
Synergistic Effect of Ribitol and Shikonin Promotes Apoptosis in Breast Cancer Cells

Abstract

1. Introduction
2. Results
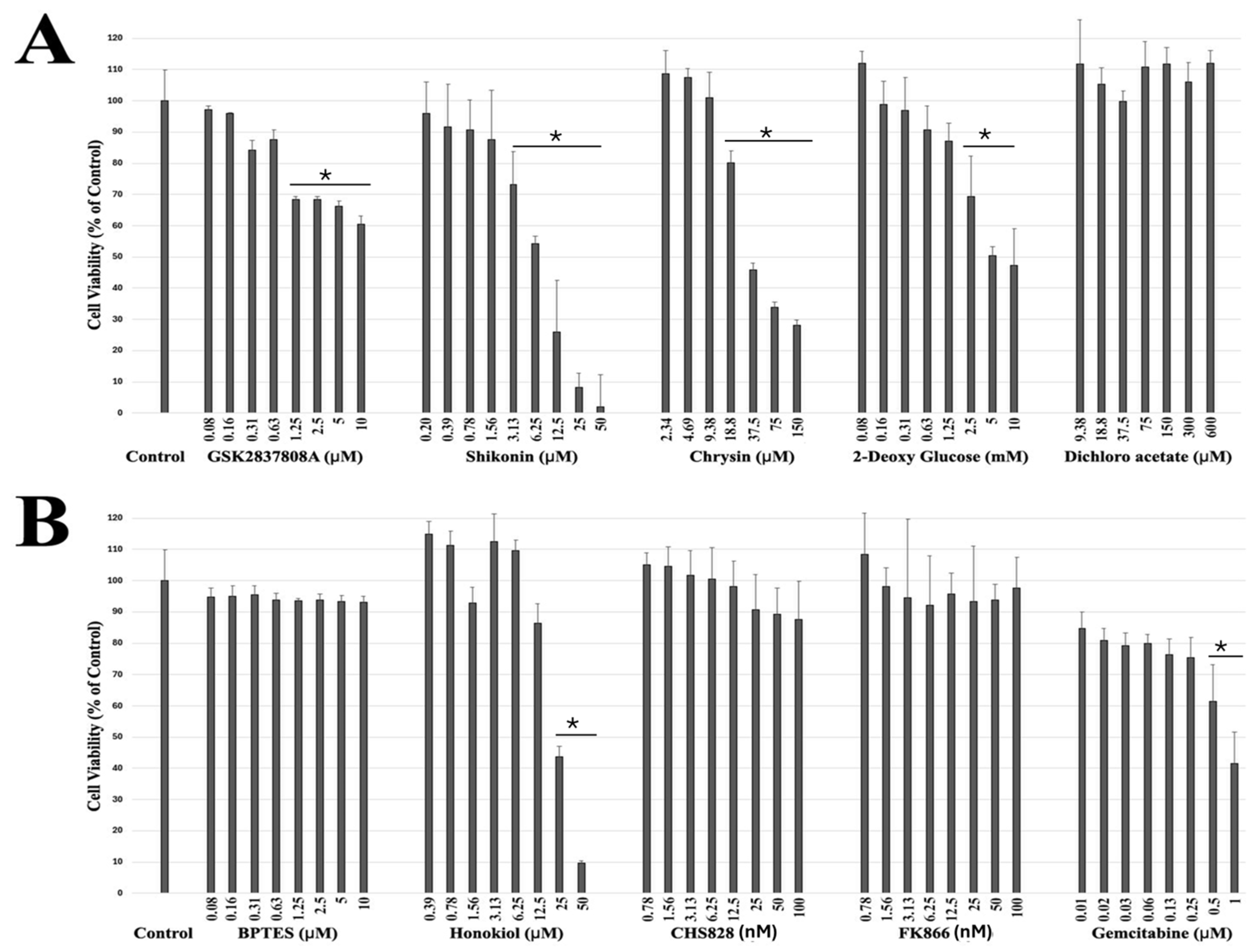
2.1. Drug Screening of Breast Cancer Cells Reveals Potential Targeted Therapeutic Candidates
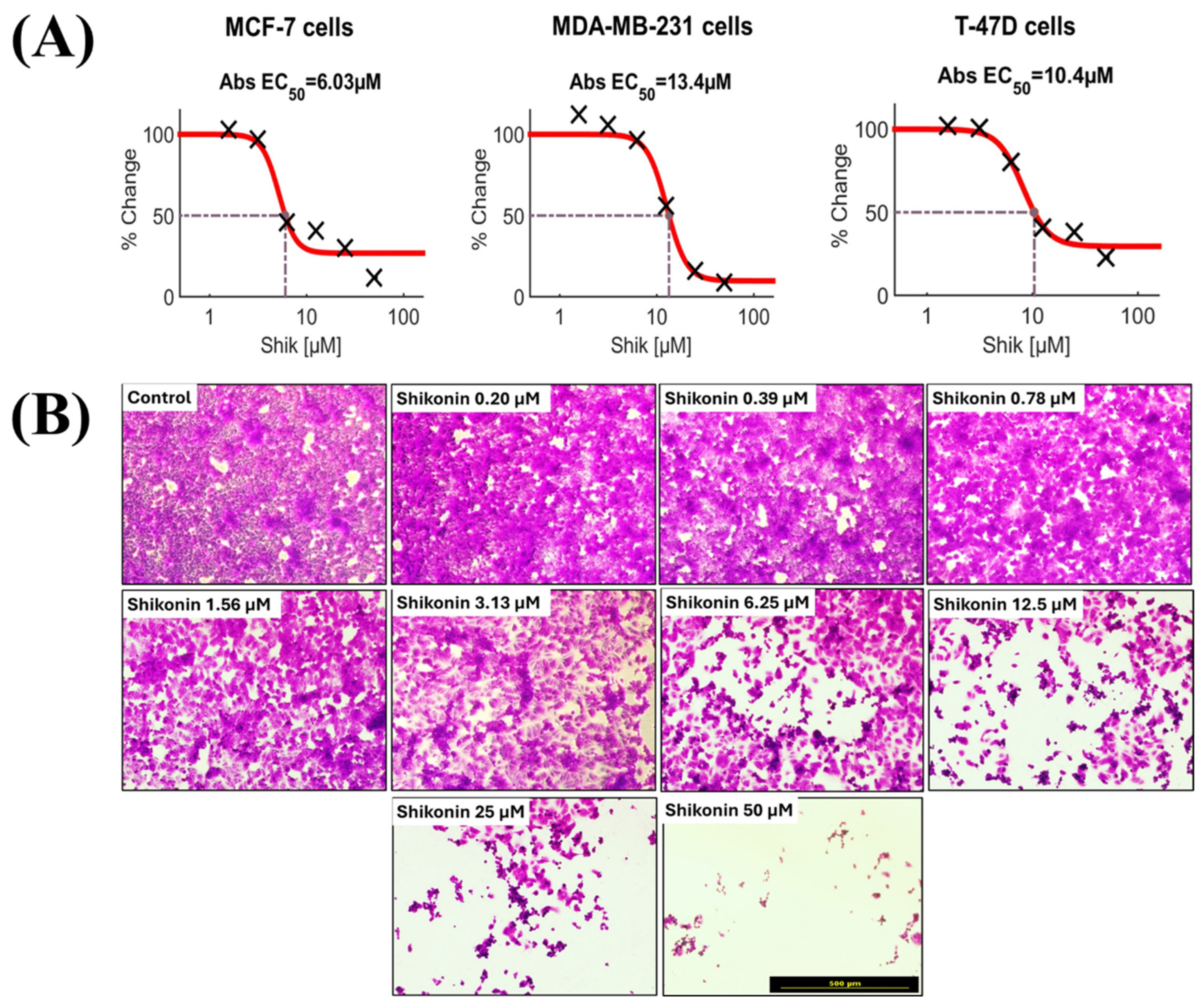
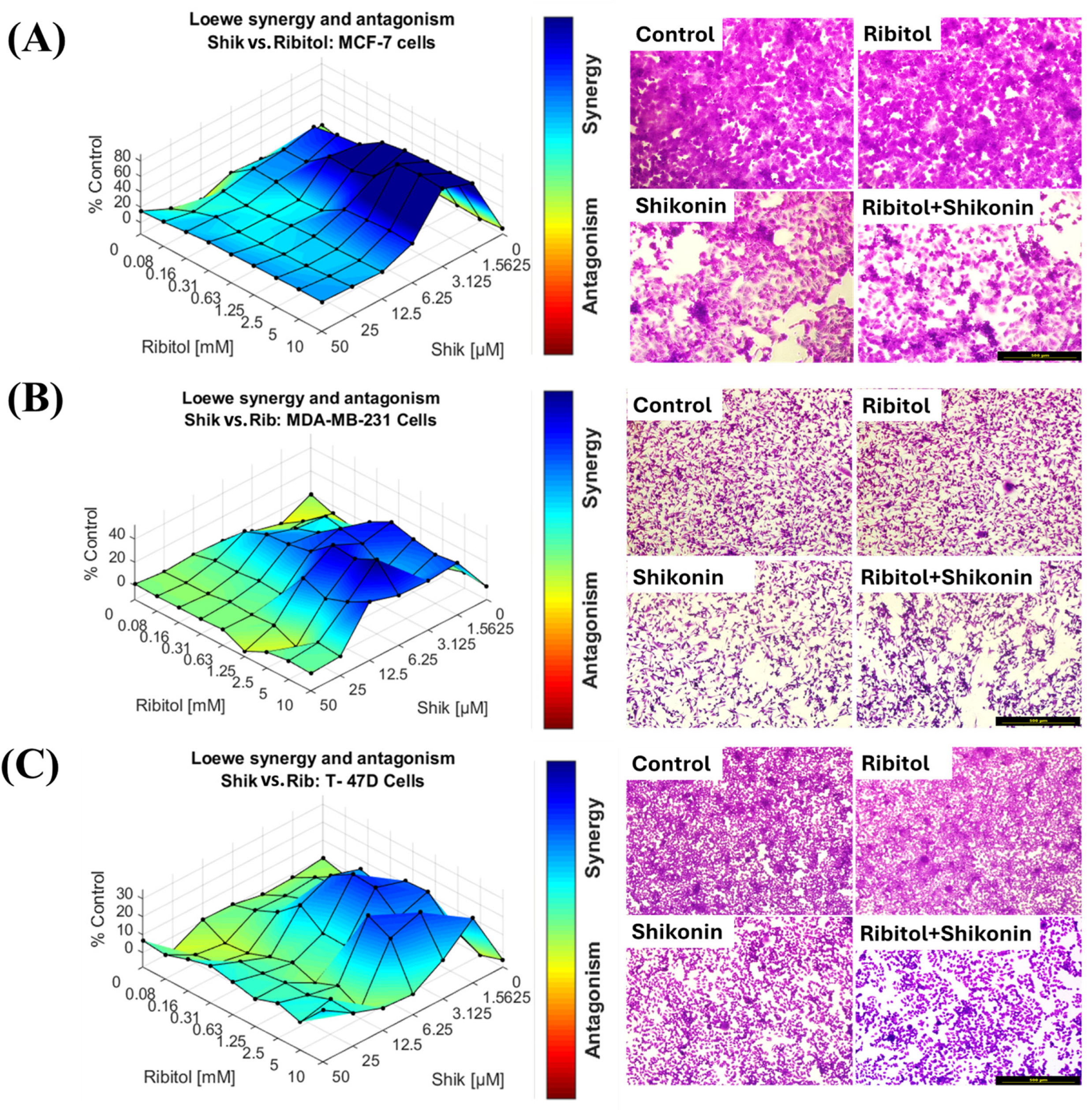
2.2. Effect of Ribitol in Combination with Shikonin on Breast Cancer Cells
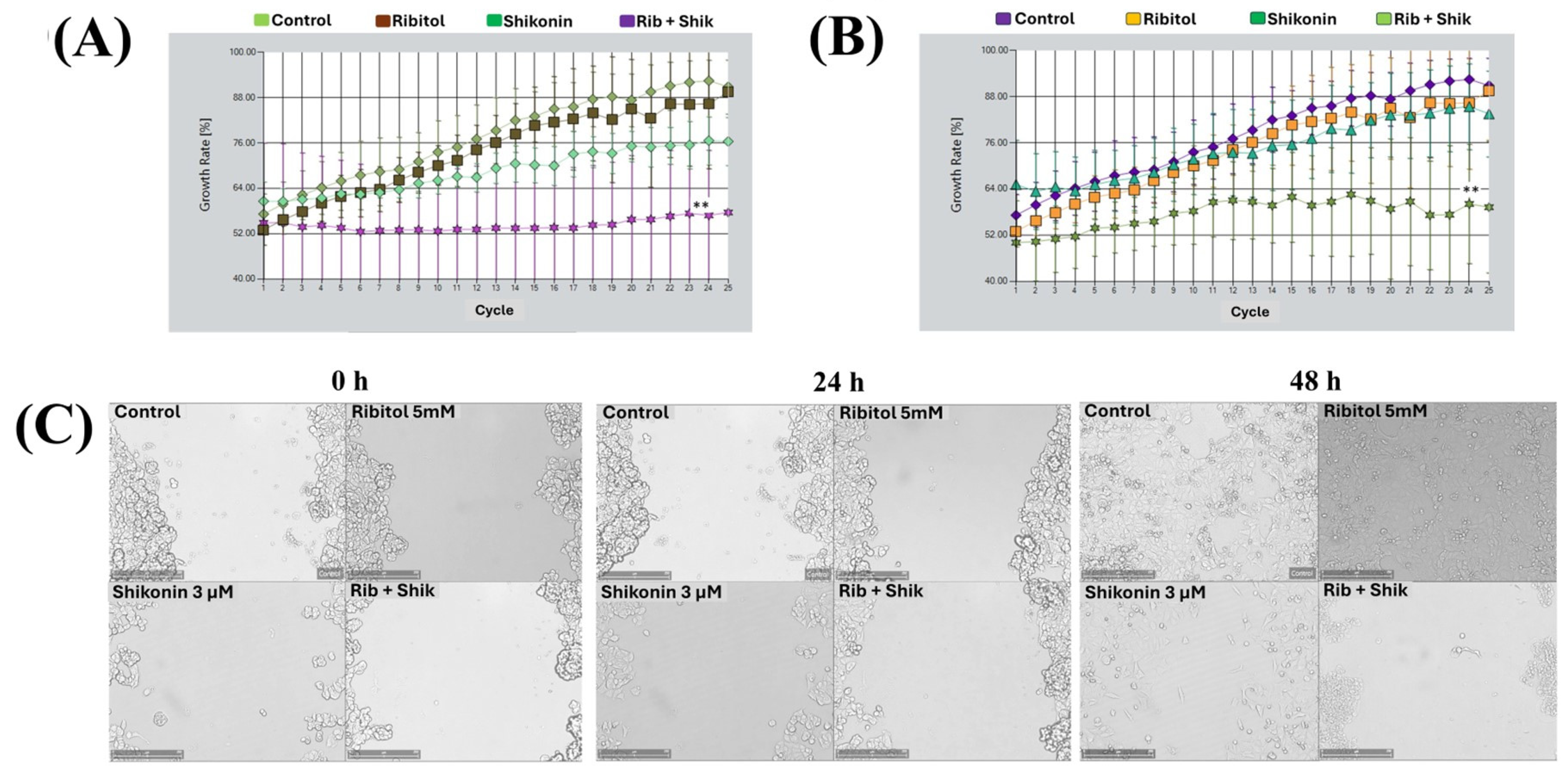
2.3. Combination of Ribitol and Shikonin Treatment Inhibits MCF-7 Cell Proliferation
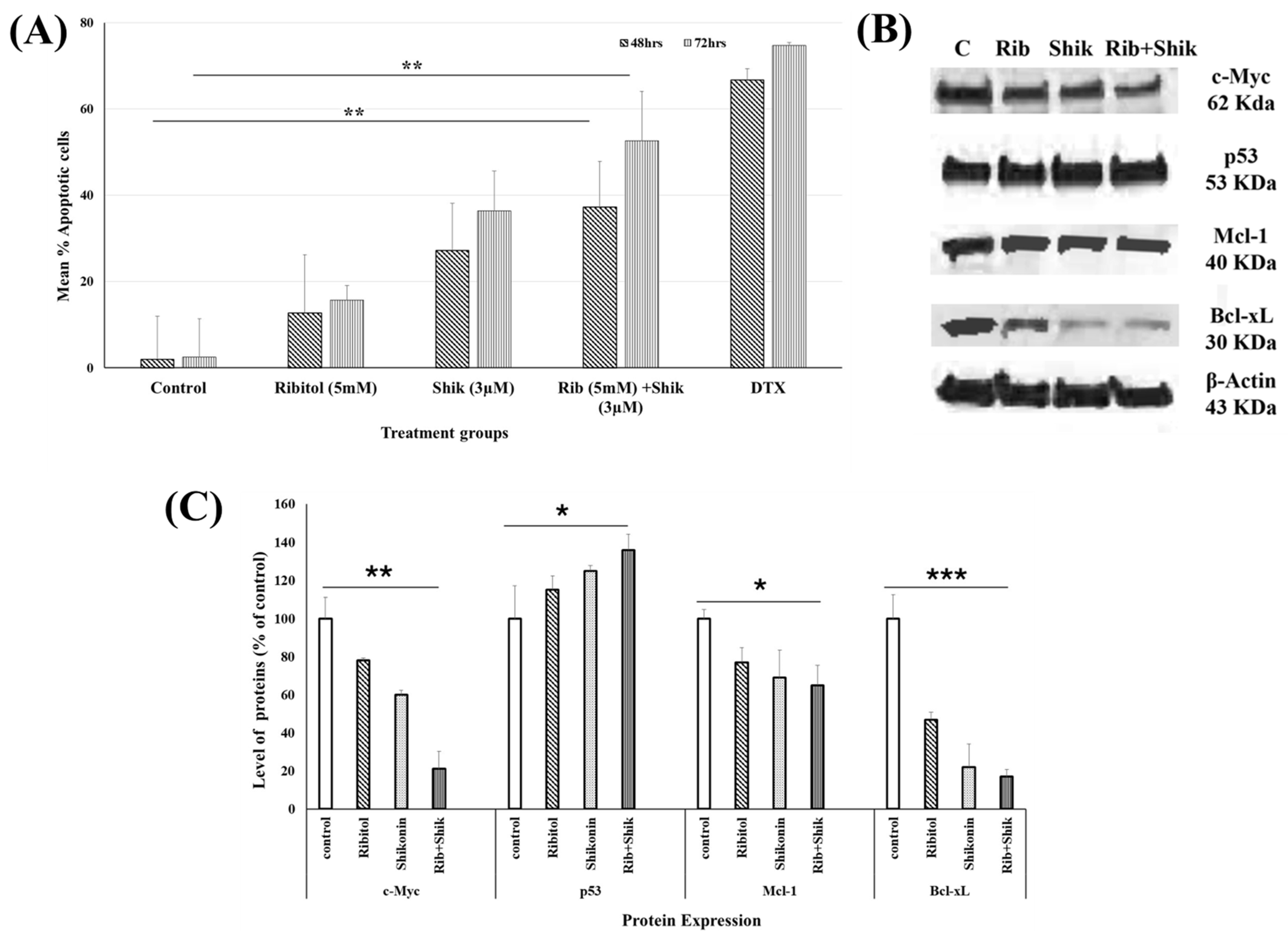
2.4. Ribitol Addition Augments Shikonin-Induced Apoptosis
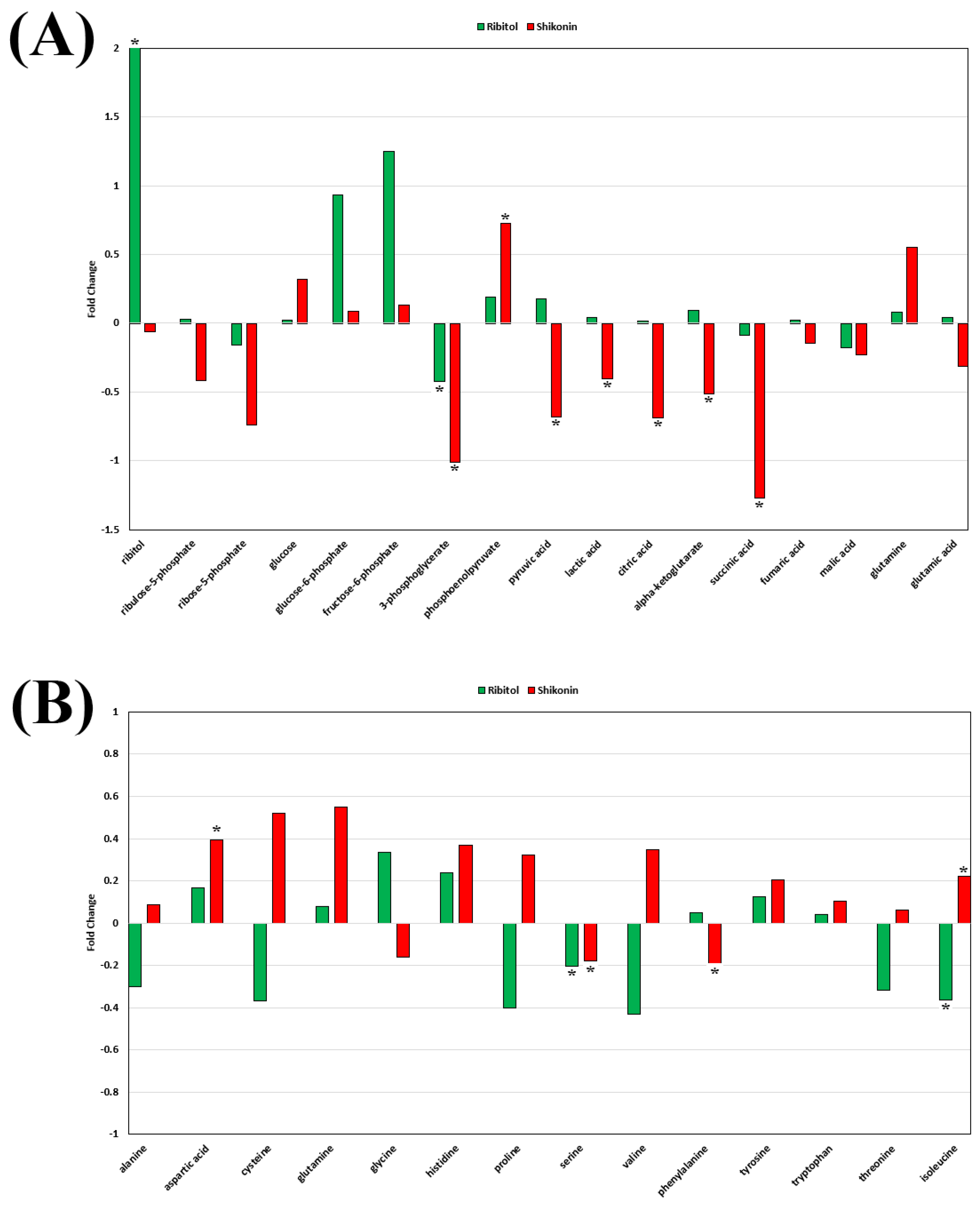
2.5. Effect of Ribitol and Shikonin on Metabolic Reprogramming of Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Compound Screening
4.3. Cell Viability Assay and Combenefit Synergy Software 2.021
4.4. Proliferation Assay
4.5. Real Time-Glo™ Annexin V Apoptosis Assay
4.6. Western Blot
4.7. Metabolomics
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Kubczak, M.; Szustka, A.; Rogalinska, M. Molecular Targets of Natural Compounds with Anti-Cancer Properties. Int. J. Mol. Sci. 2021, 22, 13659. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Su, H.; Zhou, B.; Liu, S. The function of natural compounds in important anticancer mechanisms. Front. Oncol. 2022, 12, 1049888. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in Breast Cancer Survival by Molecular Subtypes in the United States. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal 2024, 22, 109. [Google Scholar] [CrossRef]
- Saha, M.; Sarkar, A. Review on Multiple Facets of Drug Resistance: A Rising Challenge in the 21st Century. J. Xenobiot. 2021, 11, 197–214. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Di Nardo, P.; Lisanti, C.; Garutti, M.; Buriolla, S.; Alberti, M.; Mazzeo, R.; Puglisi, F. Chemotherapy in patients with early breast cancer: Clinical overview and management of long-term side effects. Expert. Opin. Drug Saf. 2022, 21, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tufail, M.; Jiang, C.H.; Li, N. Altered metabolism in cancer: Insights into energy pathways and therapeutic targets. Mol. Cancer 2024, 23, 203. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef]
- Cataldi, M.P.; Lu, P.; Blaeser, A.; Lu, Q.L. Ribitol restores functionally glycosylated alpha-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat. Commun. 2018, 9, 3448. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.P.; Vannoy, C.H.; Blaeser, A.; Tucker, J.D.; Leroy, V.; Rawls, R.; Killilee, J.; Holbrook, M.C.; Lu, Q.L. Improved efficacy of FKRP AAV gene therapy by combination with ribitol treatment for LGMD2I. Mol. Ther. 2023, 31, 3478–3489. [Google Scholar] [CrossRef]
- Lu, P.J.; Tucker, J.D.; Branch, E.K.; Guo, F.; Blaeser, A.R.; Lu, Q.L. Ribitol enhances matriglycan of alpha-dystroglycan in breast cancer cells without affecting cell growth. Sci. Rep. 2020, 10, 4935. [Google Scholar] [CrossRef]
- Tucker, J.D.; Doddapaneni, R.; Lu, P.J.; Lu, Q.L. Ribitol alters multiple metabolic pathways of central carbon metabolism with enhanced glycolysis: A metabolomics and transcriptomics profiling of breast cancer. PLoS ONE 2022, 17, e0278711. [Google Scholar] [CrossRef]
- Doddapaneni, R.; Tucker, J.D.; Lu, P.J.; Lu, Q.L. Metabolic Reprogramming by Ribitol Expands the Therapeutic Window of BETi JQ1 against Breast Cancer. Cancers 2023, 15, 4356. [Google Scholar] [CrossRef]
- Zhang, C.H.; Wang, J.; Zhang, L.X.; Lu, Y.H.; Ji, T.H.; Xu, L.; Ling, L.J. Shikonin reduces tamoxifen resistance through long non-coding RNA uc.57. Oncotarget 2017, 8, 88658–88669. [Google Scholar] [CrossRef]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Giaquinto, A.N.; Sung, H.; Newman, L.A.; Freedman, R.A.; Smith, R.A.; Star, J.; Jemal, A.; Siegel, R.L. Breast cancer statistics 2024. CA Cancer J. Clin. 2024, 74, 477–495. [Google Scholar] [CrossRef]
- Ginsburg, O.; Bray, F.; Coleman, M.P.; Vanderpuye, V.; Eniu, A.; Kotha, S.R.; Sarker, M.; Huong, T.T.; Allemani, C.; Dvaladze, A.; et al. The global burden of women’s cancers: A grand challenge in global health. Lancet 2017, 389, 847–860. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef]
- Guo, L.; Kong, D.; Liu, J.; Zhan, L.; Luo, L.; Zheng, W.; Zheng, Q.; Chen, C.; Sun, S. Breast cancer heterogeneity and its implication in personalized precision therapy. Exp. Hematol. Oncol. 2023, 12, 3. [Google Scholar] [CrossRef]
- Weigelt, B.; Reis-Filho, J.S. Histological and molecular types of breast cancer: Is there a unifying taxonomy? Nat. Rev. Clin. Oncol. 2009, 6, 718–730. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100110. [Google Scholar] [CrossRef]
- Wang, Y.; Minden, A. Current Molecular Combination Therapies Used for the Treatment of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 11046. [Google Scholar] [CrossRef]
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer Agents Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef]
- Andujar, I.; Recio, M.C.; Giner, R.M.; Rios, J.L. Traditional chinese medicine remedy to jury: The pharmacological basis for the use of shikonin as an anticancer therapy. Curr. Med. Chem. 2013, 20, 2892–2898. [Google Scholar] [CrossRef]
- Singla, R.K.; Wang, X.; Gundamaraju, R.; Joon, S.; Tsagkaris, C.; Behzad, S.; Khan, J.; Gautam, R.; Goyal, R.; Rakmai, J.; et al. Natural products derived from medicinal plants and microbes might act as a game-changer in breast cancer: A comprehensive review of preclinical and clinical studies. Crit. Rev. Food Sci. Nutr. 2023, 63, 11880–11924. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Sha, L.; Lv, Z.; Liu, Y.; Zhang, Y.; Sui, X.; Wang, T.; Zhang, H. Shikonin inhibits the Warburg effect, cell proliferation, invasion and migration by downregulating PFKFB2 expression in lung cancer. Mol. Med. Rep. 2021, 24, 560. [Google Scholar] [CrossRef]
- Iranzadeh, S.; Dalil, D.; Kohansal, S.; Isakhani, M. Shikonin in breast cancer treatment: A comprehensive review of molecular pathways and innovative strategies. J. Pharm. Pharmacol. 2024, 76, 967–982. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, P.; Huang, H.; Chen, D.; Ma, N.; Cui, Q.C.; Shen, S.; Dong, W.; Zhang, X.; Lian, W.; et al. Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int. J. Cancer 2009, 124, 2450–2459. [Google Scholar] [CrossRef]
- Hsu, P.C.; Huang, Y.T.; Tsai, M.L.; Wang, Y.J.; Lin, J.K.; Pan, M.H. Induction of apoptosis by shikonin through coordinative modulation of the Bcl-2 family, p27, and p53, release of cytochrome c, and sequential activation of caspases in human colorectal carcinoma cells. J. Agric. Food Chem. 2004, 52, 6330–6337. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef]
- Su, Q.; Luo, S.; Tan, Q.; Deng, J.; Zhou, S.; Peng, M.; Tao, T.; Yang, X. The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol. Lett. 2019, 18, 5663–5672. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Liu, Y.; Li, J.; Jin, M.; Yang, H.; Huang, G. Shikonin inhibited glycolysis and sensitized cisplatin treatment in non-small cell lung cancer cells via the exosomal pyruvate kinase M2 pathway. Bioengineered 2022, 13, 13906–13918. [Google Scholar] [CrossRef]
- Jaiswal, E.; Globisch, C.; Jain, A. Knowledge-driven design and optimization of potent symmetric anticancer molecules: A case study on PKM2 activators. Comput. Biol. Med. 2022, 151, 106313. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, T.; Liu, M.; Ren, S.; Yang, H.; Zeng, S.; Zhao, H.; Chen, L.; Ming, T.; Meng, X.; et al. Shikonin, a naphthalene ingredient: Therapeutic actions, pharmacokinetics, toxicology, clinical trials and pharmaceutical researches. Phytomedicine 2022, 94, 153805. [Google Scholar] [CrossRef]
- Tang, J.C.; Ren, Y.G.; Zhao, J.; Long, F.; Chen, J.Y.; Jiang, Z. Shikonin enhances sensitization of gefitinib against wild-type EGFR non-small cell lung cancer via inhibition PKM2/stat3/cyclinD1 signal pathway. Life Sci. 2018, 204, 71–77. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Q. A novel antiestrogen agent Shikonin inhibits estrogen-dependent gene transcription in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 233–240. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, W.; Tao, S.; Wang, Y.; Niu, J.; Zhao, P.; Rao, C.; Yang, L. ER-mediated anti-tumor effects of shikonin on breast cancer. Eur. J. Pharmacol. 2019, 863, 172667. [Google Scholar] [CrossRef]
- Tabari, A.R.; Gavidel, P.; Sabouni, F.; Gardaneh, M. Synergy between sublethal doses of shikonin and metformin fully inhibits breast cancer cell migration and reverses epithelial-mesenchymal transition. Mol. Biol. Rep. 2022, 49, 4307–4319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yin, J.; Li, M.; Shen, J.; Xiao, Z.; Zhao, Y.; Huang, C.; Zhang, H.; Zhang, Z.; Cho, C.H.; et al. Combination of shikonin with paclitaxel overcomes multidrug resistance in human ovarian carcinoma cells in a P-gp-independent manner through enhanced ROS generation. Chin. Med. 2019, 14, 7. [Google Scholar] [CrossRef]
- Lin, H.Y.; Han, H.W.; Wang, Y.S.; He, D.L.; Sun, W.X.; Feng, L.; Wen, Z.L.; Yang, M.K.; Lu, G.H.; Wang, X.M.; et al. Shikonin and 4-hydroxytamoxifen synergistically inhibit the proliferation of breast cancer cells through activating apoptosis signaling pathway in vitro and in vivo. Chin. Med. 2020, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Study to Evaluate the Efficacy and Safety of BBP-418 (Ribitol) in Patients with Limb Girdle Muscular Dystrophy 2I (LGMD2I) (Fortify by ML Bio Solutions). Available online: https://ctv.veeva.com/study/study-to-evaluate-the-efficacy-and-safety-of-bbp-418-ribitol-in-patients-with-limb-girdle-muscular (accessed on 9 September 2024).
- Nossing, C.; Ryan, K.M. 50 years on and still very much alive: ‘Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics’. Br. J. Cancer 2023, 128, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Hao, Z.; Qian, J.; Yang, J. Shikonin induces apoptosis and inhibits migration of ovarian carcinoma cells by inhibiting the phosphorylation of Src and FAK. Oncol. Lett. 2015, 9, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Cai, A.; Chen, G.; Xi, H.; Wu, X.; Cui, J.; Zhang, K.; Zhao, X.; Yu, J.; Wei, B.; et al. Shikonin induces mitochondria-mediated apoptosis and enhances chemotherapeutic sensitivity of gastric cancer through reactive oxygen species. Sci. Rep. 2016, 6, 38267. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Yu, C.R.; Li, W.H.; Li, W.X. Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 2008, 18, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, L.; Zhang, T.; Liu, Y.; Fang, F.; Wu, X.; Chen, W.; Lan, L.; Zhang, Y.; Li, N.; et al. Shikonin inhibits the proliferation of cervical cancer cells via FAK/AKT/GSK3beta signalling. Oncol. Lett. 2022, 24, 304. [Google Scholar] [CrossRef]
- Yu, C.; Xing, H.; Fu, X.; Zhang, Y.; Yan, X.; Feng, J.; He, Z.; Ru, L.; Huang, C.; Liang, J. Effect and mechanisms of shikonin on breast cancer cells in vitro and in vivo. BMC Complement. Med. Ther. 2024, 24, 389. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Drug Name | Target |
|---|---|---|
| 1 | GSK2837808A | Lactate Dehydrogenase Inhibitor II |
| 2 | Shikonin | Pyruvate kinase M2 (PKM2) inhibitor |
| 3 | Chrysin | Targets succinate dehydrogenase (SDH) |
| 4 | 2-Deoxy-D-glucose | Glycolytic inhibitor with antiviral activity. |
| 5 | Dichloroacetate | Mitochondrial pyruvate dehydrogenase kinase (PDK) inhibitor |
| 6 | BPTES | Glutaminase GLS1 (KGA) inhibitor |
| 7 | Honokiol | Inhibitor of complexes I, II, and V of ETC |
| 8 | CHS828 | Inhibitor of nicotinamide phosphoribosyltransferase (NAMPT) |
| 9 | FK866 | Nicotinamide phosphoribosyltransferase inhibitor |
| 10 | Gemcitabine, HCl | Ribonucleotide Reductase Inhibitor II |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doddapaneni, R.; Tucker, J.D.; Lu, P.J.; Lu, Q.L. Synergistic Effect of Ribitol and Shikonin Promotes Apoptosis in Breast Cancer Cells. Int. J. Mol. Sci. 2025, 26, 2661. https://doi.org/10.3390/ijms26062661
Doddapaneni R, Tucker JD, Lu PJ, Lu QL. Synergistic Effect of Ribitol and Shikonin Promotes Apoptosis in Breast Cancer Cells. International Journal of Molecular Sciences. 2025; 26(6):2661. https://doi.org/10.3390/ijms26062661
Chicago/Turabian StyleDoddapaneni, Ravi, Jason D. Tucker, Pei J. Lu, and Qi L. Lu. 2025. "Synergistic Effect of Ribitol and Shikonin Promotes Apoptosis in Breast Cancer Cells" International Journal of Molecular Sciences 26, no. 6: 2661. https://doi.org/10.3390/ijms26062661
APA StyleDoddapaneni, R., Tucker, J. D., Lu, P. J., & Lu, Q. L. (2025). Synergistic Effect of Ribitol and Shikonin Promotes Apoptosis in Breast Cancer Cells. International Journal of Molecular Sciences, 26(6), 2661. https://doi.org/10.3390/ijms26062661