Data Reveals Its Generalizability
3. Discussion
4. Materials and Methods
4.1. Data Source
4.2. Genome Mapping and Transcript Assembly
4.3. Cell Annotation and Spatial Transcriptome Analysis
4.4. Gene Set Scoring
4.5. Evolutionary Conservation Analysis
4.6. Tissue- or Cell-Specific Analysis
4.7. Gene Coexpression and Enrichment Analysis
4.8. Cell Communication Analysis
4.9. Estimating Cell Type Proportions from Bulk GBM Data
4.10. Survival Analysis
4.11. Somatic Mutation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma multiforme |
| TME | Tumor microenvironment |
| lncRNA | Long non-coding RNA |
| scRNA-seq | Single-cell RNA sequencing |
| EMT | Epithelial–mesenchymal transition |
| CAF | Cancer-associated fibroblasts |
References
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How Do lncRNAs Regulate Transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA Exploits Three-Dimensional Genome Architecture to Spread across the X Chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef]
- Guiducci, G.; Stojic, L. Long Noncoding RNAs at the Crossroads of Cell Cycle and Genome Integrity. Trends Genet. 2021, 37, 528–546. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Neha, C.V.; Bhaduri, U.; Zenia, Z.; Dutta, S.; Chidambaram, S.; Rao, M.R.S. LncRNA Mrhl Orchestrates Differentiation Programs in Mouse Embryonic Stem Cells through Chromatin Mediated Regulation. Stem Cell Res. 2021, 53, 102250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, Q.; Zhu, K.; Zhu, J.; Li, J.; Yuan, Y.; Zhang, P.; Zhou, L.; Liu, L. LncRNA LINC00265/miR-485-5p/IRF2-Mediated Autophagy Suppresses Apoptosis in Acute Myeloid Leukemia Cells. Am. J. Transl. Res. 2020, 12, 2451–2462. [Google Scholar]
- Chi, Y.; Wang, D.; Wang, J.; Yu, W.; Yang, J. Long Non-Coding RNA in the Pathogenesis of Cancers. Cells 2019, 8, 1015. [Google Scholar] [CrossRef]
- Enguita, F.J.; Leitão, A.L.; Mattick, J.S. RNA Regulatory Networks 2.0. Int. J. Mol. Sci. 2023, 24, 9001. [Google Scholar] [CrossRef]
- Park, E.-G.; Pyo, S.-J.; Cui, Y.; Yoon, S.-H.; Nam, J.-W. Tumor Immune Microenvironment lncRNAs. Brief. Bioinform. 2022, 23, bbab504. [Google Scholar] [CrossRef]
- Luo, H.; Bu, D.; Shao, L.; Li, Y.; Sun, L.; Wang, C.; Wang, J.; Yang, W.; Yang, X.; Dong, J.; et al. Single-Cell Long Non-Coding RNA Landscape of T Cells in Human Cancer Immunity. Genom. Proteom. Bioinform. 2021, 19, 377–393. [Google Scholar] [CrossRef]
- Verma, A.; Jiang, Y.; Du, W.; Fairchild, L.; Melnick, A.; Elemento, O. Transcriptome Sequencing Reveals Thousands of Novel Long Non-Coding RNAs in B Cell Lymphoma. Genome Med. 2015, 7, 110. [Google Scholar] [CrossRef]
- Fang, Y.; Xu, Y.; Wang, R.; Hu, L.; Guo, D.; Xue, F.; Guo, W.; Zhang, D.; Hu, J.; Li, Y.; et al. Recent Advances on the Roles of LncRNAs in Cardiovascular Disease. J. Cell. Mol. Med. 2020, 24, 12246–12257. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ding, N.; Li, J.; Jin, X.; Li, L.; Pan, T.; Huo, C.; Li, Y.; Xu, J.; Li, X. Landscape of the Long Non-Coding RNA Transcriptome in Human Heart. Brief. Bioinform. 2019, 20, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, N. The Emerging Role of Autophagy-Associated lncRNAs in the Pathogenesis of Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 9686. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary Dynamics and Tissue Specificity of Human Long Noncoding RNAs in Six Mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-Cell Analysis of Long Non-Coding RNAs in the Developing Human Neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef]
- Gupta, I.; Collier, P.G.; Haase, B.; Mahfouz, A.; Joglekar, A.; Floyd, T.; Koopmans, F.; Barres, B.; Smit, A.B.; Sloan, S.A.; et al. Single-Cell Isoform RNA Sequencing Characterizes Isoforms in Thousands of Cerebellar Cells. Nat. Biotechnol. 2018, 36, 1197–1202. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, Y.; Zhou, Y.; Shi, K.; Hu, X.; Li, D.; Ye, H.; Zhou, Y.; Wang, K. Full-Length Annotation with Multistrategy RNA-Seq Uncovers Transcriptional Regulation of lncRNAs in Cotton. Plant Physiol. 2021, 185, 179–195. [Google Scholar] [CrossRef]
- Goñi, E.; Mas, A.M.; Gonzalez, J.; Abad, A.; Santisteban, M.; Fortes, P.; Huarte, M.; Hernaez, M. Uncovering Functional lncRNAs by scRNA-Seq with ELATUS. Nat. Commun. 2024, 15, 9709. [Google Scholar] [CrossRef]
- Bambury, R.M.; Morris, P.G. Novel Investigational Approaches for Inhibiting Angiogenesis in Recurrent Glioblastoma. Anticancer Drugs 2014, 25, 611–613. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; de Blank, P.M.; Finlay, J.L.; Gurney, J.G.; McKean-Cowdin, R.; Stearns, D.S.; Wolff, J.E.; Liu, M.; Wolinsky, Y.; et al. American Brain Tumor Association Adolescent and Young Adult Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2016, 18, i1–i50. [Google Scholar] [CrossRef]
- Nallasamy, P.; Nimmakayala, R.K.; Karmakar, S.; Leon, F.; Seshacharyulu, P.; Lakshmanan, I.; Rachagani, S.; Mallya, K.; Zhang, C.; Ly, Q.P.; et al. Pancreatic Tumor Microenvironment Factor Promotes Cancer Stemness via SPP1-CD44 Axis. Gastroenterology 2021, 161, 1998–2013.e7. [Google Scholar] [CrossRef]
- Cheng, C.; Dong, Y.; Ru, X.; Xia, Y.; Ji, Y. LncRNA ANCR Promotes Glioma Cells Invasion, Migration, Proliferation and Inhibits Apoptosis via Interacting with EZH2 and Repressing PTEN Expression. Cancer Gene Ther. 2021, 28, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, J.; Shirasaki, T.; Shimakami, T.; Nishiyama, T.; Welsch, C.; Funaki, M.; Murai, K.; Sumiyadorj, A.; Takatori, H.; Kitamura, K.; et al. Upregulation of the Long Noncoding RNA HULC by Hepatitis C Virus and Its Regulation of Viral Replication. J. Infect. Dis. 2022, 226, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sun, X.; Zhou, X.; Han, L.; Chen, L.; Shi, Z.; Zhang, A.; Ye, M.; Wang, Q.; Liu, C.; et al. Long Non-Coding RNA HOTAIR Promotes Glioblastoma Cell Cycle Progression in an EZH2 Dependent Manner. Oncotarget 2015, 6, 537–546. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, L.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Zhou, Y.; Du, Z. Downregulation of lncRNA-MALAT1 Affects Proliferation and the Expression of Stemness Markers in Glioma Stem Cell Line SHG139S. Cell. Mol. Neurobiol. 2016, 36, 1097–1107. [Google Scholar] [CrossRef]
- Li, K.; Yao, T.; Zhang, Y.; Li, W.; Wang, Z. NEAT1 as a Competing Endogenous RNA in Tumorigenesis of Various Cancers: Role, Mechanism and Therapeutic Potential. Int. J. Biol. Sci. 2021, 17, 3428–3440. [Google Scholar] [CrossRef]
- Li, J.; An, G.; Zhang, M.; Ma, Q. Long Non-Coding RNA TUG1 Acts as a miR-26a Sponge in Human Glioma Cells. Biochem. Biophys. Res. Commun. 2016, 477, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Zuo, C.; Zhang, K.; Lei, X.; Wang, J.; Yang, Y.; Zhang, J.; Ma, K.; Wang, S.; et al. Long Non-Coding RNA H19 Regulates Glioma Cell Growth and Metastasis via miR-200a-Mediated CDK6 and ZEB1 Expression. Front. Oncol. 2021, 11, 757650. [Google Scholar] [CrossRef]
- Michel, A.M.; Fox, G.; M Kiran, A.; De Bo, C.; O’Connor, P.B.F.; Heaphy, S.M.; Mullan, J.P.A.; Donohue, C.A.; Higgins, D.G.; Baranov, P.V. GWIPS-Viz: Development of a Ribo-Seq Genome Browser. Nucleic Acids Res. 2014, 42, D859–D864. [Google Scholar] [CrossRef]
- Paz, I.; Kosti, I.; Ares, M.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A Web Server for Mapping Binding Sites of RNA-Binding Proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-Methyladenosine by IGF2BP Proteins Enhances mRNA Stability and Translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Guha, A.; Waris, S.; Nabors, L.B.; Filippova, N.; Gorospe, M.; Kwan, T.; King, P.H. The Versatile Role of HuR in Glioblastoma and Its Potential as a Therapeutic Target for a Multi-Pronged Attack. Adv. Drug Deliv. Rev. 2022, 181, 114082. [Google Scholar] [CrossRef] [PubMed]
- Pötschke, R.; Haase, J.; Glaß, M.; Simmermacher, S.; Misiak, C.; Penalva, L.O.F.; Kühnöl, C.D.; Hüttelmaier, S. MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing miRNA-Dependent Degradation. Cancers 2020, 12, 3654. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A Resource for Pan-Cancer Analysis of Epithelial-Mesenchymal Transition Genes and Signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Gulati, G.S.; Sikandar, S.S.; Wesche, D.J.; Manjunath, A.; Bharadwaj, A.; Berger, M.J.; Ilagan, F.; Kuo, A.H.; Hsieh, R.W.; Cai, S.; et al. Single-Cell Transcriptional Diversity Is a Hallmark of Developmental Potential. Science 2020, 367, 405–411. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- You, G.; Zheng, Z.; Huang, Y.; Liu, G.; Luo, W.; Huang, J.; Zhuo, L.; Tang, B.; Liu, S.; Lin, C. scRNA-Seq and Proteomics Reveal the Distinction of M2-like Macrophages between Primary and Recurrent Malignant Glioma and Its Critical Role in the Recurrence. CNS Neurosci. Ther. 2023, 29, 3391–3405. [Google Scholar] [CrossRef]
- Zhao, S.; Ni, K.; Xie, J.; Cheng, C.; Zhao, N.; Liu, J.; Ji, W.; Wang, Q.; Zhang, P.; Liu, Y. Exploring the Prognostic Value of BRMS1 + Microglia Based on Single-Cell Anoikis Regulator Patterns in the Immunologic Microenvironment of GBM. J. Neurooncol. 2024, 170, 101–117. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR Amplification, Combined Chromosome 7 Gain and Chromosome 10 Loss, and TERT Promoter Mutation in Brain Tumors and Their Potential for the Reclassification of IDHwt Astrocytoma to Glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, J.; Gu, X.; Guo, Q.; Guo, W. Single-Cell Transcriptome Sequencing Reveals SPP1-CD44-Mediated Macrophage-Tumor Cell Interactions Drive Chemoresistance in TNBC. J. Cell Mol. Med. 2024, 28, e18525. [Google Scholar] [CrossRef]
- Mahajan-Thakur, S.; Bien-Möller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme-A Systematic Review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Ippolito, T.; Mavis, C.; Gu, J.; Hernandez-Ilizaliturri, F.J.; Barth, M.J. Omipalisib (GSK458), a Dual an-PI3K/mTOR Inhibitor, Exhibits in Vitro and In Vivo Activity in Chemotherapy-Sensitive and -Resistant Models of Burkitt Lymphoma. Blood 2018, 132, 2951. [Google Scholar] [CrossRef]
- Miar, A.; Arnaiz, E.; Bridges, E.; Beedie, S.; Cribbs, A.P.; Downes, D.J.; Beagrie, R.A.; Rehwinkel, J.; Harris, A.L. Hypoxia Induces Transcriptional and Translational Downregulation of the Type I IFN Pathway in Multiple Cancer Cell Types. Cancer Res. 2020, 80, 5245–5256. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Qin, X.; Geng, H.; Zuo, D.; Zhao, Q. PI3K/AKT/mTOR Pathway-Related Long Non-Coding RNAs: Roles and Mechanisms in Hepatocellular Carcinoma. Pharmacol. Res. 2020, 160, 105195. [Google Scholar] [CrossRef]
- Cheng, M.; Liang, G.; Yin, Z.; Lin, X.; Sun, Q.; Liu, Y. Immunosuppressive Role of SPP1-CD44 in the Tumor Microenvironment of Intrahepatic Cholangiocarcinoma Assessed by Single-Cell RNA Sequencing. J. Cancer Res. Clin. Oncol. 2023, 149, 5497–5512. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Yano, H.; Pan, C.; Komohara, Y.; Fujiwara, Y.; Zhao, S.; Shinchi, Y.; Kurotaki, D.; Suzuki, M. The Significance of SPP1 in Lung Cancers and Its Impact as a Marker for Protumor Tumor-Associated Macrophages. Cancers 2023, 15, 2250. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; del Carmen Patino, M.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef]
- Zhao, H.-F.; Wang, J.; Shao, W.; Wu, C.-P.; Chen, Z.-P.; To, S.-S.T.; Li, W.-P. Recent Advances in the Use of PI3K Inhibitors for Glioblastoma Multiforme: Current Preclinical and Clinical Development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Saleem, H.; Kulsoom Abdul, U.; Küçükosmanoglu, A.; Houweling, M.; Cornelissen, F.M.G.; Heiland, D.H.; Hegi, M.E.; Kouwenhoven, M.C.M.; Bailey, D.; Würdinger, T.; et al. The TICking Clock of EGFR Therapy Resistance in Glioblastoma: Target Independence or Target Compensation. Drug Resist. Updat. 2019, 43, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Park, J.M.; Kim, D.H.; Kwon, J.; Kim, I.A. Inhibition of PI4K IIIα Radiosensitizes in Human Tumor Xenograft and Immune-Competent Syngeneic Murine Tumor Model. Oncotarget 2017, 8, 110392–110405. [Google Scholar] [CrossRef]
- Nesseler, J.P.; Schaue, D.; McBride, W.H.; Lee, M.-H.; Kaprealian, T.; Niclou, S.P.; Nickers, P. Irradiation to Improve the Response to Immunotherapeutic Agents in Glioblastomas. Adv. Radiat. Oncol. 2019, 4, 268–282. [Google Scholar] [CrossRef]
- van den Bent, M.; Azaro, A.; De Vos, F.; Sepulveda, J.; Yung, W.K.A.; Wen, P.Y.; Lassman, A.B.; Joerger, M.; Tabatabai, G.; Rodon, J.; et al. A Phase Ib/II, Open-Label, Multicenter Study of INC280 (Capmatinib) Alone and in Combination with Buparlisib (BKM120) in Adult Patients with Recurrent Glioblastoma. J. Neurooncol. 2020, 146, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.-N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool Using an Alignment-Free Logistic Regression Model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Camargo, A.P.; Sourkov, V.; Pereira, G.A.G.; Carazzolle, M.F. RNAsamba: Neural Network-Based Assessment of the Protein-Coding Potential of RNA Sequences. NAR Genom. Bioinform. 2020, 2, lqz024. [Google Scholar] [CrossRef]
- Han, S.; Liang, Y.; Ma, Q.; Xu, Y.; Zhang, Y.; Du, W.; Wang, C.; Li, Y. LncFinder: An Integrated Platform for Long Non-Coding RNA Identification Utilizing Sequence Intrinsic Composition, Structural Information and Physicochemical Property. Brief. Bioinform. 2019, 20, 2009–2027. [Google Scholar] [CrossRef] [PubMed]
- Nassar, L.R.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; et al. The UCSC Genome Browser Database: 2023 Update. Nucleic Acids Res. 2023, 51, D1188–D1195. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Domínguez Conde, C.; Xu, C.; Jarvis, L.B.; Rainbow, D.B.; Wells, S.B.; Gomes, T.; Howlett, S.K.; Suchanek, O.; Polanski, K.; King, H.W.; et al. Cross-Tissue Immune Cell Analysis Reveals Tissue-Specific Features in Humans. Science 2022, 376, eabl5197. [Google Scholar] [CrossRef]
- Ggplot2: Elegant Graphics for Data Analysis|SpringerLink. Available online: https://link.springer.com/book/10.1007/978-3-319-24277-4 (accessed on 7 May 2024).
- Xie, Y.; Allaire, J.J.; Grolemund, G. R Markdown: The Definitive Guide; Chapman and Hall/CRC: New York, NY, USA, 2023. [Google Scholar]
- Jew, B.; Alvarez, M.; Rahmani, E.; Miao, Z.; Ko, A.; Garske, K.M.; Sul, J.H.; Pietiläinen, K.H.; Pajukanta, P.; Halperin, E. Accurate Estimation of Cell Composition in Bulk Expression through Robust Integration of Single-Cell Information. Nat. Commun. 2020, 11, 1971. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily Conserved Elements in Vertebrate, Insect, Worm, and Yeast Genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative Annotation of Human Large Intergenic Noncoding RNAs Reveals Global Properties and Specific Subclasses. Genes. Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, S.; Huang, J.; Zhang, Z.; Lei, H.; Huang, Q.; Qu, L.; Zheng, L. InfoScan: A New Transcript Identification Tool Based on scRNA-Seq and Its Application in Glioblastoma. Int. J. Mol. Sci. 2025, 26, 2208. https://doi.org/10.3390/ijms26052208
Mei S, Huang J, Zhang Z, Lei H, Huang Q, Qu L, Zheng L. InfoScan: A New Transcript Identification Tool Based on scRNA-Seq and Its Application in Glioblastoma. International Journal of Molecular Sciences. 2025; 26(5):2208. https://doi.org/10.3390/ijms26052208
Chicago/Turabian StyleMei, Shiqiang, Jinjin Huang, Zhen Zhang, Haotian Lei, Qiaojuan Huang, Lianghu Qu, and Lingling Zheng. 2025. "InfoScan: A New Transcript Identification Tool Based on scRNA-Seq and Its Application in Glioblastoma" International Journal of Molecular Sciences 26, no. 5: 2208. https://doi.org/10.3390/ijms26052208
APA StyleMei, S., Huang, J., Zhang, Z., Lei, H., Huang, Q., Qu, L., & Zheng, L. (2025). InfoScan: A New Transcript Identification Tool Based on scRNA-Seq and Its Application in Glioblastoma. International Journal of Molecular Sciences, 26(5), 2208. https://doi.org/10.3390/ijms26052208