

Reactive Oxygen Species (ROS) in Metabolic Disease—Don’t Shoot the Metabolic Messenger
Abstract
1. Introduction
2. The Biochemistry and Detection of ROS
3. ROS as a Cellular Second Messenger
- Firstly, in response to stimulation, a second messenger is able to swiftly propagate and amplify the signal intracellularly.
- Secondly, it must have downstream targets that trigger an effect.
- Finally, there must be a mechanism for efficiently ending the signal and effect within an appropriately short timespan.
3.1. Sources of ROS and Signal Amplification
3.1.1. The Mitochondria
3.1.2. The Peroxisome
3.1.3. Cytosolic ROS Sources
3.1.4. The Endoplasmic Reticulum
3.2. Localisation and Termination of ROS Signalling
3.3. ROS-S Transduction
3.4. Localised Signalling vs. “Oxidative Stress”—A Matter of Magnitude?
4. Examples of Physiological ROS-S Systems Relevant to Metabolic Disease
- How does the putative agonist or physiological situation stimulate ROS production?
- What is the cellular source of the signalling ROS?
- What are the physiological targets of the ROS signal?
- What are the ROS-S system’s functional effects?
4.1. Muscular Glucose Uptake and Metabolism
4.2. Muscular and Myocardial Contractility
4.3. Angiogenesis
4.4. Glucose and Sodium Resorption in the Kidney
4.5. Circadian Metabolism
4.6. Aging Metabolism
5. “Oxidative Stress”—Where Heroes Become Villains?
6. Closing Insights
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.; Groothuis, G.M.; Olinga, P.; Boersema, M. Targeting oxidative stress for the treatment of liver fibrosis. In Reviews of Physiology, Biochemistry and Pharmacology; Nilius, B., de Tombe, P., Gudermann, T., Jahn, R., Lill, R., Eds.; Springer: Cham, Switzerland, 2018; Volume 175, pp. 71–102. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. J. Am. Med. Assoc. 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Kang, J.H.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Albert, C.M.; Grodstein, F. Vitamin E, Vitamin C, Beta carotene, and cognitive function among women with or at risk of cardiovascular disease: The women’s antioxidant and cardiovascular study. Circulation 2009, 119, 2772–2780. [Google Scholar] [CrossRef]
- Lin, J.; Cook, N.R.; Albert, C.; Zaharris, E.; Gaziano, J.M.; Van Denburgh, M.; Buring, J.E.; Manson, J.E. Vitamins C and E and beta carotene supplementation and cancer risk: A randomized controlled trial. J. Natl. Cancer Inst. 2009, 101, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cook, N.R.; Albert, C.M.; Van Denburgh, M.; Manson, J.A.E. Effects of vitamins C and E and β-carotene on the risk of type 2 diabetes in women at high risk of cardiovascular disease: A randomized controlled trial. Am. J. Clin. Nutr. 2009, 90, 429–437. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Dynamics of intracellular and intercellular redox communication. Free Radic. Biol. Med. 2024, 225, 933–939. [Google Scholar] [CrossRef]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nat. Rev. Mol. Cell Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef]
- Lerman, B.B.; Belardinelli, L.; A West, G.; Berne, R.M.; DiMarco, J.P. Adenosine-sensitive ventricular tachycardia: Evidence suggesting cyclic AMP-mediated triggered activity. Circulation 1986, 74, 270–280. [Google Scholar] [CrossRef]
- Osaki, J.; Haneda, T.; Sakai, H.; Kikuchi, K. cAMP-mediated -fos expression in pressure-overloaded acceleration of protein synthesis in adult rat heart. Cardiovasc. Res. 1997, 33, 631–640. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef] [PubMed]
- Boularan, C.; Gales, C. Cardiac cAMP: Production, hydrolysis, modulation and detection. Front. Pharmacol. 2015, 6, 203. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Li Puma, L.C.; Hedges, M.; Heckman, J.M.; Mathias, A.B.; Engstrom, M.R.; Brown, A.B.; Chicco, A.J. Experimental oxygen concentration influences rates of mitochondrial hydrogen peroxide release from cardiac and skeletal muscle preparations. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R972–R980. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Inouye, S.; Akagi, R.; Seyama, T. Local redox environment beneath biological membranes probed by palmitoylated-roGFP. Redox Biol. 2018, 14, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, A.; Ezeriņa, D.; Ivanova, J.; Guriev, N.; Pugovkina, N.; Shatrova, A.; Aksenov, N.; Messens, J.; Lyublinskaya, O. HyPer as a tool to determine the reductive activity in cellular compartments. Redox Biol. 2024, 70, 103058. [Google Scholar] [CrossRef]
- Laker, R.C.; Xu, P.; Ryall, K.A.; Sujkowski, A.; Kenwood, B.M.; Chain, K.H.; Zhang, M.; Royal, M.A.; Hoehn, K.L.; Driscoll, M.; et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014, 289, 12005–12015. [Google Scholar] [CrossRef]
- Neto, I.V.d.S.; Pinto, A.P.; de Andrade, R.V.; de Souza, F.H.V.; de Souza, P.E.N.; Assis, V.; Tibana, R.A.; Neves, R.V.P.; Rosa, T.S.; Prestes, J.; et al. Paternal exercise induces antioxidant defenses by α-Klotho/Keap1 pathways in the skeletal muscle of offspring exposed to a high fat-diet without changing telomere length. J. Nutr. Biochem. 2024, 134, 109747. [Google Scholar] [CrossRef]
- Elajaili, H.B.; Dee, N.M.; Dikalov, S.I.; Kao, J.P.Y.; Nozik, E.S. Use of Electron Paramagnetic Resonance (EPR) to Evaluate Redox Status in a Preclinical Model of Acute Lung Injury. Mol. Imaging Biol. 2023, 26, 495–502. [Google Scholar] [CrossRef]
- Janisch, K.; Schempp, H. Evaluation of the oxidative burst in suspension cell culture of Phaseolus vulgaris. Z. Naturforschung C 2004, 59, 849–855. [Google Scholar] [CrossRef]
- Endo, M.; Tanaka, M.; Ogawa, Y. Calcium Induced release of calcium from the Sarcoplasmic Reticulum of Skinned Skeletal Muscle Fibres. Nature 1970, 228, 34–36. [Google Scholar] [CrossRef]
- Bridge, J.H.B.; Smolley, J.R.; Spitzer, K.W. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science 1979, 248, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Gielecińska, A.; Kciuk, M.; Kontek, R. The Impact of Calcium Overload on Cellular Processes: Exploring Calcicoptosis and Its Therapeutic Potential in Cancer. Int. J. Mol. Sci. 2024, 25, 13727. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, X.; Liang, X.; Meng, M.; Zhang, H.; Li, Z.; Lin, Y.; Li, J.; Ma, C. Verapamil inhibits ferroptosis in septic acute lung injury by blocking L-type calcium channels. Biochem. Biophys. Res. Commun. 2024, 744, 151202. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Lou, H.; Dong, X.; Hao, S.; Sun, Z.; Dou, Z.; Li, H.; Zhao, W.; Sun, X.; et al. Cardiomyocyte-specific long noncoding RNA Trdn-as induces mitochondrial calcium overload by promoting the m6A modification of calsequestrin 2 in diabetic cardiomyopathy. Front. Med. 2025, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef]
- Pavlaki, N.; Froese, A.; Li, W.; A De Jong, K.; Geertz, B.; Subramanian, H.; Mohagaonkar, S.; Luo, X.; Schubert, M.; Wiegmann, R.; et al. Gene therapy with phosphodiesterases 2A and 4B ameliorates heart failure and arrhythmias by improving subcellular cAMP compartmentation. Cardiovasc. Res. 2024, 120, 1011–1023. [Google Scholar] [CrossRef]
- Chandel, N.S.; Budinger, G.R.S.; Choe, S.H.; Schumacker, P.T. Cellular respiration during hypoxia: Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 1997, 272, 18808–18816. [Google Scholar] [CrossRef]
- Kanaan, A.; Farahani, R.; Douglas, R.M.; LaManna, J.C.; Haddad, G.G.; Ducsay, C.A.; Goyal, R.; Pearce, W.J.; Wilson, S.; Hu, X.-Q.; et al. Effect of chronic continuous or intermittent hypoxia and reoxygenation on cerebral capillary density and myelination. Am. J. Physiol. Integr. Comp. Physiol. 2006, 290, R1105–R1114. [Google Scholar] [CrossRef]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, N.; Kukidome, D.; Sada, K.; Motoshima, H.; Furukawa, N.; Matsumura, T.; Nishikawa, T.; Araki, E. Low glucose induces mitochondrial reactive oxygen species via fatty acid oxidation in bovine aortic endothelial cells. J. Diabetes Investig. 2017, 8, 750–761. [Google Scholar] [CrossRef]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain Suppressors of mitochondrial superoxide and hydrogen peroxide production. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar]
- Pryde, K.R.; Hirst, J. Superoxide Is Produced by the Reduced Flavin in Mitochondrial Complex I—A Single Unified Mechanism That Applies During Both Forward and Reverse Electron Transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef]
- Mito, M.S.; Constantin, J.; de Castro, C.V.; Yamamoto, N.S.; Bracht, A. Effects of ranolazine on fatty acid transformation in the isolated perfused rat liver. Mol. Cell. Biochem. 2010, 345, 35–44. [Google Scholar] [CrossRef]
- Mollica, M.P.; Iossa, S.; Liverini, G.; Soboll, S. Oxygen Consumption and Biosynthetic Function in Perfused Liver from Rats at Different Stages of Development. Cell. Mol. Life Sci. 1998, 54, 1277–1282. [Google Scholar] [CrossRef]
- How, O.-J.; Aasum, E.; Kunnathu, S.; Severson, D.L.; Myhre, E.S.; Larsen, T.S. Influence of substrate supply on cardiac efficiency, as measured by pressure-volume analysis in ex vivo mouse hearts Downloaded from. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, 2979–2985. [Google Scholar] [CrossRef] [PubMed]
- Mjøs, O.D.; Kjekshus, J.K.; Lekven, J. Importance of free fatty acids as a determinant of myocardial oxygen consumption and myocardial ischemic injury during norepinephrine infusion in dogs. J. Clin. Investig. 1974, 53, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Baudhuin, P. Peroxisomes (Microbodies and Related Particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B.; De Duve, C. Cell Biology A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug (lipid metabolism/atherosclerosis/microbodies/cell fractionation). Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046. [Google Scholar] [CrossRef]
- Fry, D.W.; Richardson, K. Isolation and characterization of glycolic acid oxidase from human liver. Biochim. Biophys. Acta Enzym. 1979, 568, 135–144. [Google Scholar] [CrossRef]
- Jorns, M.S. Glycolic Acid Oxidase from Pig Liver. Methods Enzym. 1975, 41, 337–343. [Google Scholar]
- Chen, H.; Li, X.; Sun, Y.; Du, Y.; Wu, S.; Wu, Y.; Liu, H.; Liu, Y.; Wang, Y.; Zhao, Q.; et al. HAO1 negatively regulates liver macrophage activation via the NF-κB pathway in alcohol-associated liver disease. Cell Signal 2022, 99, 110436. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; del Río, L.A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell. Biochem. 2013, 69, 231–255. [Google Scholar]
- Corpas, F.J.; Palma, J.M.; Sandalio, L.M.; Valderrama, R.; Barroso, J.B.; del Río, L.A. Peroxisomal xanthine oxidoreductase: Characterization of the enzyme from pea (Pisum sativum L.) leaves. J. Plant Physiol. 2008, 165, 1319–1330. [Google Scholar] [CrossRef]
- del Río, L.A.; Fernández, V.M.; Rupérez, F.L.; Sandalio, L.M.; Palma, J.M. NADH Induces the Generation of Superoxide Radicals in Leaf Peroxisomes. Plant Physiol. 1989, 89, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Sandalio, L.M.; Fernández, V.M.; Rupérez, F.L.; Del Río, L.A. Superoxide Free Radicals Are Produced in Glyoxysomes. Plant Physiol. 1988, 87, 1–4. [Google Scholar] [CrossRef]
- Sonani, R.R.; Blat, A.; Dubin, G. Crystal structures of apo- and FAD-bound human peroxisomal acyl-CoA oxidase provide mechanistic basis explaining clinical observations. Int. J. Biol. Macromol. 2022, 205, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.; Klug, Y.A.; Dimitrov, L.; Erez, Z.; Chuartzman, S.G.; Elinger, D.; Yofe, I.; Soliman, K.; Gärtner, J.; Thoms, S.; et al. Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol. Biosyst. 2014, 10, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Ušaj, M.M.; Brložnik, M.; Kaferle, P.; Žitnik, M.; Wolinski, H.; Leitner, F.; Kohlwein, S.; Zupan, B.; Petrovič, U. Genome-wide localization study of yeast pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J. Mol. Biol. 2015, 427, 2072–2087. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.-S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef]
- Wang, B.; Van Veldhoven, P.P.; Brees, C.; Rubio, N.; Nordgren, M.; Apanasets, O.; Kunze, M.; Baes, M.; Agostinis, P.; Fransen, M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic. Biol. Med. 2013, 65, 882–894. [Google Scholar] [CrossRef]
- Baumgart, E.; Vanhorebeek, I.; Grabenbauer, M.; Borgers, M.; Declercq, P.E.; Fahimi, H.D.; Baes, M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol. 2001, 159, 1477–1494. [Google Scholar] [CrossRef]
- Iyer, G.Y.; Questel, J.H. NADPH and NADH oxidation by guinea pig polymorphonuclear leucocytes. Can. J. Biochem. Physiol. 1963, 41, 427–434. [Google Scholar] [CrossRef]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2014, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Then, A.; Goenawan, H.; Lesmana, R.; Christoper, A.; Sylviana, N.; Gunadi, J.W. Exploring the potential regulation of DUOX in thyroid hormone autophagy signaling via IGF 1 in the skeletal muscle. Biomed. Rep. 2024, 22, 39. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Zuo, L.; Ikeda, S.; Tojo, T.; Patrushev, N.A.; Alexander, R.W. cAbl tyrosine kinase mediates reactive oxygen species- and caveolin-dependent AT1 receptor signaling in vascular smooth muscle: Role in vascular hypertrophy. Circ. Res. 2005, 97, 829–836. [Google Scholar] [CrossRef]
- Lacy, F.; Gough, D.A.; Schmid-Schönbein, G.W. Role of xanthine oxidase in hydrogen peroxide production. Free Radic. Biol. Med. 1998, 25, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Galbusera, C.; Orth, P.; Fedida, D.; Spector, T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem. Pharmacol. 2006, 71, 1747–1752. [Google Scholar] [CrossRef]
- Bonini, M.G.; Miyamoto, S.; Di Mascio, P.; Augusto, O. Production of the carbonate radical anion during xanthine oxidase turnover in the presence of bicarbonate. J. Biol. Chem. 2004, 279, 51836–51843. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. The Underrated Carbonate Radical (CO3)-Detoxification and Reduced Formation by Melatonin. Biomed. J. Sci. Tech. Res. 2017, 1, 634–637. [Google Scholar] [CrossRef]
- Medinas, D.B.; Cerchiaro, G.; Trindade, D.F.; Augusto, O. The carbonate radical and related oxidants derived from bicarbonate buffer. IUBMB Life 2007, 59, 255–262. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxidants Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Goldberger, R.F.; Epstein, C.J.; Anfinsen, C.B. Acceleration of Reactivation of Reduced Bovine Pancreatic Ribonuclease by a Microsomal System from Rat Liver. J. Biol. Chem. 1963, 238, 628–635. [Google Scholar] [CrossRef]
- Cabibbo, A.; Pagani, M.; Fabbri, M.; Rocchi, M.; Farmery, M.R.; Bulleid, N.J.; Sitia, R. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Frand, A.R.; Kaiser, C.A. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 1999, 4, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Chin, K.-T.; Blais, J.; Harding, H.P.; Ron, D. ERO1-β, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 2010, 188, 821–832. [Google Scholar] [CrossRef]
- Alarcon, C.; Boland, B.B.; Uchizono, Y.; Moore, P.C.; Peterson, B.; Rajan, S.; Rhodes, O.S.; Noske, A.B.; Haataja, L.; Arvan, P.; et al. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes 2015, 65, 438–450. [Google Scholar] [CrossRef]
- Kang, T.; Boland, B.B.; Jensen, P.; Alarcon, C.; Nawrocki, A.; Grimsby, J.S.; Rhodes, C.J.; Larsen, M.R. Characterization of Signaling Pathways Associated with Pancreatic β-cell Adaptive Flexibility in Compensation of Obesity-linked Diabetes in db/db Mice. Mol. Cell. Proteom. 2020, 19, 971–993. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and β-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Robertson, R.P. β-Cell deterioration during diabetes: What’s in the gun? Trends Endocrinol. Metab. 2009, 20, 388–393. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R.; et al. Ero1α regulates Ca2+ fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Li, X.; Zhao, X.; Qin, Z.; Li, J.; Sun, B.; Liu, L. Regulation of calcium homeostasis in endoplasmic reticulum-mitochondria crosstalk: Implications for skeletal muscle atrophy. Cell Commun. Signal. 2025, 23, 17. [Google Scholar] [CrossRef]
- Zaar, K.; Völkl, A.; Fahimi, H. Association of isolated bovine kidney cortex peroxisomes with endoplasmic reticulum. Biochim. Biophys. Acta Biomembr. 1987, 897, 135–142. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Mitochondrial Superoxide Dismutase Site of Synthesis and Intramitochondrial Localisation. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar] [CrossRef]
- Mills, G.C. Hemoglobin Catabolism. I. Glutathione Peroxidase, an Erythrocyte Enzyme which Protects Hemoglobin from Oxidative Breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [CrossRef]
- Cao, Z.; Bhella, D.; Lindsay, J.G. Reconstitution of the Mitochondrial PrxIII Antioxidant Defence Pathway: General Properties and Factors Affecting PrxIII Activity and Oligomeric State. J. Mol. Biol. 2007, 372, 1022–1033. [Google Scholar] [CrossRef]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997, 272, 16644–16651. [Google Scholar] [CrossRef]
- Putnam, C.D.; Arvai, A.S.; Bourne, Y.; A Tainer, J. Active and inhibited human catalase structures: Ligand and NADPH binding and catalytic mechanism. J. Mol. Biol. 2000, 296, 295–309. [Google Scholar] [CrossRef]
- Wilcke, M.; Hultenby, K.; Alexson, S.E. Novel peroxisomal populations in subcellular fractions from rat liver. Implications for peroxisome structure and biogenesis. J. Biol. Chem. 1995, 270, 6949–6958. [Google Scholar] [CrossRef]
- Espeel, M.; Brière, N.; De Craemer, D.; Jauniaux, E.; Roels, F. Catalase-negative peroxisomes in human embryonic liver. Cell Tissue Res. 1993, 272, 89–92. [Google Scholar] [CrossRef]
- Oikawa, I.; Novikoff, P.M. Catalase-negative peroxisomes: Transient appearance in rat hepatocytes during liver regeneration after partial hepatectomy. Am. J. Pathol. 1995, 146, 673–687. [Google Scholar] [PubMed]
- Klucis, E.; Crane, D.I.; Hughes, J.L.; Poulos, A.; Masters, C.J. Identification of a catalase-negative sub-population of peroxisomes induced in mouse liver by clofibrate. Biochim. Biophys. Acta Gen. Subj. 1991, 1074, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Flatmark, T.; Christiansen, E.; Kryvi, H. Polydispersity of rat liver peroxisomes induced by the hypolipidemic and carcinogenic agent clofibrate. Eur. J. Cell Biol. 1981, 24, 62–69. [Google Scholar]
- Fahimi, H.D.; Reinicke, A.; Sujatta, M.; Yokota, S.; Ozel, M.; Hartig, F.; Stegmeier, K. The Short- and Long-Term Effects of Bezafibrate in the Rat. Ann. N. Y. Acad. Sci. 1982, 386, 111–135. [Google Scholar] [CrossRef]
- Pérez-Estrada, J.R.; Hernández-García, D.; Leyva-Castro, F.; Ramos-León, J.; Cuevas-Benítez, O.; Díaz-Muñoz, M.; Castro-Obregón, S.; Ramírez-Solís, R.; García, C.; Covarrubias, L. Reduced lifespan of mice lacking catalase correlates with altered lipid metabolism without oxidative damage or premature aging. Free Radic. Biol. Med. 2019, 135, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef]
- Tavender, T.J.; Sheppard, A.M.; Bulleid, N.J. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 2008, 411, 191–199. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.; Vercellotti, G. Ferritin: A Cytoprotective Antioxidant Strategem of Endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin: Structure and functions. Trends Biochem. Sci. 1981, 6, 26–29. [Google Scholar] [CrossRef]
- Ubersax, J.A.; Ferrell, J.E. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef]
- Hoshi, T.; Heinemann, S.H. Regulation of cell function by methionine oxidation and reduction. J. Physiol. 2001, 531, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors Affecting Protein Thiol Reactivity and Specificity in Peroxide Reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef]
- Hall, A.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Structural Evidence that Peroxiredoxin Catalytic Power is Based on Transition-State Stabilization. J. Mol. Biol. 2010, 402, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.L.; Carroll, K.S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef]
- Finkel, T. From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal. 2012, 5, pe10. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.S.P.; Møller, I.M.; Thelen, J.J.; Miernyk, J.A. Convergent signaling pathways—Interaction between methionine oxidation and serine/threonine/tyrosine O-phosphorylation. Cell Stress Chaperon. 2015, 20, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, M.; Pace, P.E.; Cheng, Q.; Frijhoff, J.; Östman, A.; Arnér, E.S.J.; Hampton, M.B.; Winterbourn, C.C. Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J. Biol. Chem. 2017, 292, 14371–14380. [Google Scholar] [CrossRef]
- Kil, I.S.; Ryu, K.W.; Lee, S.K.; Kim, J.Y.; Chu, S.Y.; Kim, J.H.; Park, S.; Rhee, S.G. Circadian Oscillation of Sulfiredoxin in the Mitochondria. Mol. Cell 2015, 59, 651–663. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A Nuclear Factor Induced by Hypoxia via De Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.R.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Lee, G.; Won, H.S.; Lee, Y.M.; Choi, J.W.; Oh, T.I.; Jang, J.H.; Choi, D.-K.; Lim, B.-O.; Kim, Y.J.; Park, J.-W.; et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1α Activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef]
- Fialho, M.d.L.S.; Jamil, A.H.A.; Stannard, G.A.; Heather, L.C. Hypoxia-inducible factor 1 signalling, metabolism and its therapeutic potential in cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1865, 831–843. [Google Scholar] [CrossRef]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1α-PPARγ Axis Underlies the Integration of Glycolytic and Lipid Anabolic Pathways in Pathologic Cardiac Hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef]
- Belanger, A.J.; Luo, Z.; Vincent, K.A.; Akita, G.Y.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-inducible factor 1 mediates hypoxia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated receptor α/retinoid X receptor. Biochem. Biophys. Res. Commun. 2007, 364, 567–572. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Liu, Q.; Unger, T.L.; Rha, J.; Davidoff, O.; Keith, B.; Epstein, J.A.; Moores, S.L.; Erickson-Miller, C.L.; Haase, V.H. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010, 116, 3039–3048. [Google Scholar] [CrossRef]
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwon, K.-S.; Kim, S.-R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Rudolph, J. Redox regulation of MAP kinase phosphatase 3. Biochemistry 2006, 45, 8476–8487. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.C.; Shafiq, M.; Briggs, D.C.; Knowles, P.P.; Collister, M.; Didmon, M.J.; Makrantoni, V.; Dickinson, R.J.; Hanrahan, S.; Totty, N.; et al. Redox-mediated substrate recognition by Sdp1 defines a new group of tyrosine phosphatases. Nature 2007, 447, 487–492. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef]
- Brodie, A.E.; Reed, D.J. Cellular recovery of glyceraldehyde-3-phosphate dehydrogenase activity and thiol status after exposure to hydroperoxides. Arch. Biochem. Biophys. 1990, 276, 212–218. [Google Scholar] [CrossRef]
- Reddy, S.; Jones, A.D.; Cross, C.E.; Wong, P.S.-Y.; Van Der Vliet, A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem. J. 2000, 347, 821–827. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Zhao, T.J.; Yan, Y.-B.; Liu, Y.; Zhou, H.-M. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J. Biol. Chem. 2007, 282, 12022–12029. [Google Scholar] [CrossRef]
- Niggli, E. Localised Intracellular Calcium Signalling in Muscle: Calcium Sparks and Calcium Quarks. Annu. Rev. Physiol. 1999, 61, 311–335. [Google Scholar] [CrossRef]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef]
- Gordienko, D.V.; Bolton, T.B.; Cannell, M.B. Variability in spontaneous subcellular calcium release in guinea-pig ileum smooth muscle cells. J. Physiol. 1998, 507, 707–720. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy is a HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 2008, 294, C460–C466. [Google Scholar] [CrossRef]
- Higuchi, M.; Honda, T.; Proske, R.J.; Yeh, E.T. Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene 1998, 17, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Kozlovsky, N.; Rudich, A.; Potashnik, R.; Bashan, N. Reactive oxygen species activate glucose transport in L6 myotubes. Free Radic. Biol. Med. 1997, 23, 859–869. [Google Scholar] [CrossRef]
- Kim, J.S.; Saengsirisuwan, V.; Sloniger, J.A.; Teachey, M.K.; Henriksen, E.J. Oxidant stress and skeletal muscle glucose transport: Roles of insulin signaling and p38 MAPK. Free Radic. Biol. Med. 2006, 41, 818–824. [Google Scholar] [CrossRef]
- Sandström, M.E.; Zhang, S.; Bruton, J.; Silva, J.P.; Reid, M.B.; Westerblad, H.; Katz, A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J. Physiol. 2006, 575, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Tanaka, S.; Ebihara, K.; Masuzaki, H.; Hosoda, K.; Sato, K.; Fushiki, T.; Nakao, K.; Hayashi, T. Low-intensity contraction activates the α1-isoform of 5′-AMP-activated protein kinase in rat skeletal muscle. Am. J. Physiol. Metab. 2006, 290, E583–E590. [Google Scholar] [CrossRef]
- Toyoda, T.; Hayashi, T.; Miyamoto, L.; Yonemitsu, S.; Nakano, M.; Tanaka, S.; Ebihara, K.; Masuzaki, H.; Hosoda, K.; Inoue, G.; et al. Possible involvement of the α1 isoform of 5′AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am. J. Physiol. Metab. 2004, 287, E166–E173. [Google Scholar] [CrossRef] [PubMed]
- Fryer, L.G.; Foufelle, F.; Barnes, K.; Baldwin, S.A.; Woods, A.; Carling, D. Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem. J. 2002, 363, 167–174. [Google Scholar] [CrossRef]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 1993, 75, 1081–1087. [Google Scholar] [CrossRef]
- Gao, Y.J.; Lee, R.M.K.W. Hydrogen peroxide induces a greater contraction in mesenteric arteries of spontaneously hypertensive rats through thromboxane A2 production. Br. J. Pharmacol. 2001, 134, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, G.; Pedrozo, Z.; Domenech, R.J.; Hidalgo, C.; Donoso, P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J. Mol. Cell. Cardiol. 2005, 39, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef]
- Hidalgo, C.; Sánchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Close, G.L.; Kayani, A.; McArdle, A.; Vina, J.; Jackson, M.J. Effect of xanthine oxidase-generated extracellular superoxide on skeletal muscle force generation. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R2–R8. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.S.; Powers, S.K.; Rowell, B.; Hamilton, K.L.; Dodd, S.L.; Shanely, R.A.; Sen, C.K.; Packer, L. Effects of vitamin E and α-lipoic acid on skeletal muscle contractile properties. J. Appl. Physiol. 2001, 90, 1424–1430. [Google Scholar] [CrossRef]
- Khawli, F.A.; Reid, M.B. N-acetylcysteine depresses contractile function and inhibits fatigue of diaphragm in vitro. J. Appl. Physiol. 1994, 77, 317–324. [Google Scholar] [CrossRef]
- Reid, M.B.; Stokic, D.; Koch, S.M.; Khawli, F.A.; Leis, A.A. N-acetylcysteine inhibits muscle fatigue in humans. J. Clin. Investig. 1994, 94, 2468–2474. [Google Scholar] [CrossRef]
- McClellan, G.; Weisberg, A.; Winegrad, S. Endothelin regulation of cardiac contractility in absence of added endothelin. Am. J. Physiol. Circ. Physiol. 1995, 268, H1621–H1627. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhou, Q.; Yao, F.; O’rourke, S.T.; Sun, C. Endothelin-1 regulates cardiac L-type calcium channels via NAD(P)H oxidase-derived superoxide. J. Pharmacol. Exp. Ther. 2008, 326, 732–738. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Fukuda, S.; Otani, H.; Zhu, L.; Yamaura, G.; Engelman, R.M.; Das, D.K.; Maulik, N. Hypoxic preconditioning triggers myocardial angiogenesis: A novel approach to enhance contractile functional reserve in rat with myocardial infarction. J. Mol. Cell. Cardiol. 2002, 34, 335–348. [Google Scholar] [CrossRef]
- Koch, A.; Cho, M.; Burrows, J.; Polverini, P.; Leibovich, S. Inhibition of production of monocyte/macrophage-derived angiogenic activity by oxygen free-radical scavengers. Cell Biol. Int. Rep. 1992, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Cho, M.; Burrows, J.; Leibovich, S.; Polverini, P.J. Inhibition of production of macrophage-derived angiogenic activity by the anti-rheumatic agents gold sodium thiomalate and auranofin. Biochem. Biophys. Res. Commun. 1988, 154, 205–212. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, K.E.; Koh, G.Y.; Ho, Y.-S.; Lee, K.J. Hydrogen peroxide produced by angiopoietin-1 mediates angiogenesis. Cancer Res. 2006, 66, 6167–6174. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-X.; Zeng, H.; Lawrence, M.L.; Blackwell, T.S.; Meyrick, B. Angiopoietin-1-induced angiogenesis is modulated by endothelial NADPH oxidase. Am. J. Physiol. Circ. Physiol. 2006, 291, H1563–H1572. [Google Scholar] [CrossRef]
- Seshiah, P.N. Angiotensin II stimulation of NAD(P)H oxidase activity: Upstream mediators. Circ. Res. 2002, 91, 406–413. [Google Scholar] [CrossRef]
- Hao, L.; Nishimura, T.; Wo, H.; Fernandez-Patron, C. Vascular responses to α1-adrenergic receptors in small rat mesenteric arteries depend on mitochondrial reactive oxygen species. Arter. Thromb. Vasc. Biol. 2006, 26, 819–825. [Google Scholar] [CrossRef]
- Bleeke, T.; Zhang, H.; Madamanchi, N.; Patterson, C.; Faber, J.E. Catecholamine-Induced Vascular Wall Growth Is Dependent on Generation of Reactive Oxygen Species. Circ. Res. 2004, 94, 37–45. [Google Scholar] [CrossRef]
- Mueller, C.; Baudler, S.; Welzel, H.; Böhm, M.; Nickenig, G. Identification of a Novel Redox-Sensitive Gene, Id3, Which Mediates Angiotensin II–Induced Cell Growth. Circulation 2002, 105, 2423–2428. [Google Scholar] [CrossRef]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, R.W. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 2005, 111, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Norton, C.E.; Broughton, B.R.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxidants Redox Signal. 2013, 18, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.R.S.; Jernigan, N.L.; Norton, C.E.; Walker, B.R.; Resta, T.C. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L232. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Fernandes, D.C.; Amanso, A.M.; Lopes, L.R.; Santos, C.X. Novel role of protein disulfide isomerase in the regulation of NADPH oxidase activity: Pathophysiological implications in vascular diseases. Antioxidants Redox Signal. 2008, 10, 1101–1113. [Google Scholar] [CrossRef]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.C.; Laurindo, F.R.M. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef]
- Fernandes, D.C.; Heloisa, A.; Manoel, O.; Wosniak, J.; Laurindo, F.R. Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: Effects of nitrosothiol exposure. Arch. Biochem. Biophys. 2009, 484, 197–204. [Google Scholar] [CrossRef]
- Wosniak, J.; Santos, C.X.C.; Kowaltowski, A.J.; Laurindo, F.R.M. Cross-talk between mitochondria and NaDPH oxidase: Effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in nox Isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 2009, 11, 1265–1278. [Google Scholar] [CrossRef]
- Greger, R. Physiology of renal sodium transport. Am. J. Med. Sci. 2000, 319, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, P.A.; Garvin, J.L. Interaction of O2− and NO in the Thick Ascending Limb. Hypertension 2002, 39, 591–596. [Google Scholar] [CrossRef]
- Ortiz, P.A.; Garvin, J.L. Superoxide stimulates NaCl absorption by the thick ascending limb. Am. J. Physiol. Physiol. 2002, 283, F957–F962. [Google Scholar] [CrossRef]
- Plato, C.F.; Stoos, B.A.; Wang, D.; Garvin, J.L. Endogenous nitric oxide inhibits chloride transport in the thick ascending limb. Am. J. Physiol. Ren. Physiol. 1999, 276, F159–F163. [Google Scholar] [CrossRef]
- Silva, G.B.; Garvin, J.L. Rac1 mediates NaCl-induced superoxide generation in the thick ascending limb. Am. J. Physiol. Ren. Physiol. 2010, 298, F421–F425. [Google Scholar] [CrossRef]
- Liu, R.; Garvin, J.L.; Ren, Y.; Pagano, P.J.; Carretero, O.A. Depolarization of the macula densa induces superoxide production via NAD(P)H oxidase. Am. J. Physiol. Ren. Physiol. 2007, 292, F1867–F1872. [Google Scholar] [CrossRef]
- Liu, R.; Juncos, L.A. GTPase-Rac enhances depolarization-induced superoxide production by the macula densa during tubuloglomerular feedback. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R453–R458. [Google Scholar] [CrossRef]
- Mori, T.; Cowley, A.W. Renal oxidative stress in medullary thick ascending limbs produced by elevated NaCl and glucose. Hypertension 2004, 43, 341–346. [Google Scholar] [CrossRef]
- Garvin, J.L.; Hong, N.J. Cellular stretch increases superoxide production in the thick ascending limb. Hypertension 2008, 51, 488–493. [Google Scholar] [CrossRef]
- Abe, M.; O’Connor, P.; Kaldunski, M.; Liang, M.; Roman, R.J.; Cowley, A.W. Effect of sodium delivery on superoxide and nitric oxide in the medullary thick ascending limb. Am. J. Physiol. Physiol. 2006, 291, 350–357. [Google Scholar] [CrossRef]
- Hong, N.J.; Garvin, J.L. Flow increases superoxide production by NADPH oxidase via activation of Na-K-2Cl cotransport and mechanical stress in thick ascending limbs. Am. J. Physiol. Physiol. 2007, 292, F993–F998. [Google Scholar] [CrossRef]
- Hong, N.J.; Garvin, J.L. Endogenous Flow-Induced superoxide stimulates Na/H exchange activity via PKC in thick ascending limbs. Am. J. Physiol. Physiol. 2014, 307, F800–F805. [Google Scholar] [CrossRef]
- Hong, N.J.; Silva, G.B.; Garvin, J.L. PKC-α mediates flow-stimulated superoxide production in thick ascending limbs. Am. J. Physiol. Physiol. 2010, 298, F885–F891. [Google Scholar] [CrossRef]
- Silva, G.B.; Ortiz, P.A.; Hong, N.J.; Garvin, J.L. Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension 2006, 48, 467–472. [Google Scholar] [CrossRef]
- Juncos, R.; Garvin, J.L. Superoxide enhances Na-K-2Cl cotransporter activity in the thick ascending limb. Am. J. Physiol. Physiol. 2005, 288, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Klann, E.; Roberson, E.D.; Knapp, L.T.; Sweatt, J.D. A Role for Superoxide in Protein Kinase C Activation and Induction of Long-term Potentiation. J. Biol. Chem. 1998, 273, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Knapp, L.T.; Klann, E. Superoxide-induced Stimulation of Protein Kinase C via Thiol Modification and Modulation of Zinc Content. J. Biol. Chem. 2000, 275, 24136–24145. [Google Scholar] [CrossRef]
- Hannemann, A.; Flatman, P.W. Phosphorylation and Transport in the Na-K-2Cl Cotransporters, NKCC1 and NKCC2A, Compared in HEK-293 Cells. PLoS ONE 2011, 6, e17992. [Google Scholar] [CrossRef]
- Liu, R.; Ren, Y.; Garvin, J.L.; Carretero, O.A. Superoxide enhances tubuloglomerular feedback by constricting the afferent arteriole. Kidney Int. 2004, 66, 268–274. [Google Scholar] [CrossRef]
- Ren, Y.; Carretero, O.A.; Garvin, J.L. Mechanism by Which Superoxide Potentiates Tubuloglomerular Feedback. Hypertension 2002, 39, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Harding, P.; Garvin, J.L.; Juncos, R.; Peterson, E.; Juncos, L.A.; Liu, R. Isoforms and functions of NAD(P)H oxidase at the macula densa. Hypertension 2009, 53, 556–563. [Google Scholar] [CrossRef]
- Lőrincz, Á.M.; Szarvas, G.; Smith, S.M.; Ligeti, E. Role of Rac GTPase activating proteins in regulation of NADPH oxidase in human neutrophils. Free Radic. Biol. Med. 2014, 68, 65–71. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Welch, W.J. Interaction between nitric oxide and oxygen radicals in regulation of tubuloglomerular feedback. in Acta Physiol. Scand. 2000, 168, 119–124. [Google Scholar] [CrossRef]
- Milev, N.B.; Reddy, A.B. Circadian redox oscillations and metabolism. Trends Endocrinol. Metab. 2015, 26, 430–437. [Google Scholar] [CrossRef]
- Edgar, R.S.; Green, E.W.; Zhao, Y.; van Ooijen, G.; Olmedo, M.; Qin, X.; Xu, Y.; Pan, M.; Valekunja, U.K.; Feeney, K.A.; et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature 2012, 485, 459–464. [Google Scholar] [CrossRef]
- Kil, I.S.; Lee, S.K.; Ryu, K.W.; Woo, H.A.; Hu, M.-C.; Bae, S.H.; Rhee, S.G. Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol. Cell 2012, 46, 584–594. [Google Scholar] [CrossRef]
- Luther, J.M. Effects of aldosterone on insulin sensitivity and secretion. Steroids 2014, 91, 54–60. [Google Scholar] [CrossRef]
- Wada, T.; Ohshima, S.; Fujisawa, E.; Koya, D.; Tsuneki, H.; Sasaoka, T. Aldosterone Inhibits Insulin-Induced Glucose Uptake by Degradation of Insulin Receptor Substrate (IRS) 1 and IRS2 via a Reactive Oxygen Species-Mediated Pathway in 3T3-L1 Adipocytes. Endocrinology 2008, 150, 1662–1669. [Google Scholar] [CrossRef]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Chen, Q.; Samidurai, A.; Thompson, J.; Hu, Y.; Das, A.; Willard, B.; Lesnefsky, E.J. Endoplasmic reticulum stress-mediated mitochondrial dysfunction in aged hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165899. [Google Scholar] [CrossRef]
- Zarse, K.; Schmeisser, S.; Groth, M.; Priebe, S.; Beuster, G.; Kuhlow, D.; Guthke, R.; Platzer, M.; Kahn, C.R.; Ristow, M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012, 15, 451–465. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Brewer, G.J. Epigenetic oxidative redox shift (EORS) theory of aging unifies the free radical and insulin signaling theories. Exp. Gerontol. 2009, 45, 173–179. [Google Scholar] [CrossRef]
- Zámbó, V.; Simon-Szabó, L.; Szelényi, P.; Kereszturi, É.; Bánhegyi, G.; Csala, M. Lipotoxicity in the liver. World J. Hepatol. 2013, 5, 550–557. [Google Scholar] [CrossRef]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef]
- Prentki, M.; Corkey, B.E. Are the β-cell signaling molecules malonyl-CoA and cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes 1996, 45, 273–283. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Brinkmann, J.F.; Abumrad, N.A.; Ibrahimi, A.; Vandervusse, G.J.; Glatz, J.F. New insights into long-chain fatty acid uptake by heart muscle: A crucial role for fatty acid translocase/CD36. Biochem. J. 2002, 367, 561–570. [Google Scholar] [CrossRef]
- Befroy, D.E.; Perry, R.J.; Jain, N.; Dufour, S.; Cline, G.W.; Trimmer, J.K.; Brosnan, J.; Rothman, D.L.; Petersen, K.F.; I Shulman, G. Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13 C magnetic resonance spectroscopy. Nat. Med. 2013, 20, 98–102. [Google Scholar] [CrossRef]
- Magnusson, I.; Schumann, W.C.; E Bartsch, G.; Chandramouli, V.; Kumaran, K.; Wahren, J.; Landau, B.R. Noninvasive tracing of Krebs cycle metabolism in liver. J. Biol. Chem. 1991, 266, 6975–6984. [Google Scholar] [CrossRef] [PubMed]
- Nieth, H.; Schollmeyer, P. Substrate Utilization of the Human Kidney. Nature 1966, 209, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Gorp, P.J.; Bieghs, V.; Gijbels, M.J.; Duimel, H.; Lütjohann, D.; Kerksiek, A.; van Kruchten, R.; Maeda, N.; Staels, B.; et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 2008, 48, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Pang, P.S. Acute Heart Failure Syndromes. J. Am. Coll. Cardiol. 2009, 53, 557–573. [Google Scholar] [CrossRef]
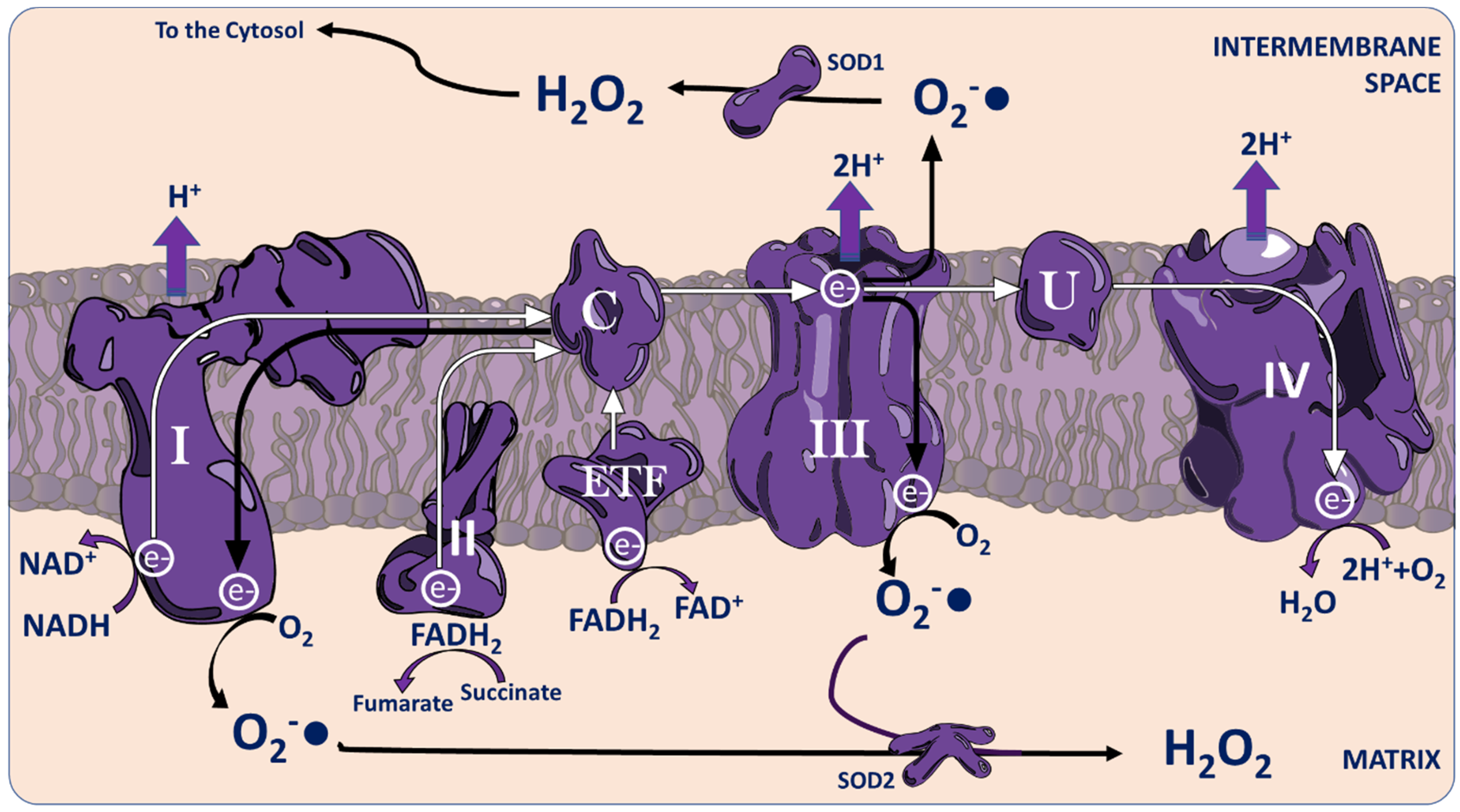
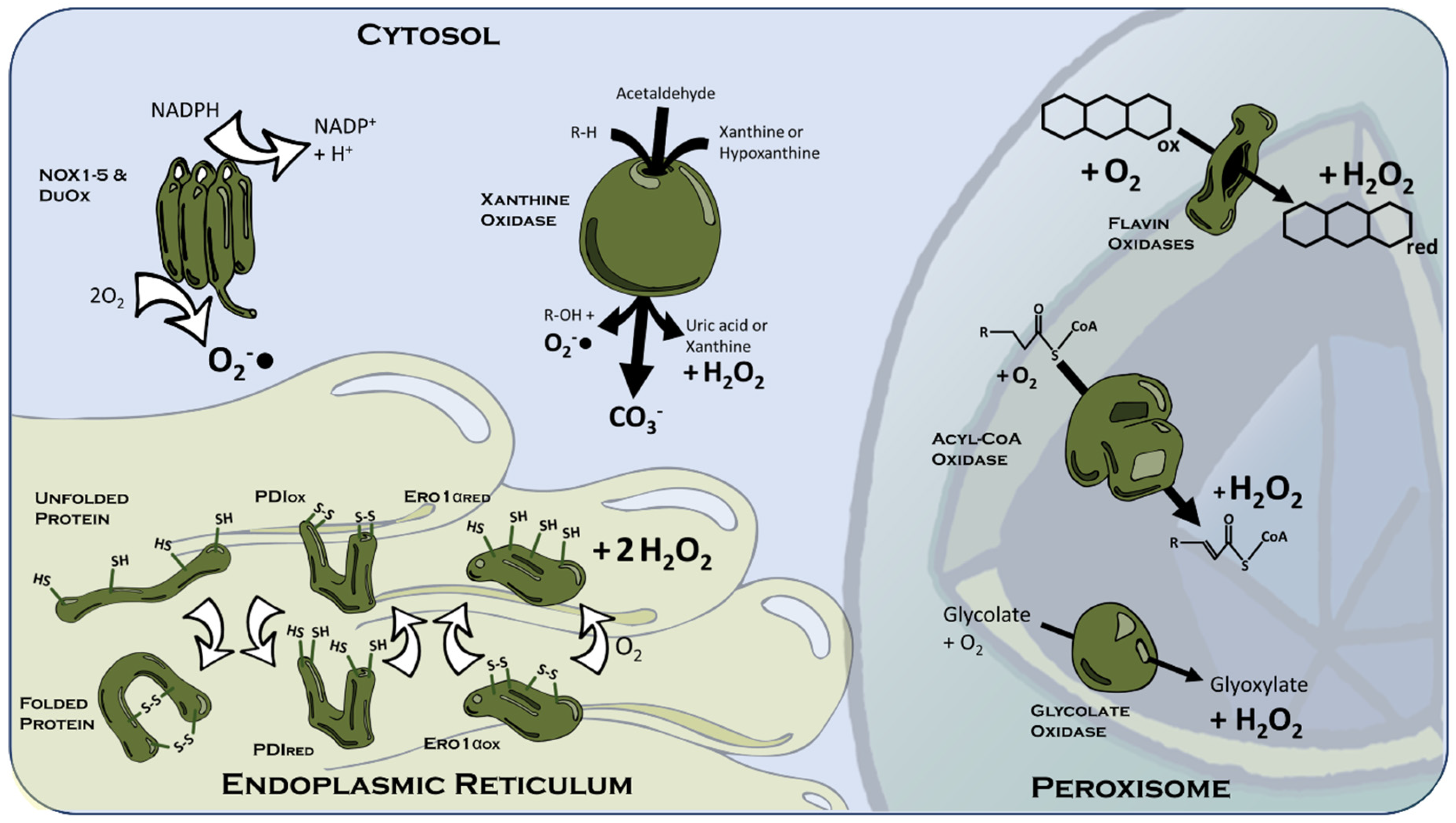


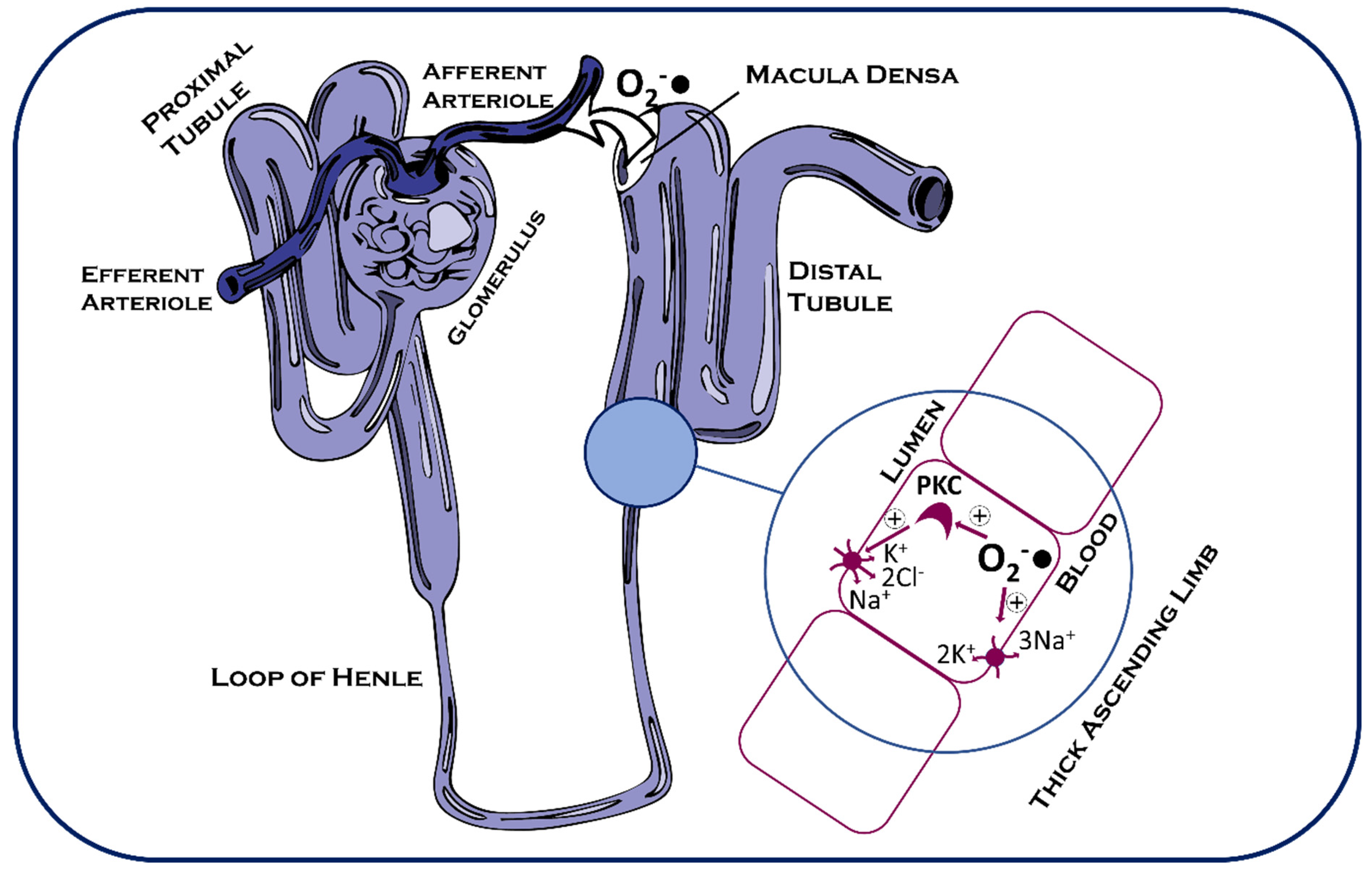

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ROS | Chemical Composition | Reduction Potential (V) |
|---|---|---|
| Oxygen | O2 | −0.35 |
| Superoxide | O2−● | 0.94 |
| Hydroxyl radical | OH● | 2.31 |
| Hydrogen peroxide | H2O2 | 0.32 |
| Carbonate radical | CO3−● | 1.78 |
| Alkoxyl radical | R-O● | 1.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindsay, R.T.; Rhodes, C.J. Reactive Oxygen Species (ROS) in Metabolic Disease—Don’t Shoot the Metabolic Messenger. Int. J. Mol. Sci. 2025, 26, 2622. https://doi.org/10.3390/ijms26062622
Lindsay RT, Rhodes CJ. Reactive Oxygen Species (ROS) in Metabolic Disease—Don’t Shoot the Metabolic Messenger. International Journal of Molecular Sciences. 2025; 26(6):2622. https://doi.org/10.3390/ijms26062622
Chicago/Turabian StyleLindsay, Ross T., and Christopher J. Rhodes. 2025. "Reactive Oxygen Species (ROS) in Metabolic Disease—Don’t Shoot the Metabolic Messenger" International Journal of Molecular Sciences 26, no. 6: 2622. https://doi.org/10.3390/ijms26062622
APA StyleLindsay, R. T., & Rhodes, C. J. (2025). Reactive Oxygen Species (ROS) in Metabolic Disease—Don’t Shoot the Metabolic Messenger. International Journal of Molecular Sciences, 26(6), 2622. https://doi.org/10.3390/ijms26062622