
In Silico Validation of OncoOrigin: An Integrative AI Tool for Primary Cancer Site Prediction with Graphical User Interface to Facilitate Clinical Application

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
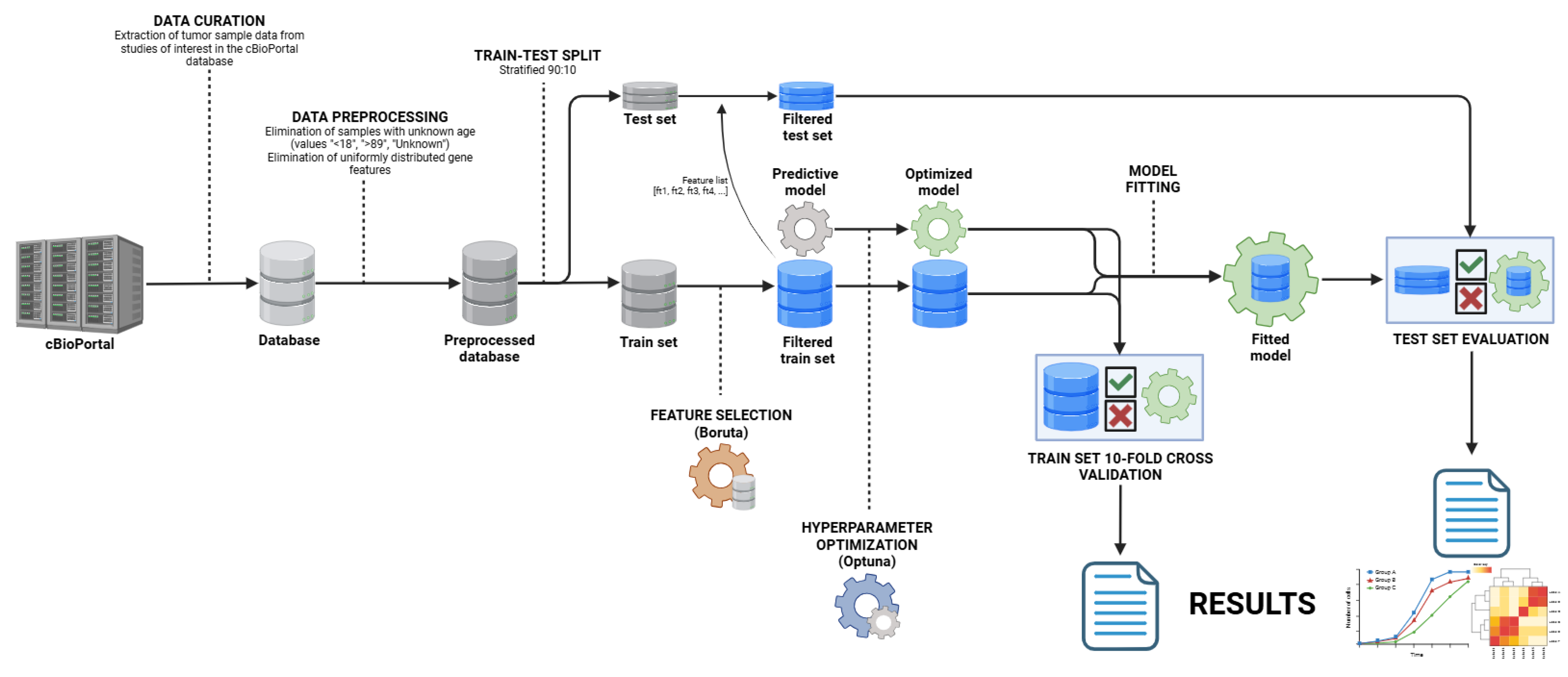
2.1. Data Preprocessing
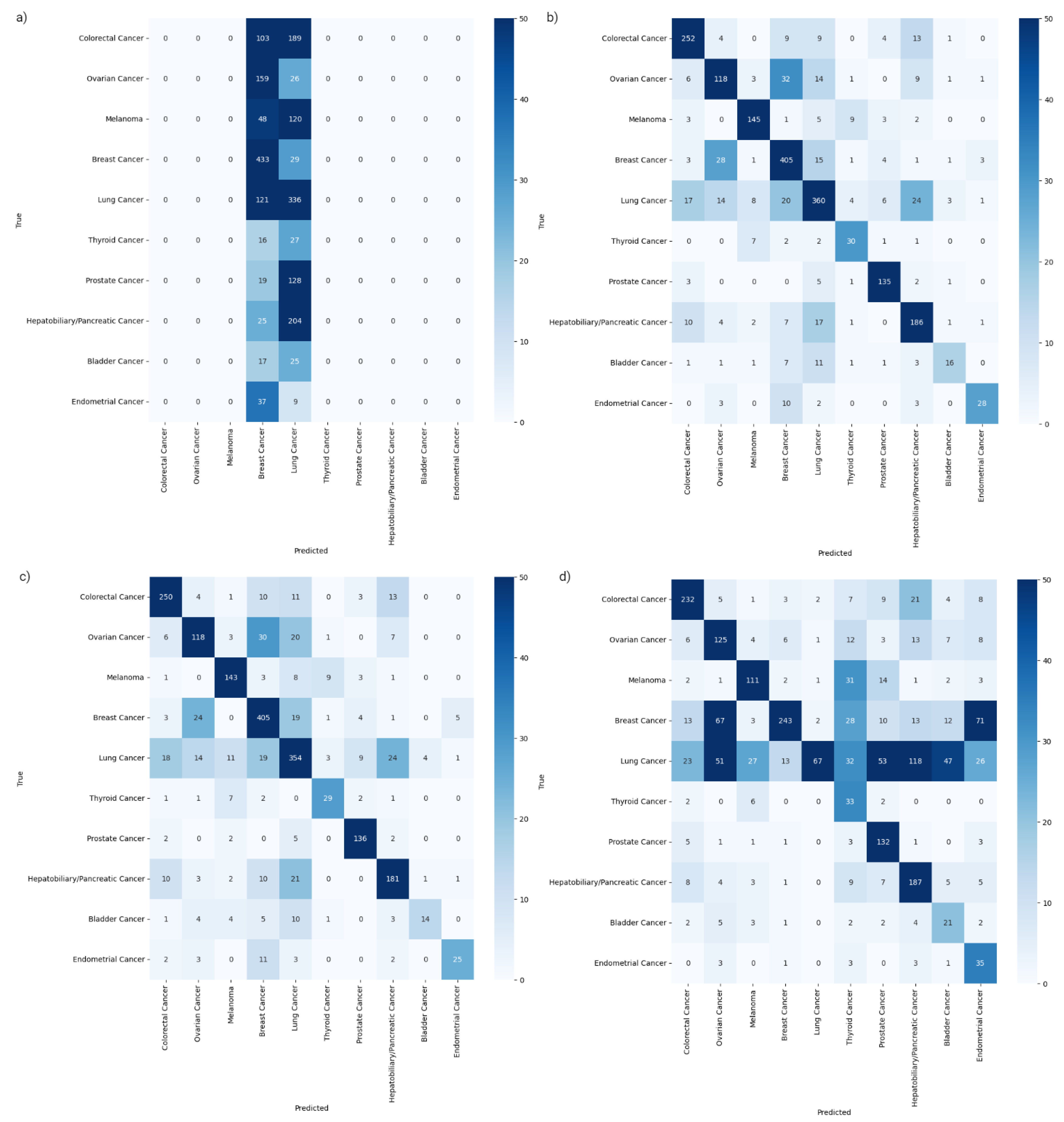
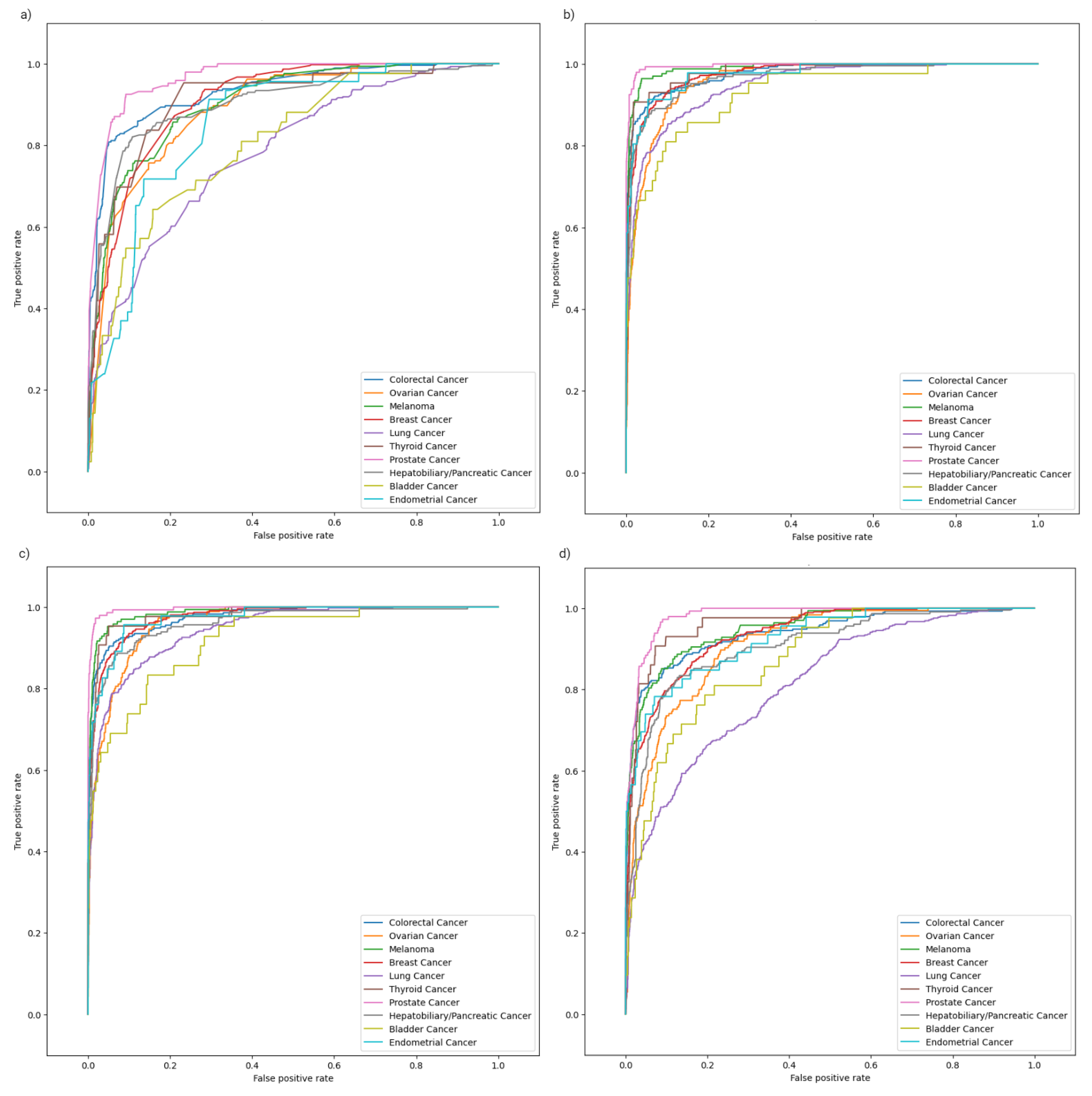
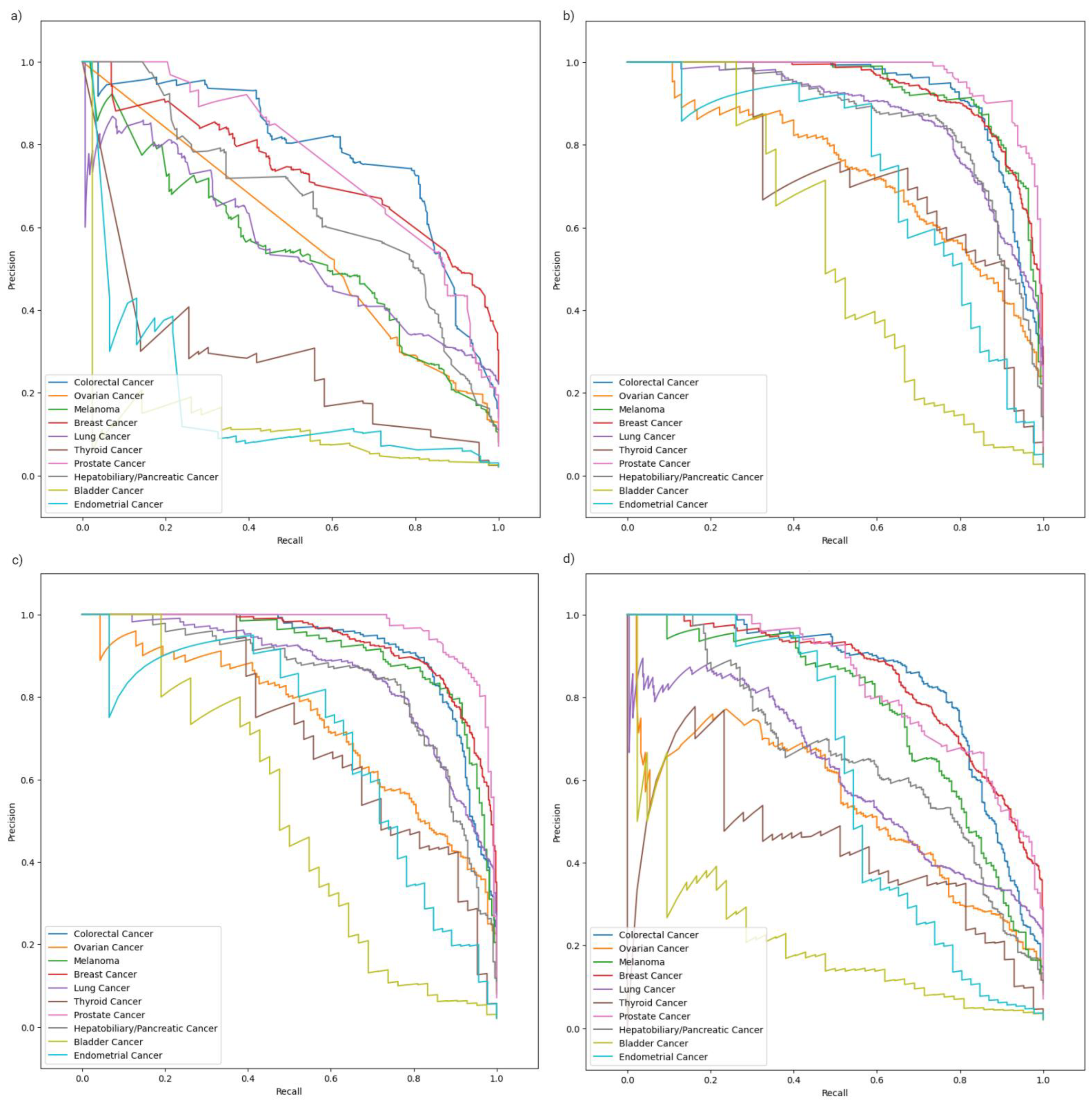
2.2. Model Evaluation
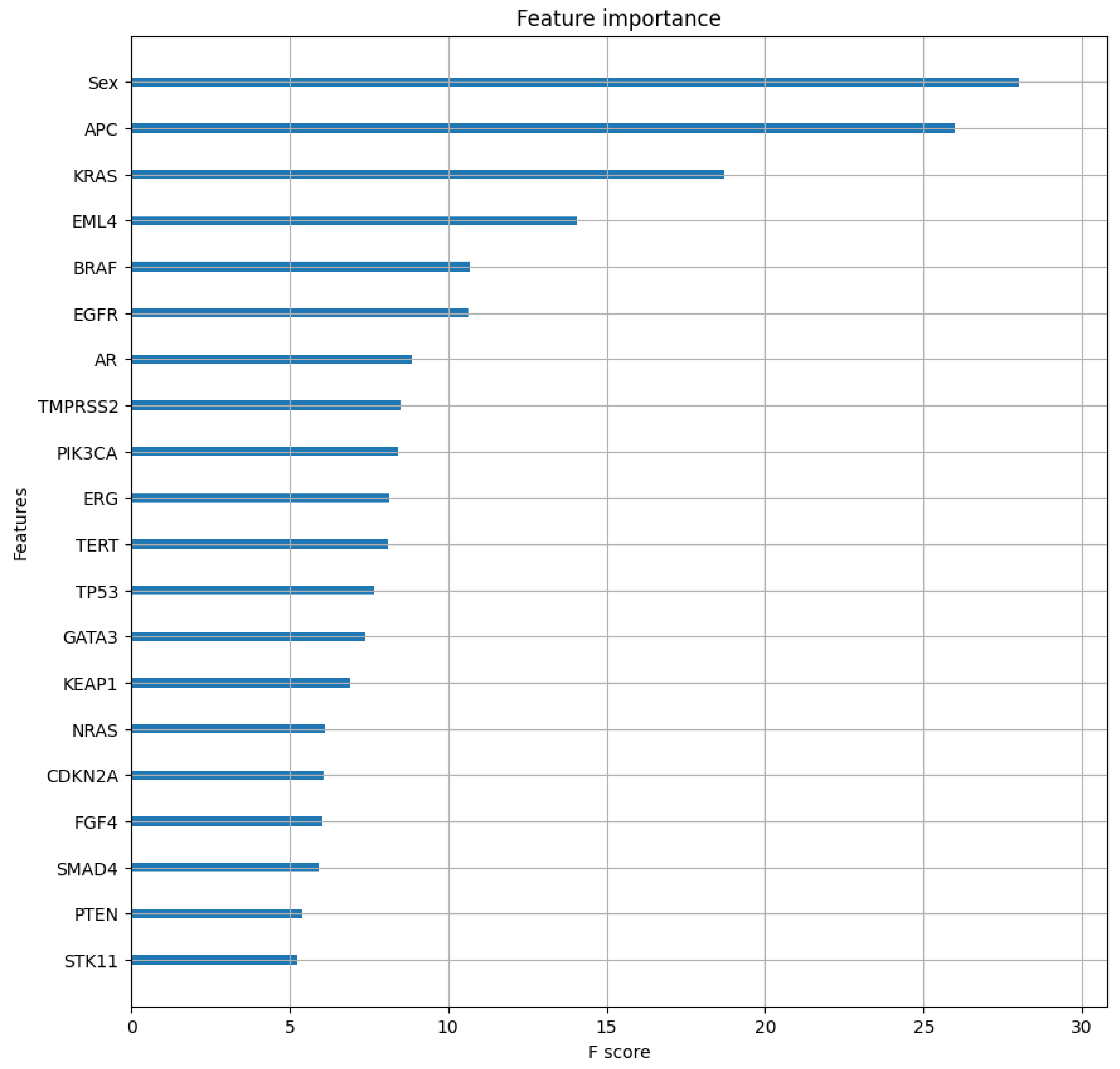
2.3. Class-Specific Evaluation and Feature Analysis
3. Discussion
3.1. Software Quality Evaluation and Comparison
3.2. Software Limitations
3.3. Feature Importance Analysis Implications
3.4. Practical Applications of OncoOrigin in Precision Oncology
4. Materials and Methods
4.1. Study Design
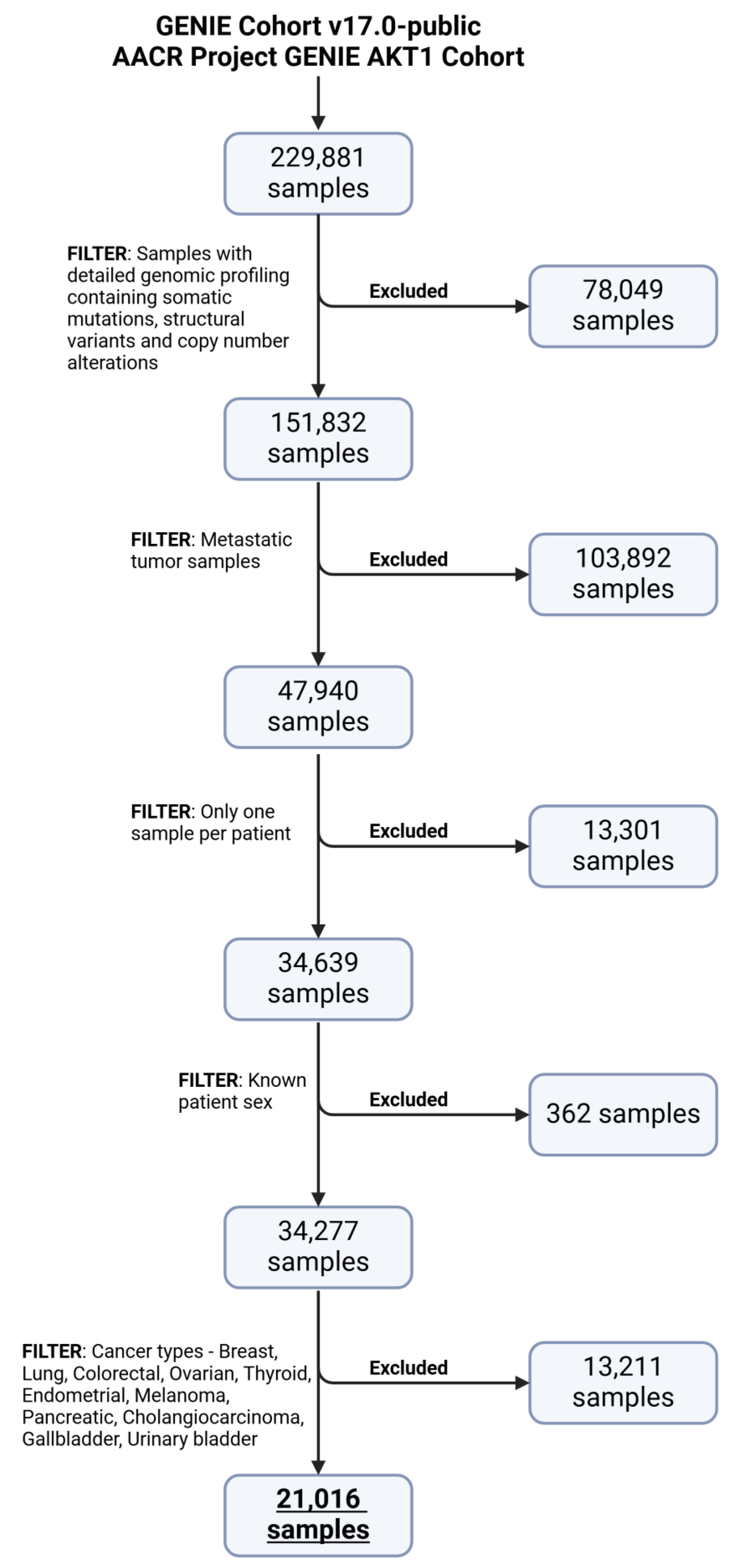
4.2. Data Curation
4.3. Data Preprocessing
4.4. Model Development
4.5. Model Evaluation
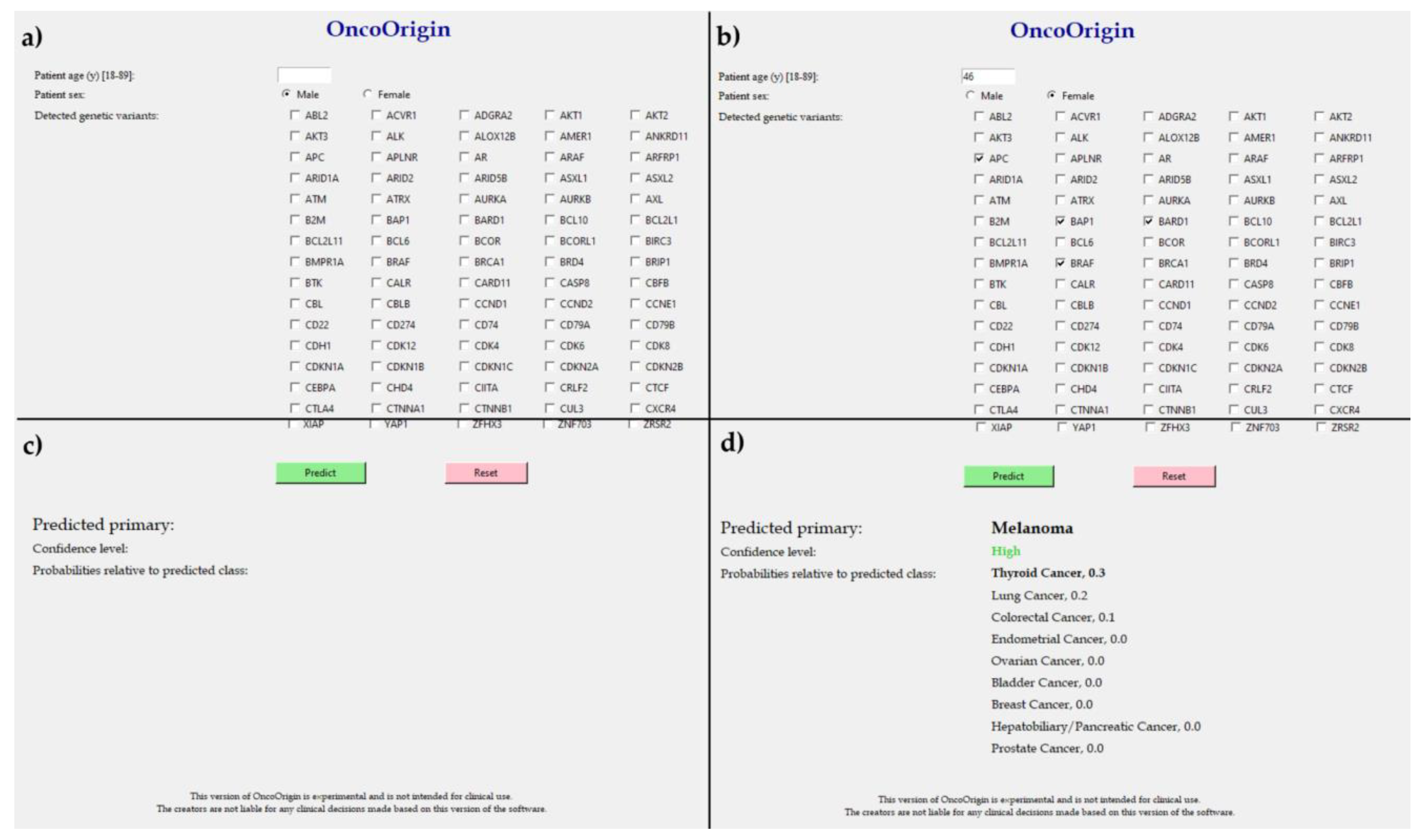
4.6. User Interface Development
4.7. Programming Implementation
5. Conclusions
Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Repetto, M.; Fernandez, N.; Drilon, A.; Chakravarty, D. Precision Oncology: 2024 in Review. Cancer Discov. 2024, 14, 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.S.; Hughes, E.G.; Sukhadia, S.S.; Green, D.C.; Houde, B.E.; Tsongalis, G.J.; Tafe, L.J. Validation and Implementation of a Somatic-Only Tumor Exome for Routine Clinical Application. J. Mol. Diagn. 2024, 26, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid biopsy: Current technology and clinical applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Zolotykh, M.A.; Mingazova, L.A.; Filina, Y.V.; Blatt, N.L.; Nesterova, A.I.; Sabirov, A.G.; Rizvanov, A.A.; Miftakhova, R.R. Cancer of unknown primary and the «seed and soil» hypothesis. Crit. Rev. Oncol. Hematol. 2024, 196, 104297. [Google Scholar] [CrossRef] [PubMed]
- Cancer of Unknown Primary Statistics. Cancer Research UK. 2015. Updated 3 July 2024. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/cancer-of-unknown-primary (accessed on 9 September 2024).
- Board, P.A. Cancer of Unknown Primary (CUP) Treatment (PDQ®). In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2024. [Google Scholar]
- Brlek, P.; Bulić, L.; Bračić, M.; Projić, P.; Škaro, V.; Shah, N.; Shah, P.; Primorac, D. Implementing Whole Genome Sequencing (WGS) in Clinical Practice: Advantages, Challenges, and Future Perspectives. Cells 2024, 13, 504. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Gouin, K.H., 3rd; Ing, N.; Ho, A.; Basho, R.; Shah, A.; Mebane, R.H.; Zitser, D.; Martinez, A.; Mevises, N.Y.; et al. Single-cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell 2024, 42, 70–84.e8. [Google Scholar] [CrossRef]
- Pounraj, S.; Chen, S.; Ma, L.; Mazzieri, R.; Dolcetti, R.; Rehm, B.H.A. Targeting Tumor Heterogeneity with Neoantigen-Based Cancer Vaccines. Cancer Res. 2024, 84, 353–363. [Google Scholar] [CrossRef]
- Ting Sim, J.Z.; Fong, Q.W.; Huang, W.; Tan, C.H. Machine learning in medicine: What clinicians should know. Singap. Med. J. 2023, 64, 91–97. [Google Scholar]
- Moon, I.; LoPiccolo, J.; Baca, S.C.; Sholl, L.M.; Kehl, K.L.; Hassett, M.J.; Liu, D.; Schrag, D.; Gusev, A. Machine learning for genetics-based classification and treatment response prediction in cancer of unknown primary. Nat. Med. 2023, 29, 2057–2067, Erratum in Nat. Med. 2024, 30, 607. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar]
- Xiong, Z.; Raphael, I.; Olin, M.; Okada, H.; Li, X.; Kohanbash, G. Glioblastoma vaccines: Past, present, and opportunities. EBioMedicine 2024, 100, 104963. [Google Scholar] [CrossRef]
- Bicakci, N. Diagnostic and prognostic value of F-18 FDG PET/CT in patients with carcinoma of unknown primary. North. Clin. Istanb. 2022, 9, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Chen, T.Y.; Williamson, D.F.K.; Zhao, M.; Shady, M.; Lipkova, J.; Mahmood, F. AI-based pathology predicts origins for cancers of unknown primary. Nature 2021, 594, 106–110. [Google Scholar] [CrossRef]
- Bastian, G.; Baker, G.H.; Limon, A. Bridging the divide between data scientists and clinicians. Intell.-Based Med. 2022, 6, 100066. [Google Scholar] [CrossRef]
- Peng, H.; Ying, J.; Zang, J.; Lu, H.; Zhao, X.; Yang, P.; Wang, X.; Li, J.; Gong, Z.; Zhang, D.; et al. Specific Mutations in APC, with Prognostic Implications in Metastatic Colorectal Cancer. Cancer Res. Treat. 2023, 55, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Xue, W.; Yang, L.; Chen, C.; Ashrafizadeh, M.; Tian, Y.; Sun, R. Wnt/β-catenin-driven EMT regulation in human cancers. Cell Mol. Life Sci. 2024, 81, 79. [Google Scholar] [CrossRef]
- Bukovac, A.; Kafka, A.; Raguž, M.; Brlek, P.; Dragičević, K.; Müller, D.; Pećina-Šlaus, N. Are We Benign? What Can Wnt Signaling Pathway and Epithelial to Mesenchymal Transition Tell Us about Intracranial Meningioma Progression. Cancers 2021, 13, 1633. [Google Scholar] [CrossRef]
- Kafka, A.; Bukovac, A.; Brglez, E.; Jarmek, A.M.; Poljak, K.; Brlek, P.; Žarković, K.; Njirić, N.; Pećina-Šlaus, N. Methylation Patterns of DKK1, DKK3 and GSK3β Are Accompanied with Different Expression Levels in Human Astrocytoma. Cancers 2021, 13, 2530. [Google Scholar] [CrossRef]
- Akhoundova, D.; Rubin, M.A. Clinical application of advanced multi-omics tumor profiling: Shaping precision oncology of the future. Cancer Cell 2022, 40, 920–938. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.B.; Hou, L.K.; Yu, F.; Zhang, J.; Wu, W.; Tang, X.-M.; Sun, F.; Lu, H.-M.; Deng, J.; et al. Liquid biopsy in lung cancer: Significance in diagnostics, prediction, and treatment monitoring. Mol. Cancer 2022, 21, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fang, Y.; Yan, W.-P. Considerations for multidisciplinary management of synchronous primary breast cancer and primary lung cancer—Analysis of thirty-one patients. Thorac. Cancer 2024, 15, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Brlek, P.; Kafka, A.; Bukovac, A.; Pećina-Šlaus, N. Integrative cBioPortal Analysis Revealed Molecular Mechanisms That Regulate EGFR-PI3K-AKT-mTOR Pathway in Diffuse Gliomas of the Brain. Cancers 2021, 13, 3247. [Google Scholar] [CrossRef]
- De Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comp. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Soft. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Dorogush, A.V.; Ershov, V.; Gulin, A. CatBoost: Gradient boosting with categorical features support. arXiv 2018, arXiv:1810.11363v1. [Google Scholar]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-generation Hyperparameter Optimization Framework. arXiv 2019, arXiv:1907.10902v1. [Google Scholar]
- Lundh, F. An Introduction to Tkinter. 1999. Available online: https://ftp.math.utah.edu/u/ma/hohn/linux/tcl/an-introduction-to-tkinter.pdf (accessed on 21 September 2024).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | XGBC | CBC | ETC | RFC | |
|---|---|---|---|---|---|
| TRAIN SET 10-FOLD CROSS-VALIDATION | |||||
| Mean accuracy | 0.79 | 0.78 | 0.55 | 0.37 | |
| Accuracy 95-CI | 0.782–0.793 | 0.771–0.784 | 0.543–0.561 | 0.365–0.375 | |
| Mean weighted F1 | 0.79 | 0.78 | 0.54 | 0.23 | |
| Weighted F1 95-CI | 0.781–0.791 | 0.769–0.782 | 0.533–0.552 | 0.225–0.232 | |
| Mean weighted ROC-AUC | 0.97 | 0.96 | 0.90 | 0.87 | |
| Weighted ROC-AUC 95-CI | 0.965–0.968 | 0.962–0.966 | 0.895–0.902 | 0.868–0.877 | |
| TEST SET EVALUATION | |||||
| Accuracy | 0.81 | 0.80 | 0.57 | 0.37 | |
| Top-2 accuracy | 0.91 | 0.90 | 0.74 | 0.47 | |
| Weighted F1 | 0.81 | 0.80 | 0.56 | 0.23 | |
| Weighted ROC-AUC | 0.97 | 0.97 | 0.91 | 0.88 | |
| Weighted PR-AUC | 0.88 | 0.87 | 0.71 | 0.63 | |
| Sensitivity | Colorectal Cancer | 0.86 | 0.86 | 0.79 | 0.00 |
| Ovarian Cancer | 0.64 | 0.64 | 0.68 | 0.00 | |
| Melanoma | 0.86 | 0.85 | 0.66 | 0.00 | |
| Breast Cancer | 0.88 | 0.88 | 0.53 | 0.94 | |
| Lung Cancer | 0.79 | 0.77 | 0.15 | 0.74 | |
| Thyroid Cancer | 0.70 | 0.67 | 0.77 | 0.00 | |
| Prostate Cancer | 0.92 | 0.93 | 0.90 | 0.00 | |
| Hepatobiliary/Pancreatic Cancer | 0.81 | 0.79 | 0.82 | 0.00 | |
| Bladder Cancer | 0.38 | 0.33 | 0.50 | 0.00 | |
| Endometrial Cancer | 0.61 | 0.54 | 0.76 | 0.00 | |
| Specificity | Colorectal Cancer | 0.98 | 0.98 | 0.97 | 1.00 |
| Ovarian Cancer | 0.97 | 0.97 | 0.93 | 1.00 | |
| Melanoma | 0.99 | 0.98 | 0.97 | 1.00 | |
| Breast Cancer | 0.95 | 0.94 | 0.98 | 0.66 | |
| Lung Cancer | 0.95 | 0.94 | 1.00 | 0.53 | |
| Thyroid Cancer | 0.99 | 0.99 | 0.94 | 1.00 | |
| Prostate Cancer | 0.99 | 0.99 | 0.95 | 1.00 | |
| Hepatobiliary/Pancreatic Cancer | 0.97 | 0.97 | 0.91 | 1.00 | |
| Bladder Cancer | 1.00 | 1.00 | 0.96 | 1.00 | |
| Endometrial Cancer | 1.00 | 1.00 | 0.94 | 1.00 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brlek, P.; Bulić, L.; Shah, N.; Shah, P.; Primorac, D. In Silico Validation of OncoOrigin: An Integrative AI Tool for Primary Cancer Site Prediction with Graphical User Interface to Facilitate Clinical Application. Int. J. Mol. Sci. 2025, 26, 2568. https://doi.org/10.3390/ijms26062568
Brlek P, Bulić L, Shah N, Shah P, Primorac D. In Silico Validation of OncoOrigin: An Integrative AI Tool for Primary Cancer Site Prediction with Graphical User Interface to Facilitate Clinical Application. International Journal of Molecular Sciences. 2025; 26(6):2568. https://doi.org/10.3390/ijms26062568
Chicago/Turabian StyleBrlek, Petar, Luka Bulić, Nidhi Shah, Parth Shah, and Dragan Primorac. 2025. "In Silico Validation of OncoOrigin: An Integrative AI Tool for Primary Cancer Site Prediction with Graphical User Interface to Facilitate Clinical Application" International Journal of Molecular Sciences 26, no. 6: 2568. https://doi.org/10.3390/ijms26062568
APA StyleBrlek, P., Bulić, L., Shah, N., Shah, P., & Primorac, D. (2025). In Silico Validation of OncoOrigin: An Integrative AI Tool for Primary Cancer Site Prediction with Graphical User Interface to Facilitate Clinical Application. International Journal of Molecular Sciences, 26(6), 2568. https://doi.org/10.3390/ijms26062568